Topology of silica supported vanadium–titanium oxide catalysts for oxidative dehydrogenation of propane†
Neil
Hamilton
a,
Till
Wolfram
a,
Genka
Tzolova Müller
a,
Michael
Hävecker
b,
Jutta
Kröhnert
a,
Carlos
Carrero
c,
Reinhard
Schomäcker
c,
Annette
Trunschke
*a and
Robert
Schlögl
a
aDepartment of Inorganic Chemistry, Fritz Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
bDepartment of Solar Energy Research, Helmholtz-Zentrum Berlin/BESSY II, Albert-Einstein-Str. 15, 12489 Berlin, Germany
cTechnical University Berlin, Department of Chemistry, Strasse des 17. Juni 124, 10623 Berlin, Germany
First published on 6th March 2012
Abstract
Two-dimensional vanadia and titania surface clusters were hosted on the walls of the hierarchical pore system of mesoporous silica SBA-15. The topology of the catalyst surface was varied by sequential grafting of vanadium and titanium alkoxides generating an extended library of mixed (VOx)n–(TiOx)n/SBA-15 catalysts. The surface of the catalysts was analyzed by FTIR, UV-vis, Raman, and NEXAFS spectroscopy. Electron microscopy, X-ray fluorescence, X-ray diffraction, and nitrogen adsorption have been applied to characterize chemical composition, micro- and meso-structure of the materials. Segregation of nano-crystalline vanadia and titania particles was excluded by UV-vis, Raman and NEXAFS spectroscopy. Monolayer coverage of titanium oxide surface species has been achieved in the range between 17 and 19 wt% Ti loading corresponding to 6–8 Ti atoms per nmcat2 and Si/Ti ratios between 3.3 and 2.8. Up to a critical total metal loading, vanadia is grafted on both the silica surface and surface titania species yielding tetrahedrally coordinated vanadium oxo-species characterized by low nuclearity and moderate catalytic activity. A volcano-type dependency with respect to the propylene space-time yield has been observed in the oxidative dehydrogenation of propane. The maximum in productivity of propylene is attributed to a particular surface topology that is characterized by (VOx)n islands embedded in a matrix of dispersed titania species forming an almost complete combined vanadia–titania monolayer on the silica surface.
1. Introduction
Oxidative dehydrogenation (ODH) of light alkanes is an attractive alternative to the conventional cracking and dehydrogenation pathways for production of olefins that are currently applied in industry since the energy demand is substantially lower.1 Supported vanadia shows a comparatively high yield in oxidative dehydrogenation of propane (ODP).2,3 The role of the support in these catalysts is a topic that has provoked stimulated discussions in the literature, e.g.refs. 3–14. Apparent activation energies calculated considering the rate of propane consumption follow the trend VOx/SiO2 > VOx/Al2O3 > VOx/ZrO2 > VOx/CeO2 > VOx/TiO2.12 Vanadia supported on silica is found to be significantly more selective than that supported on alumina, which, in turn, is more selective than that supported on titania.6–8,10In an attempt to explain these observations, the contribution of support surface sites to the conversion of propane has been discussed. In particular, non-selective primary and secondary combustion reactions have been attributed to areas that are not covered by vanadia species.5,7 Differences in dispersion and local structure of vanadia surface clusters of different size9,15,16 are related to the abundance of vanadium–oxygen-support bonds, which have been considered as catalytic active sites9,13 in the rate determining step that involves the abstraction of the methylene hydrogen atom from the propane molecule.17 Acting as a ligand, the support has been supposed to affect the basicity of the bridging oxygen in S–O–V moieties (S = Si, Al, Ti, etc.) by differences in the electronegativity and therefore their reactivity.13 Given that the oxidative dehydrogenation of propane requires the transfer of four electrons, the electronic properties of the surface vanadia species, which are integrated by chemical bonds in the collective catalyst system, are of importance. It is known that the support controls the ability of the active oxygen atoms to mediate the electron transfer to the metal.14,18 In addition, specific electronic properties of the support facilitate the reaction. Titanium dioxide, for example, is involved by acting as an electron sink forming TiIII and/or trapped O2(−) surface species under reaction conditions of oxidative dehydrogenation of propane.18
In the present work we investigate the effect of the abundance of Ti–O ligands on the reactivity of vanadium oxo-species in oxidative dehydrogenation of propane. The environment of dispersed vanadium oxide species was modified by synthetic means applying mesoporous silica (SBA-15) as a host for highly dispersed titania and vanadia surface species. SBA-15 has been selected due to its high surface area and density of hydroxyl groups. A library of mixed (VOx)n–(TiOx)n/SBA-15 catalysts was prepared by grafting alkoxides on Si–OH groups with the objective of exploring the interactions between supported titania and vanadia guest species with each other and the silica support as a function of metal loading while strictly avoiding the formation of segregated, bulk-like transition metal oxide particles. In this manner, the metal oxide loading was incrementally adjusted resulting in sub-monolayer, monolayer and multilayer catalysts with varying V/Ti ratio (Fig. 1). Complementary spectroscopic techniques were applied to analyze the topology of vanadia–titania surface patterns and the local environment of vanadium in surface oxide clusters aiming to elucidate an improved understanding of ligand effects in oxidative dehydrogenation of propane to propylene.

2. Experimental
2.1 Catalyst synthesis
The details of support and catalyst synthesis will be described below. Firstly, to exclude batch effects, mesoporous silica SBA-15 was synthesized in large quantities allowing a single batch of SBA-15 to be used to prepare catalyst series in sufficient quantities (ca. 8 g per catalyst) required for extensive catalytic measurements and comprehensive characterization. Vanadium and titanium oxide species were introduced by grafting alkoxide precursors. Table S1 in the ESI† summarizes the results of chemical analysis and microstructural details of the catalysts and supports. The table also contains the catalyst identification numbers (ID) used in our laboratory to identify each catalyst and its precursors with absolute certainty.SBA-15 was synthesized by adopting and up-scaling the original synthesis method described in the literature.19 The preparation was performed in an automated laboratory reactor (LabMax, Mettler-Toledo) that allows the synthesis of 350 g of the non-calcined, template containing SBA-15 precursor in a single batch. In detail, 256.5 g of the triblock co-polymer EO20PO70EO20 (EO = ethylene glycol, PO = propylene glycol, BASF Pluronic P123, Aldrich) were dissolved at 308 K in 4800 ml of 1.6 M HCl (CARL ROTH), yielding a clear solution after 6 hours. Subsequently, 552.9 g of tetraethoxysilane (TEOS, >99%, Alfa Aesar) were added within 3 minutes. The solution quickly turned cloudy and was stirred for 12 h at 308 K. After aging for 24 h at 358 K, the white suspension was filtered over a P2 glass frit without washing. The resulting white powder was dried at 353 K for 12 h and pestled afterwards. The polymer was removed by calcination in static air in a muffle furnace applying the following temperature program: heating rate 0.5 K min−1 to 473 K, 2 h holding time, heating rate 1 K min−1 to 823 K, 4 h holding time. After cooling down to room temperature, 165 g fine white powder of SBA-15 was obtained.
The (TiOx)n/SBA-15 supports were prepared by grafting titanium(IV) isopropoxide ((Ti(OCH(CH3)2)4, Acros Organics >98%) on the silica surface via reaction with the surface silanol groups of SBA-15 adding an appropriate amount of an alkoxide stock solution. For preparation of the stock solution, 250 g of Ti(OCH(CH3)2)4 were mixed with 750 g of dry isopropanol yielding a bright yellow solution containing 25 wt% titanium(IV) isopropoxide. The radius of one Ti(OCH(CH3)2)4 molecule is approximately 1 nm. The maximum loading that can be achieved in one grafting step whilst avoiding undesired segregation of titania is approximately 8 wt% Ti. For titanium loadings higher than 8 wt%, the procedure was repeated in a sequential manner with intermediate calcination. Dehydration of the sample between grafting cycles is important to avoid hydrolysis of the alkoxide precursor by water trapped in micropores, which can lead to formation of an unsupported crystalline TiO2 phase. Lower Ti loadings were similarly obtained in a single step by adjusting the concentration of the titanium isopropoxide solution by dilution with isopropanol. One grafting step was carried out as follows. SBA-15 was initially dried for 16 h at 403 K to remove physisorbed water. 120 g of the dried SBA-15 were suspended in 2000 ml dry isopropanol. The alkoxide was then allowed to react with the silica surface by transferring 712.5 g Ti stock solution to the SBA-15 suspension and stirring for 2 h at room temperature. Titanium was added in excess corresponding to a nominal loading of 20 wt% Ti on SBA-15. The opacity of the white suspension intensified after several minutes. After 2 h, the suspension was filtered over a P2 glass frit applying vacuum and washed twice with 500 ml dry isopropanol. The white filter cake was dried at 353 K and 80 mbar dynamic vacuum for 2 h and pestled. Organic residues were removed by calcination in static air in a muffle furnace applying the following temperature program: heating rate 0.5 K min−1 to 473 K, 2 h holding time, heating rate 1.5 K min−1 to 823 K, 2 h holding time. After cooling down to room temperature, 133 g fine white powder of (TiOx)n/SBA-15 were obtained. For synthesis of, e.g., 13 wt% Ti on SBA-15, three grafting steps have been carried out. The final (TiOx)n/SBA-15 powder is more compact and whiter than the SBA-15 mother.
Grafting of vanadium oxide species to the SBA-15 and (TiOx)n/SBA-15 supports was performed in a similar manner using vanadium(V) triisopropoxide (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) V(OCH(CH3)2)3, Alfa Aesar 96%) as the precursor. For preparation of a bright orange vanadium(V) triisopropoxide stock solution that contains 20 wt% of the alkoxide, 20 g OV(OCH(CH3)2)3 was mixed with 80 g dry isopropanol. The V loading was varied by adjusting the volume of the stock solution added to the support suspension. Exemplarily, we describe here the synthesis of the catalyst 4V/13Ti/SBA-15. In the first step, 8 g of the support were dried for 16 h at 403 K and suspended in 300 ml dry isopropanol. 10.09 g of the V stock solution were transferred to the suspension of 13Ti/SBA-15 in isopropanol and stirred for 2 h at room temperature. Again, an excess of V was applied corresponding to a nominal loading of 5 wt% V on 13Ti/SBA-15. Afterwards isopropanol was removed by evaporation at 323 K at a residual pressure of 50 mbar. The obtained light orange powder was pestled. Organic residues were removed by calcination in static air in a muffle furnace applying the following temperature program: heating rate 0.5 K min−1 to 473 K, 2 h holding time, heating rate 1.5 K min−1 to 823 K, 2 h holding time. After cooling to room temperature, 8.5 g fine powder of 4V/13Ti/SBA-15 was obtained. The colour of the pale yellow product changed with time after exposing the sample to air from very pale yellow to darker yellow-orange and finally to dark orange.
V(OCH(CH3)2)3, Alfa Aesar 96%) as the precursor. For preparation of a bright orange vanadium(V) triisopropoxide stock solution that contains 20 wt% of the alkoxide, 20 g OV(OCH(CH3)2)3 was mixed with 80 g dry isopropanol. The V loading was varied by adjusting the volume of the stock solution added to the support suspension. Exemplarily, we describe here the synthesis of the catalyst 4V/13Ti/SBA-15. In the first step, 8 g of the support were dried for 16 h at 403 K and suspended in 300 ml dry isopropanol. 10.09 g of the V stock solution were transferred to the suspension of 13Ti/SBA-15 in isopropanol and stirred for 2 h at room temperature. Again, an excess of V was applied corresponding to a nominal loading of 5 wt% V on 13Ti/SBA-15. Afterwards isopropanol was removed by evaporation at 323 K at a residual pressure of 50 mbar. The obtained light orange powder was pestled. Organic residues were removed by calcination in static air in a muffle furnace applying the following temperature program: heating rate 0.5 K min−1 to 473 K, 2 h holding time, heating rate 1.5 K min−1 to 823 K, 2 h holding time. After cooling to room temperature, 8.5 g fine powder of 4V/13Ti/SBA-15 was obtained. The colour of the pale yellow product changed with time after exposing the sample to air from very pale yellow to darker yellow-orange and finally to dark orange.
Though the guest phases are present as oxides we label the samples according to the loading in terms of percentage weight loading of metallic vanadium or titanium (rounded to the nearest 0.5%). In the case of mixed V/Ti/SBA-15 samples the label for titanium refers to the percentage weight loading of the Ti/SBA-15 mother from which it was prepared. For example, 17Ti/SBA-15 refers to SBA-15 with 17.1 wt% Ti while 4V/17Ti/SBA-15 refers to the same 17Ti/SBA-15 sample with 3.9 wt% vanadium although the addition of vanadium has adjusted the relative titanium loading to 16.3 wt%.
2.2 Catalyst characterization
Nitrogen sorption was performed at 77 K on an AUTOSORB-6B analyzer (Quantachrome). Surface areas were determined using the isotherm method of Brunauer, Emmett, and Teller (BET) assuming a cross-sectional area of 16.2 Å2 for adsorbed nitrogen. The microporous fraction of the surface area was determined using t-plot analysis and pore size distributions were calculated from the adsorption branch of the isotherms using an NLDFT method. All calculations were performed using the software package AUTOSORB-1 (Quantachrome, 1-software AS1Win V2.11).Interpore spacing, a0, was determined from small angle X-ray diffraction patterns measured using a transmission diffractometer (STOE STADI P) equipped with a primary focusing Ge monochromator (Cu Kα1 radiation) and a scintillation counter. Powder X-ray diffraction measurements (not presented), using the same diffractometer type and applying a position sensitive detector, excluded the presence of large crystalline V2O5 and TiO2 domains from all samples. The wall thickness of deposited metal oxide guest phases (Table S1, ESI†) was estimated by comparing the average pore diameter determined by nitrogen adsorption and the interpore spacing, a0, of the bare support and the loaded catalysts.
FTIR spectra were collected using a Perkin-Elmer PE 100 spectrometer fitted with a deuterated triglycine sulfate (DTGS) pyroelectric detector (32 accumulated scans, 4 cm−1 resolution). Self-supporting sample wafers with an areal density of ca. 10 mg cm−2 prepared by pressing at 13.5 MPa were located within a stainless steel infrared transmission cell fitted with CaF2 windows. Samples were dehydrated at 723 K for a period of 1 h in 200 mbar of O2. The oxygen atmosphere was exchanged several times during this procedure to remove evolved water. For CO adsorption measurements, the activated sample was evacuated, and backfilled with 2 mbar He (Westfalen, 99.999%), then cooled to 77 K using liquid nitrogen before recording a spectrum of the dehydrated catalyst sample. The pressure of CO (Westfalen, 99.97%) was sequentially increased and spectra acquired at each pressure. Data are presented as difference spectra where the spectrum of the dehydrated catalyst (at 77 K) has been subtracted from that of the sample in the presence of an equilibrium pressure of ca. 0.04 mbar CO.
Raman spectra were recorded by using a triple filter Raman spectrometer (TriVista S&I GmbH) with a CCD camera (Princeton Instruments) as the detector attached to a confocal microscope (Olympus, 10× long-working distance objective), using an Ar+ laser with 488 nm excitation wavelength (1 mW on the sample). The spectrometer was operated in triple subtractive mode and each spectrum was integrated for 2–10 min as required to produce spectra with an acceptable signal to noise ratio. A two point wavelength calibration was used (laser wavelength and first-order Stokes phonon of Si at 520 cm−1).
UV-Vis spectra were measured with a Perkin-Elmer Lambda 650 instrument using a diffuse reflectance accessory (Harrick Praying Mantis) complete with environmental chamber attachment (Harrick HVC-VUV-4). To avoid saturation on strongly absorbing signals samples were diluted 20-fold with calcined bare SBA-15 from the same batch used to prepare the catalyst. Spectra were recorded at room temperature after the mixtures were dehydrated in synthetic air (20% O2, 80% Ar, total flow of 60 ml min−1) at 723 K for a period of 60 min. Dehydrated SBA-15 was used as a white reference. Tungsten–halogen and deuterium lamps were used as light sources allowing spectra to be recorded in the range 200–800 nm. Reflectance data were converted to Kubelka–Munk units.
A FEI Titan 80–300 Cs-corrected microscope equipped with a Gatan Tridiem Image Filter was used to acquire HRTEM and STEM images of the SBA-15-samples.
Thermal analysis was performed on a Netzsch Jupiter STA 449C thermobalance equipped with a Pfeiffer QMS200 OmniStar quadrupole mass spectrometer for analysing the exhausted gases. Around 30–70 mg of SBA-15 were heated in corundum TG-DTA-crucibles under 100 ml min−1 Ar with a step-wise temperature program up to 1573 K. Desorption of physically adsorbed water occurs while heating from room temperature to 403 K with 5 K min−1. After a holding time of 120 min no further mass loss was observed and the temperature program was allowed to continue with 10 K min−1 to 723 K followed by an isothermal stage for 60 min and further heating with 20 K min−1 to 1573 K and a final isothermal stage for 30 min. The concentration of surface hydroxyl groups was calculated based on the mass loss between 403 K and 1573 K assuming that one water molecule is formed by condensation of two hydroxyl groups.
In-situ near edge X-ray absorption fine structure (NEXAFS) measurements were performed at the synchrotron radiation facility BESSY II of the Helmholtz-Zentrum Berlin, Germany (HZB) using monochromatic radiation of the ISISS (Innovative Station for In-situ Spectroscopy) beamline as a tuneable X-ray source. High-pressure soft X-ray absorption was measured in the presence of oxygen using the high pressure station designed and constructed at the Fritz Haber Institute, Berlin. Details of the setup are described elsewhere.20 The catalyst powders have been pressed into self-supporting discs (1 t, 8 mm diameter) and mounted inside a cell onto a sapphire sample holder approximately 1.4 mm in front of the first aperture of a differentially pumped electrostatic lens system. The home-built electron lens serves as the input system for a modified commercial hemispherical electron analyzer (PHOIBOS 150, Specs-GmbH). Oxygen is introduced into the cell via a calibrated mass flow controller, heating is provided by a NIR laser at the rear of the sample, and the temperature is monitored by a thermocouple attached directly to the sample surface. NEXAFS spectra of the dehydrated samples were obtained in 50 Pa O2 at 673 K by heating the material in-situ in the XAS cell with 5 K min−1 up to the final temperature. Oxygen K-edge excitation spectra have been recorded in the Auger electron yield mode by operating the electron spectrometer with a pass energy of 100 eV as an X-ray absorption spectroscopy (XAS) detector to minimize contributions from the gas phase to the spectra. O K-edge spectra of the sample surface have been corrected for the remaining effects of O2 gas phase absorption. In order to increase the signal to noise ratio a data reduction by a factor 2 has been applied to the raw spectra (containing about 1000 pts per scan) by averaging adjacent points. Three scans have been averaged and the X-ray spot position on the sample has been changed after each scan to avoid damage of the surface by the brilliant synchrotron X-ray beam. Absolute energy calibration has been achieved by setting the π* resonance of the O2 gas phase signal to 530.9 eV and the spectral resolution was about 150 meV. Further details of the methodology and data treatment are described elsewhere.21 Additionally, Ti L-edge spectra and V L-edge spectra have been obtained in the total electron yield (TEY) mode using a biased wire collector (U = + 90 V). Reference NEXAFS spectra of anatase TiO2 (Alfa Aesar, 99.9%) and V2O5 (RIEDEL, 99.5%) have been obtained at the Ti L-, V L-, and O K-edges, respectively, from pressed powders in the TEY mode.
The Ti L-NEXAFS was simulated using the semi-empirical charge-transfer multiplet (CTM) approach in order to study general effects of modification in the geometric arrangement of titanium oxide species on SBA-15.22–24 The calculations have been carried out by applying the CTM4XAS vs3.1 code.25 The Slater integrals have been reduced to 90% of their atomic values except for the simulation of the isolated Ti4+ ion, where the atomic values have been used. The crystal field (10 Dq), the charge transfer energy, Δ, and the difference between the core hole potential, Upd, and the Hubbard 3d–3d repulsion energy, Udd, have been adjusted to fit the experimental spectra. Otherwise, default values of the program have been used. A constant Gaussian broadening of 0.2 eV has been applied to account for the instrumental resolution and the Lorentzian broadening was 0.2 eV for the L3-edge and 0.5 eV for the L2-edge to consider the different core hole lifetime due to extra Auger decay channels for the L2-edge (Coster–Kronig Auger decay).
2.3 Activity test
Catalytic measurements were performed in U-shaped fixed bed quartz reactors (6 mm internal diameter) operated at ambient pressure at 773 K. Catalyst samples were pressed at 50 bar and sieved to a particle size range of 200–300 μm and diluted twofold with silicon carbide of the same particle size. The feed consisted of synthetic air (20.5% O2 in N2, Air Liquide, 99.95%) and propane (99.9% C3H8, Air Liquide) delivered by independent electronic mass flow controllers (Bronkhost Hi-Tech, E1-flow), applying a propane to oxygen ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Propane conversion was limited to <10% to ensure differential reaction conditions. Catalyst mass and total flow rates were varied from 1.5 to 300 mg and from 20 to 100 ml min−1, respectively. Exhaust gases were analyzed by on-line gas chromatography (GC, Shimadzu 2014) using two separation columns (HayeSep Q and molecular sieve 13X) for the separation of O2, N2, CO, CO2, and hydrocarbon gases, respectively. Oxygen and nitrogen were detected by a thermal conductivity detector, while hydrocarbons and methanized carbon oxides were detected by a flame ionization detector. Blank measurements carried out over SiC eliminated significant contributions from homogeneous gas phase reactions at the applied temperature. Carbon balances for all the tested catalysts were closed up to 100 ± 5%.
:1. Propane conversion was limited to <10% to ensure differential reaction conditions. Catalyst mass and total flow rates were varied from 1.5 to 300 mg and from 20 to 100 ml min−1, respectively. Exhaust gases were analyzed by on-line gas chromatography (GC, Shimadzu 2014) using two separation columns (HayeSep Q and molecular sieve 13X) for the separation of O2, N2, CO, CO2, and hydrocarbon gases, respectively. Oxygen and nitrogen were detected by a thermal conductivity detector, while hydrocarbons and methanized carbon oxides were detected by a flame ionization detector. Blank measurements carried out over SiC eliminated significant contributions from homogeneous gas phase reactions at the applied temperature. Carbon balances for all the tested catalysts were closed up to 100 ± 5%.
3. Results and discussion
3.1 Grafting of titanium and vanadium oxide guest species
Incorporation of titania and vanadia into SBA-15 by grafting occurs via reaction of surface silanol groups with the alkoxide precursors forming Si–O–(Ti,V) bonds. Coordination of the alkoxide is demonstrated by the depletion of the ν(OH) stretching mode of Si hydroxyl groups (silanols) upon increasing metal loading, as observed in infrared spectrometric measurements (Fig. S1 and S2, ESI†). Due to the large dynamic dipole moment associated with hydroxyl stretching vibrations that yields intense infrared absorption bands, infrared spectroscopy can be used to sensitively probe the interaction between support and deposited surface oxide species. In agreement with the previous literature, the infrared spectra for both the (TiOx)n/SBA-15 (Fig. S1, ESI†) and (VOx)n/SBA-15 (Fig. S2, ESI†) sample series exhibit a general trend of decreasing intensity in the O–H stretching vibration region (νOH = 3800–3400 cm−1) with increasing loading.11,26–30 All spectra exhibit a band with a peak maximum located at 3745 cm−1, which is attributed to the νOH stretching mode of isolated silanol groups associated with the SBA-15 support, that becomes broader and less intense as metal loading is increased. In pure SBA-15 this band is sufficiently broad to include a contribution from isolated/geminal silanol couples, which yield a signal located at 3742 cm−1.31 The shoulder at 3670 cm−1 is assigned to vicinal and H-bonded OH groups. As vanadium loading is increased, an additional band located at 3660 cm−1, assigned to νOH stretching modes of hydroxyl ligands located on vanadium centres, is developed.26,28,32 At high titanium loadings additional features attributed to TiO–H species are observed.33,34 A shoulder that is developed at ca. 3720 cm−1 assigned to isolated (terminal) Ti–OH groups is accompanied by a comparatively low intensity signal located at 3670 cm−1 for bridging OH groups. On this basis, we conclude that the supported titania species are sufficiently dispersed so that isolated hydroxyl groups are the dominant species.A detailed infrared investigation of siliceous materials clearly established that changes in the surface hydroxyl density perturb hydrogen bonding interactions between OH groups resulting in a distortion of the ν(OH) band profile, which subsequently leads to a deviation of the integrated intensity from Beer–Lambert conditions.35 Gallas et al. proposed that the hydroxyl stretching–deformation combination band (ν + δ) or the first overtone of the OH stretch (2νOH) are more suitable alternatives for the quantification of hydroxyl groups residing in hydrogen bonded environments since they are less sensitive to hydrogen bonding interaction than the νOH fundamental.36 However, due to the inherent broadness of combination bands in infrared spectroscopy, such a combination band is likely to include contributions from any hydroxyl group native to the metal oxide guest phase(s) and is therefore unsuitable for the selective quantification of silanol groups present in the samples considered here. In order to explore the interaction of silanol groups with the titania and vanadia guest phases in first approximation, the integrated intensity of the OH stretching massif was determined in the energy range 3680–3765 cm−1. In this manner, both isolated and hydrogen bonded silanol groups are considered in the integration procedure, while hydroxyl groups associated with vanadia surface species, located at 3660 cm−1, are excluded. However, at high titania loadings the aforementioned features located at 3720 and 3670 cm−1 will contribute to the integrated intensity. Fig. 2 presents the relationship between the ratio of the integrated areas of the νOH massif of each (VOx)n/SBA-15 and (TiOx)n/SBA-15 sample and that of the relevant SBA-15 mother, i.e. A(νOHsample):A(νOHSBA-15), versus the ratio of the number of moles of deposited metal and silanol groups present on the mother SBA-15, i.e. mol(deposited metal):mol(OHSBA-15). By this process, we compare the interaction of titanium and vanadium with surface silanol groups. The trend for vanadia deposition is approximately linear, which, when extrapolated, intersects the x-axis at a value of approximately 0.5 moles of deposited vanadium per mole of silanol groups. This indicates that at theoretical surface saturation, i.e. where every silanol group is consumed, approximately two silanol groups are required to graft each vanadium atom.
 | ||
| Fig. 2 Relationship between titanium loading (blue triangles), vanadium loading (orange squares) and OH density as revealed by IR spectroscopy. The dotted lines have been inserted to guide the eye. | ||
In contrast, the trend for the (TiOx)n/SBA-15 series is represented by a curve, which is in agreement with Cozzolino et al. who performed a similar analysis on a series of Ti/SiO2 catalysts prepared by multistep grafting of titanium isopropoxide.29 The asymptotic nature of the curve indicates the contribution of TiO–H stretching modes located at 3720 and 3670 cm−1 towards the integrated intensity of the νOH massif at higher Ti loadings. Clearly, the curve for the Ti/SBA-15 series represented in Fig. 2 intersects the x-axis at a very high ratio that corresponds to a titanium loading of 23 wt%. However, based on linear extrapolation of the data points at low Ti loading, the upper limit that can reasonably be proposed for the stoichiometry of a theoretical titania monolayer is one mole of deposited Ti per mole of silanol groups. Indeed, Santacesaria et al. presented an adsorption isotherm for titanium alkoxide grafted onto silica, which indicated the monolayer coverage to have a stoichiometry of approximately one hydroxyl per titanium atom.37 But, if it is assumed that mainly silanol groups and not the newly formed Ti–OH groups contribute to the IR signal used for analysis, any ratio mol(deposited metal):mol(OHSBA-15) higher than one may also indicate that incoming titanium isopropoxide molecules interact with titania domains already present on the catalyst surface forming Ti–O–Ti bonds leading to formation of titania multilayers. However, Raman, UV-vis and NEXAFS spectroscopies, as presented in the following section, clearly show that this is not the case for loadings up to 19 wt% Ti on SBA-15.
3.2 Dispersion of titanium oxide surface species
Fig. 3 presents Raman spectra for (TiOx)n/SBA-15 prepared by sequential multistep grafting of titanium alkoxide. Due to its exceptionally high sensitivity towards phonon vibrations we employ Raman spectroscopy in order to identify the gradual transformation of highly dispersed, isolated titanium oxide species into three-dimensional, bulk-like arrangements. At a Ti loading of 17 wt%, the first weak signs of phonon bands appear (Fig. 3e). At 19 wt% Ti loading, very weak signals can be identified at 159, 343, 474, and 598 cm−1 (Fig. 3f). With increasing Ti content, these broad and ill-resolved features become more intense and shift to 155, 400, 451, 510 and 626 cm−1 at loadings of 20 wt% and above (Fig. 3g). No indication for titanyl stretching vibrations ν(TiO), which are expected in the range between 900 and 1000 cm−1,38 has been found. The Raman spectra of the crystalline TiO2 modifications anatase and rutile show a common band at 144 cm−1 corresponding to the Eg and B1g mode of lattice vibrations, respectively.39,40 Other collective vibrations appear at 447 cm−1 (Eg), 612 cm−1 (A1g), and 826 cm−1 (B2g) for rutile, and at 197 cm−1 (Eg), 399 cm−1 (B1g), 513 cm−1 (A1g), 519 cm−1 (B1g), and 639 cm−1 (Eg) for anatase. For reference, the Raman spectrum of anatase is shown in Fig. 3k. The broadness and relative low intensity of the bands emerging in the phonon range with increasing titania loading indicate the transition from well-defined, isolated titanium oxide species into two- and three-dimensional precursors of the solid state, which are characterized by a wide range of bond angles and distances. Based on the Raman spectra presented in Fig. 3, we conclude that at loadings of 17 wt% and below the multi-step grafting procedure described here yields mainly dispersed titania surface species on SBA-15, while at loadings greater than 17–19 wt% the titanium oxide species exist in the form of two- and three-dimensional networks. XRD excludes the presence of larger crystalline particles in all samples studied in the present work (results not shown). This was confirmed by high resolution TEM imaging of 19Ti/SBA-15 (Fig. 4, left). The varying contrast visible in the HAADF STEM image (Fig. 4, right) could indicate small clusters of titania in the walls, i.e. within the micropores of SBA-15.
 | ||
| Fig. 3 Raman spectra for SBA-15 (11780) (a), 7Ti/SBA-15 (11807) (b), 11.5Ti/SBA-15 (11815) (c), 14.7Ti/SBA-15 (11817) (d), 17Ti/SBA-15 (11827) (e), 19Ti/SBA-15 (11836) (f), 20Ti/SBA-15 (11838) (g), 21Ti/SBA-15 (11840) (h), 22.5Ti/SBA-15 (11876) (i), 23Ti/SBA-15 (11886) (j), and commercially acquired TiO2 (anatase) (k). The intensity of each spectrum has been normalised between 0 and 1 to facilitate comparison. | ||
 | ||
| Fig. 4 High resolution TEM (left) and HAADF STEM (right) images of 19Ti/SBA-15 (11836). | ||
Interestingly, the titanium loading of 19 wt% corresponds to a calculated surface density of 2.95 Ti per nmSBA-152. In comparison, the surface silanol density of the corresponding SBA-15 mother, determined by TGA, is 3.1 OH nm−2, which implies a near 1:1 ratio of silanol groups and deposited Ti atoms in agreement with our IR spectroscopic result and previous findings in the literature.29
Fig. S3 (ESI†) shows the UV-vis spectra of (TiOx)n/SBA-15 including the reference spectrum of anatase. The ligand-to-metal charge transfer (LMCT) transitions that are responsible for absorption in the UV range of the spectrum are strongly influenced by the number of ligands surrounding the central metal ion in the first coordination sphere and, therefore, provide information on its local coordination environment. As the titanium loading is increased, the peak maxima shift to higher wavelengths. For the (TiOx)n/SBA-15 with low Ti loading (3 wt% Ti) the peak maximum was found at 210 nm. In reference to UV-vis spectroscopic studies of amorphous TiO2–SiO2 mixed oxides, the band is assigned to isolated Ti ions in tetrahedral coordination.41–44 It is assumed that Ti substitutes for Si in the silica network. With increasing titania loading the absorption maximum shifts to lower energies (240 nm for 8 wt% Ti, and 276 nm for 23 wt% Ti), which has been attributed to progressive cross-linking by Ti–O–Ti bridges and changes in the coordination environment that finally result in the electronic structure of bulk-like titanium oxide. Gao et al. reported that both dimeric and one-dimensional polymeric, i.e. ribbon-like, titania species may yield maxima located at ca. 246 nm, while a shift toward higher wavelength indicates formation of two-dimensional polymerized TiO5 units.45 The ligand to metal charge transfer (LMCT) band for titanium in octahedral coordination in anatase is located at 340 nm (Fig. S3, ESI†).42 The spectra of (TiOx)n/SBA-15 presented in Fig. S3 (ESI†) exhibit a tail towards higher wavelength that includes only a very minor contribution at ca. 340 nm for loadings higher than 17 wt% Ti. On this basis, the presence of titanium oxide nano-domains can therefore be excluded for loadings up to 17 wt% Ti in agreement with Raman spectroscopy.
The Ti L3,2-NEXAFS measurements support these findings. Ti L-edge spectra of dehydrated (TiOx)n/SBA-15 with 3 and 13 wt% Ti at 773 K in oxygen are compared to bulk anatase TiO2 in Fig. 5A. The Ti L-edge is dominated by Ti 2p to Ti 3d transitions and shows a spin–orbit splitting into the L3 edge below 462 eV and the L2 edge above, respectively. NEXAFS as an element and site selective probe is sensitive to the local symmetry, bonding, and coordination of the absorbing atom and its nearest neighbors. The position and intensity ratio of Ti L-edge resonances are closely related to the Ti site geometry.46,47 The bulk spectrum of TiO2 shows a strong pre-peak at 458.1 eV and a splitting in the main peak of the L3 edge at about 459.9 eV and 460.7 eV, respectively. These structures are also visible although blurred at the L2 edge because the intrinsic broadening is significantly increased due to an extra Auger decay channel (Coster–Kronig Auger decay). The Ti L-NEXAFS of SBA-15 supported titania differs remarkably from the bulk oxide indicating that no Ti bulk-like oxide structures are present on the studied catalysts in agreement with UV-vis and Raman measurements. However, the evolution of a pre-peak at 458.1 eV photon energy and a shoulder around 460.7 eV that are also present in the bulk TiO2 oxide suggest an increased linkage via Ti–O–Ti bonds with increasing Ti loading, which is also reflected in the UV-vis spectra by the shift of the absorption maximum to lower energies. Thus, Ti L-NEXAFS supports the view of isolated TiO4 species at low Ti loading (i.e., 3 wt%) with an increasing formation of Ti–O–Ti bonds at higher Ti loading (e.g., 13 wt%) without the formation of bulk TiO2.
 | ||
| Fig. 5 (A) Ti L3,2-NEXFAS of dehydrated 3Ti/SBA-15 (7569), 13Ti/SBA-15 (9240), and 4V13Ti/SBA-15 (9507). Spectra have been taken in O2 at 773 K. The spectrum of anatase TiO2 is shown for comparison. In (B) the results of CTM simulations of the Ti L-absorption edges are presented. Parameters used for the simulations are: (a) isolated Ti4+ ion, e.g. 10 Dq = 0; (b) tetrahedral symmetry (Td), 10 Dq = −1.6 eV, Δ = 0.5 eV, Upd–Udd = 1 eV; (c) octahedral symmetry (Oh), 10 Dq = 1.6 eV, Δ = 0.5 eV, Upd–Udd = 2 eV; (d) octahedral symmetry (Oh), 10 Dq = 1.3 eV, Δ = 0.5 eV, Upd–Udd = 5 eV. The theoretical spectra are shifted so as to align the main peak of the L3-edge to 459.9 eV. Spectra are offset for clarity. | ||
In order to study the impact of the geometric arrangement around the absorbing Ti atom on the Ti L-NEXAFS we performed some basic simulations of the absorption edges by the charge transfer multiplet approach (CTM).22–24 In contrast to other first-principle codes, e.g.ref. 42, and references therein,48 several semi-empirical parameters need to be adjusted. Nevertheless, since the present work is not aiming to obtain the best match with the experimental spectrum but rather to study general changes in the spectral shape when the local chemical bonding is modified, this approach is justified. Results of these simulations are shown in Fig. 5B. The X-ray absorption spectrum of an isolated Ti4+ ion (e.g., without ligands) shows two strong peaks and a very weak pre-peak (around 457.1 eV). In the CTM approach solid state effects are introduced to the atomic multiplets by parameters like the crystal field strength (10 Dq) that is projected to different symmetries (e.g., Td or Oh) thus reflecting the electronic charge distribution surrounding the absorbing atom and the charge transfer effect from the ligands to the metal. Inclusion of a small crystal field (10 Dq = 1.6 eV) and charge transfer (Δ = 0.5 eV, Upd–Udd = 1 eV) that is expected to be relevant for the NEXAFS of covalently bound high valent Ti compounds causes an increase of the pre-peak at the L3- and L2-edge (457.7 eV and 463.5 eV in spectrum b). The resulting simulated spectrum resembles the experimental spectrum of the catalyst with 3 wt% Ti in Fig. 5A. Using the same parameters but changing the symmetry of the crystal field from Td to Oh, e.g. by a modification of the local geometric arrangement of the ligands around the central Ti atom, results in a strong increase of the pre-peak and also a slight decrease in the distance between the pre-peak and the main peak at the Ti L3-edge. Similar changes can be observed when comparing the experimental NEXAFS of 3 wt% Ti and 13 wt% Ti (Fig. 5A). Thus, the intensity and position of the pre-peak of the L3-edge around 458 eV are strongly affected by variations of the local geometry. The shoulder in the main peak of the L3 edge at 460.7 eV in the experimental spectrum of 13 wt% Ti is not reproduced by the simulations. The interpretation of this feature in anatase is controversially debated in the literature. It has been suggested to be the result of a non-cubic ligand field49,50 or as a non-local, long range band-structure effect.48,51 In spectrum d in Fig. 5B it is shown that a variation in the ligand field parameters (10 Dq = 1.3 eV, Δ = 0.5 eV, Upd–Udd = 5 eV) does induce a shoulder around 461 eV at the main L3-resonance and decreases the separation of the L3-main peak and the pre-peak to about 1.9 eV, which is comparable to the value observed in the experimental spectrum. Furthermore, a strong charge transfer satellite at high photon energies (hν = 470.3 eV) appears in the simulated spectrum that is much less pronounced in the experimental spectrum of 13 wt% Ti in Fig. 5A.
In conclusion, the CTM simulations have shown the sensitivity of the Ti L-NEXAFS to modification of the local bonding (geometry/bond strength) between Ti and oxygen, in particular of the intensity and position of the pre-peak.
In summary, titanium oxide species were grafted on the walls of the meso- and micropores of SBA-15 in sub-mono- and monolayer quantities. The coated layer is composed of highly dispersed, two-dimensional titania surface species. Mesoporous silica SBA-15 allows grafting of exceptionally high amounts of titanium due to the high specific surface area and the density of anchoring OH groups. Monolayer coverage without noticeable segregation of titanium oxide nano-particles has been achieved in the range between 17 and 19 wt% Ti loading corresponding to 6–8 Ti atoms per nmcat2 and Si/Ti ratios between 3.3 and 2.8.
3.3 Location of vanadium oxide surface species
After covering the pore walls with titania, vanadium oxide species have been subsequently grafted on the (TiOx)n/SBA-15 supports. On sub-monolayer TiOx/SiO2 catalysts, vanadia may be located on either, or both, of the titania and silica components.52–61 Accessible titania species are identified by infrared spectroscopic measurement of adsorbed carbon monoxide (Fig. 6 and Fig. S4, ESI†). Titanium containing catalysts exhibit two bands located at ca. 2180 and 2158 cm−1, assigned to CO chemisorbed on coordinatively unsaturated TiIV species62 and surface hydroxyl groups,63,64 respectively. The signal corresponding to the accessible coordinatively unsaturated titanium ions is compared in Fig. S4 (ESI†) for a number of catalysts. The band is absent from the spectra of 4V/SBA-15 and SBA-15. When the Ti loading is increased from 3 wt% to 8 wt%, the signal intensity on the (TiOx)n/SBA-15 catalysts increases indicating that the number of coordinatively unsaturated TiIV sites on the surface is higher at higher Ti loadings. However, the trend is reversed when the titanium content is further increased to 13 wt%, which might be an expression of progressive oligomerization of surface titanium oxide species. The intensity of the band is significantly attenuated upon addition of vanadium to the (TiOx)n/SBA-15 supports. In Fig. 6, the percentage of residual free titania surface after grafting vanadium has been plotted as a function of the V/Ti ratio. Comparison of the spectra of 1V/3Ti/SBA-15 and 4V/3Ti/SBA-15 (Fig. S4, ESI†) reveals that vanadia species progressively cover the surface titania species reducing the fraction of accessible titania from 60% for 1 wt% V to 40% for 4 wt% V (Fig. 6). Interestingly, even when vanadium is present in relative excess compared with previously grafted titanium oxide species (such as in, e.g., 4V/3Ti/SBA-15) the TiIV signal is still present. This indicates that vanadium species coordinate to both titanium centres and silanol groups unless the titanium oxide is present in monolayer concentration. An interesting phenomenon becomes apparent from the comparison of the spectra of 1V/3Ti/SBA-15 and 4V/13Ti/SBA-15, respectively. The two catalysts are characterized by similar V/Ti ratios but different total metal loading. For 3Ti/SBA-15, grafting of 1 wt% vanadia results in a substantial decrease of the signal belonging to the accessible, coordinatively unsaturated titanium ions at the surface, indicating that grafting of V leads to coverage of approximately 40% of the Ti oxide surface species, leaving 60% of the titania surface free. In contrast, the signal is less attenuated by grafting of the same relative amount of V on 13Ti/SBA-15 leaving 90% of the titania uncovered (Fig. 6) and suggesting that vanadia is grafted in this case to a larger extent on the residual free silica surface. At this point, it is interesting to note that the total metal loading in 4V/13Ti/SBA-15 corresponds to 17 wt%, which is close to the monolayer coverage of this particular catalyst. Apparently, the V–Ti interaction changes with titania loading. The reason for this unexpected observation might be related to the different structure of the surface titanium oxide species in the two supports, since titanium in 3Ti/SBA-15 occurs mainly isolated in a silica matrix in tetrahedral coordination, whereas polymerized titania species prevail in 13Ti/SBA-15 (Fig. 5 and Fig. S3, ESI†). | ||
| Fig. 6 Fraction of uncovered titania surface as a function of the V/Ti ratio comprising 4V3Ti/SBA-15 (7620), 4V8Ti/SBA-15 (7624), 1V3Ti/SBA-15 (7606), and 4V13Ti/SBA-15 (9954). The free titania surface was estimated from the ratio of the height of the peaks at 2180 cm–1 in the FTIR spectrum of CO adsorbed on the V-containing catalyst and the corresponding support. The peak is attributed to CO adsorbed on coordinatively unsaturated TiIV ions. | ||
The presence of differently coordinated vanadium oxide surface species is confirmed by UV-vis spectroscopy. The UV-vis spectra indicate that vanadium ions are present on SBA-15 mainly in tetrahedral coordination (Fig. S5, ESI†). Absorption maxima between 270 and 350 nm have been attributed to vanadium in tetrahedral coordination both as monomeric, isolated species for very low loadings and small oligomers with increasing loading.5,52,57,65–67 A band near 400 nm has been assigned to vanadium in a square pyramidal environment in ribbon-like two-dimensional surface patches.52,57 The spectrum of crystalline vanadium pentoxide shows absorption features between 220 and 580 nm (Fig. S5, ESI†). The degree of polymerization increases with increasing loading, which is reflected in the shift of the absorption maximum from 245 nm to 295 nm (Fig. S5, ESI†). The additional occurrence of pentagonal coordinated vanadium ions cannot be excluded.
For vanadia supported on (TiOx)n/SBA-15, the bands in the UV-vis spectra (Fig. 7) originate from superimposed LMCT transitions of both vanadium and titanium surface oxide species. Apparently, titania in sub-monolayer abundance stabilizes surface vanadium oxide species in tetrahedral coordination, because the peak maxima in the spectra of the catalysts 4V/3Ti/SBA-15 and 4V/8Ti/SBA-15 are shifted to higher energy compared to 4V/SBA-15. The interpretation of the 4V/13Ti/SBA-15 spectrum is not straightforward. The maximum in absorption is shifted again to 278 nm and the shape of the signal resembles very much the spectrum of 4 wt% vanadium on SBA-15. The total metal loading in this catalyst is close to the capacity of a monolayer (ca. 17 wt% metal). This might lead to the formation of a mixed two-dimensional V–Ti surface oxide monolayer with unique absorption properties resulting in a peak maximum energetically close to that of 4V/SBA-15. The peculiar spectroscopic appearance of the 4V/13Ti/SBA-15 catalyst is in agreement with the results of infrared spectroscopy of adsorbed CO, which illustrates that vanadium preferentially replenishes the residual free silica surface on 13Ti/SBA-15. Raman spectroscopy confirms two-dimensional dispersion of surface vanadia and titania species in 4V/13Ti/SBA-15 (Fig. S6, ESI†).
 | ||
| Fig. 7 UV-vis spectra of 4V/SBA-15 (7815), 4V3Ti/SBA-15 (7620), 4V8Ti/SBA-15 (7624), and 4V13Ti/SBA-15 (9954) after dehydration in synthetic air at 723 K. The spectra were taken at 313 K. The spectra of 13Ti/SBA-15 (9846), and 17Ti/SBA-15 (11827) are shown for reference. | ||
The interpretation of the O K-NEXAFS of vanadia supported on Ti/SBA-15 is complicated by the overlap of absorption features related to oxygen bonded only to V, only to Ti or both to V and Ti. The Ti L3-edge pre-peak intensity is further increased in the mixed system (VOx)n–(TiOx)n/SBA-15 (refer to top spectrum in Fig. 5A). This is the result of a modification of Ti–O bonds by the anchorage of V species at the titania clusters. Interestingly, this spectroscopic fingerprint for an enhanced linkage at the Ti site is the same either when the Ti loading is increased or when the Ti site configuration is modified by adding V. This points to the fact that the newly formed V–O–Ti bonds create a bonding configuration at the Ti site that is similar to the formation of Ti–O–Ti bonds when the Ti loading is increased. This similarity might facilitate the preferential anchorage of V on titania sites instead on the SBA-15 support. There is work in progress to obtain a sound understanding of the mixed (VOx)n–(TiOx)n/SBA-15 system by comparison of experimental absorption spectra with theoretical DFT cluster calculations of the O K-NEXAFS.
3.4 Mesostructure of the catalysts
Vanadium oxide supported on mesostructured silica shows appreciable yields of propylene in oxidative dehydrogenation of propane.68–70 To analyze whether the incorporation of high loadings of the titania and vanadia guest species degrade the mesopore structure of SBA-15, nitrogen adsorption has been performed. The profiles shown in Fig. 8 are representative for all of the prepared catalysts and conform to type IV isotherm maintaining H1 hysteresis following the categorization stipulated by IUPAC. For pure (TiOx)n/SBA-15 samples this observation is valid up to a loading of 20 wt% titanium. Grafting of metal oxide species is accompanied by an associated decrease in specific surface area (Table S1, ESI†). For (TiOx)n/SBA-15 samples, the fact that mesoporosity is maintained while the total surface area decreases with loading indicates that, up to a loading of 20 wt%, the titanium guest species line the walls of the mesopores without filling or plugging them. This is also in agreement with the estimated thickness of the metal oxide layers within the mesopores of SBA-15 that are in the order of magnitude of a few Å (Table S1, ESI†). At loadings greater than 20 wt% the quality of the previously well-ordered pore system appears to be diminished, as indicated by the presence of partial H2 character in the hysteresis loop of a 23.5 wt% Ti/SBA-15 catalyst (not shown). Binary (VOx)n–(TiOx)n/SBA-15 samples also yield H1 hysteresis loops indicating that subsequent addition of vanadium does not cause degradation of the well-defined mesoporous network. | ||
| Fig. 8 Nitrogen adsorption isotherms of selected catalyst samples recorded at 77 K. Profiles are offset for clarity. | ||
The total volume of nitrogen taken up by the (TiOx)n/SBA-15 samples is observed to decrease with increasing titanium loading indicating a reduction in surface area with each impregnation step. In addition, the percentage of specific surface area due to microporosity, as determined by t-plot analysis, is also observed to decrease with increasing loading (Fig. S7, ESI†). With respect to molar metal loading, the filling of micropores is more facile for vanadia deposition than for titania deposition, indicating that vanadia has a greater affinity for micropores than titania.
3.5 Catalytic activity in oxidative dehydrogenation of propane
The catalytic properties of (VOx)n species anchored on sub-monolayer quantities of titania dispersed on silica are complex due to various options in the distribution of the two elements that affect the intrinsic catalytic properties of both, vanadia and titania surface species.56Fig. 9A presents surface area corrected integral rates of propane consumption and propylene formation calculated based on conversion and/or selectivity at the specific contact time for catalysts containing 4 wt% V as a function of titanium loading. All data are collected at modified residence times in the range 9–30 kg s m−3, with the exception of 3Ti/SBA-15, which was collected at 165 kg s m−3. The promotional effect of titania species on deposited vanadium oxide clusters is well demonstrated by comparison of each V containing catalyst with its associated mother (TiOx)n/SBA-15 support and pure 4V/SBA-15. The rates of propane consumption and propylene formation are significantly greater only for high titania loadings. The enhanced activity of the mixed 4V/Ti/SBA-15 catalysts comes with a penalty in propylene selectivity, which is reflected by the fact that the propylene formation rate remains almost constant, i.e. within error scattering, at Ti loadings of 0–7 wt%. At higher Ti loadings there is a dramatic increase in both propane conversion and propylene formation. At these higher Ti loadings the rate of propylene formation is approximately constant, while the rate of propane conversion varies. Comparison of the selectivity at the same propane conversion shows that the effect of titania on the selectivity is observable, but not dramatic. For the catalysts containing 4 wt% V, a decrease of the propylene selectivity from 64.5% for the Ti-free SBA-15 support to 53.2% for 23 wt% Ti was registered at 6% propane conversion. The 4V/13Ti/SBA-15 catalyst offers the highest productivity in terms of space time yield of propylene (Fig. 9B). The volcano type behavior of the catalyst performance in oxidative dehydrogenation of propane may be rationalized by considering the changing topology patterns within the investigated catalyst library as illustrated in Scheme 1. At low titania loadings, surface vanadia species are anchored preferentially on isolated titanium oxide species or dispersed two-dimensional titanium oxide oligomers as evidenced by UV-vis and infrared spectroscopy. A considerable fraction of vanadium atoms is located on the residual titanium-free silica surface in the form of highly dispersed species characterized by low nuclearity. Hence, sub-monolayer surface titanium oxide species clearly improve the dispersion of vanadium, which has also been observed for V–Ti oxide supported on macroporous silica.27 According to the UV-vis spectra of, e.g. the catalysts 4V/3Ti/SBA-15 and 4V/8Ti/SBA-15 shown in Fig. 7 that contain titania in sub-monolayer concentration, vanadia species appear to exist predominantly in the form of monomers, which have been shown to be less efficient catalytic species in oxidative dehydrogenation of propane to propylene.68,71 Although many of the vanadium atoms in the catalysts 4V/3Ti/SBA-15 and 4V/8Ti/SBA-15 are anchored via V–O–Ti bonds to the support, the rate of propane consumption is not much increased compared to 4V/SBA-15. These experimental results suggest that, apparently, the ligand effect of vanadium and titanium is comparable when vanadium is either arranged in the form of two-dimensional oligomers on the surface of silica with high abundance of V–O–V bonds or as monomer on highly dispersed surface titania species with high abundance of Ti–O–V bonds. Still, vanadia supported on monolayer or multilayer titania, such as in the catalysts 4V/17Ti/SBA-15 and 4V/23Ti/SBA-15, respectively, present increased consumption rates of propane, indicating that not only the nature of the S–O–V bond, but also other factors are operative. Elucidation of the molecular structure of vanadia clusters supported on monolayer titanium oxide is, however, challenging due to the similarities in the spectroscopic features of highly dispersed vanadia and titania species. | ||
| Fig. 9 Consumption rate of propane and rate of propylene formation (A) and space time yield of propylene (B) in the oxidative dehydrogenation of propane at T = 773 K over catalysts that contain 4 wt% V and varying loading of Ti. | ||
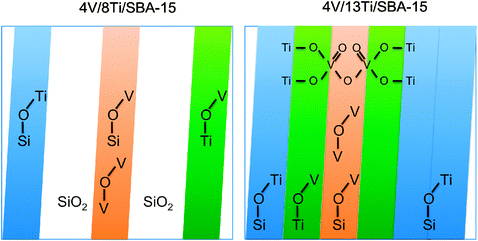 | ||
| Scheme 1 Schematic illustration of the different surface topologies of V–Ti sub-monolayer catalysts, like 4V/8Ti/SBA-15, and V–Ti monolayer catalysts, like 4V/13Ti/SBA-15, including all topological features evidenced by spectroscopic techniques (white area: free silica surface, blue area: surface area covered by titanium oxide surface species, green area: vanadium oxide species supported on dispersed titania species, orange area: dispersed vanadia species on the silica surface). | ||
A unique situation exists on the surface of the catalyst 4V/13Ti/SBA-15 that is distinguished by a maximum in propylene productivity. Under the conditions applied in the current experiment the space time yield of more than 5 gpropylene gcat−1 h−1 exceeds the performance of most of the catalysts studied in ODH of propane so far and leaves room for optimization.2 An extended kinetic investigation is in progress that includes studies of this catalyst in a broader range of reaction conditions. The surface of 4V/13Ti/SBA-15 is characterized by a monolayer that covers the silica almost entirely. The concentration of the residual silanol groups is low. As outlined above, UV-vis spectroscopy and NEXAFS reveal unique spectroscopic features that may be interpreted in terms of a joint V–Ti oxide monolayer. In this joint monolayer the abundance of V–O–V and V–O–Ti bonds is maximized and oligomers with mixed nuclearity are formed, which is reflected in peculiar electronic and catalytic properties of the material. In the present study, the maximization has been achieved by systematic synthetic means. An exact quantification of the corresponding active sites is difficult. A starting point might be the intensity of the Ti L3-edge pre-peak in the NEXAFS spectra, which will be tackled in future investigations.
The surface topology of the V/Ti/SBA-15 catalysts is likely to be governed by the differences in the reactivity of vanadyl(V) triisopropoxide OV(OCH(CH3)2)3 and titanium(IV) isopropoxide Ti(OCH(CH3)2)4 with the silica support. The titanium alkoxide is grafted on silanol groups in a 1:1 stoichiometry consuming only one of the four alkoxy ligands (eqn (1)). This stoichiometry allows cross-linking and the formation of ribbons and extended carpets by self-condensation via the remaining propoxy ligands and bears the potential for building three-dimensional structures as well. The high surface density and homogeneous distribution of silanol groups in SBA-15 certainly promotes the self-organization of titania in two-dimensional structures. In contrast, grafting of vanadium propoxide on silica consumes two of the three alkoxy ligands, which restricts the maximum nuclearity of vanadium clusters to two in a single grafting step, if the two V atoms are grafted to silica (eqn (2)). Reaction of vanadium isopropoxide with Ti–OH groups in the sequential grafting procedure may be responsible for cross-linking of two titania ribbons and formation of (Ti–O)2–VO–O–VO–(O–Ti)2 bridges, as may occur in the exceptional case of the V–Ti mixed monolayer catalyst 4V/13Ti/SBA-15 (Scheme 1).
 | (1) |
 | (2) |
4. Summary and conclusions
Using a multi-step grafting procedure, titania was deposited in high concentration within the pore network of a SBA-15 silica support. An unsurpassed loading of titanium was deposited without compromising the well-defined mesoporous architecture of the silica support. Surface coverage of almost the entire silica surface has been achieved due to the high surface density of silanol groups, which is characteristic for mesoporous silica SBA-15 and which was the motivation for selecting this type of support in the present study. The titanium oxide surface layers are composed of highly dispersed, two-dimensional titania species resulting in monolayer coverage without noticeable segregation of titanium oxide nano-particles at a Ti loading of 17 wt% that corresponds to a surface density of 5.8 Ti atoms per nmcat2 and a Si/Ti ratio of 3.3.Compared to vanadia supported on bulk titanium oxide, the V/Ti/SBA-15 catalysts show promising productivities.2,6,72 One reason might be the high specific surface area that can be achieved by using mesoporous silica as the support. In addition, the geometrical and electronic structure of surface titanium oxide species is different from bulk titania. Consequently, the interaction of vanadium oxide with bulk titanium oxide differs from the interaction with highly dispersed titania. Sub-monolayer titania promotes the dispersion of surface vanadia species up to a critical concentration. In the particular case, when the total metal loading approaches monolayer concentration, vanadium preferentially replenishes the residual free silica surface instead of being anchored on titania and/or acts as an end group or bridging ligand between two surface titania domains. The resulting V–Ti mixed monolayer catalyst is characterized by maximum productivity in the formation of propylene, which is attributed to the enhanced abundance of V–O–Ti bonds. The present study, which was aimed at synthesis and analysis of model catalysts, yielded an excellent catalyst for oxidative dehydrogenation of propane to propylene due to an unexpected enhancement of the catalytic activity through self-organization of the active phase.
Acknowledgements
The authors thank Daniel Brennecke for his support in catalyst synthesis, Gisela Lorenz and Maike Hashagen for their help with the N2 physisorption measurements, Dr. Frank Girgsdies for analysis by X-ray diffraction, and Achim Klein-Hoffmann for chemical analysis by X-ray fluorescence. The HZB staff is acknowledged for their continued support of the high-pressure electron spectroscopy activities of the FHI at BESSY II. We thank Professor Israel E. Wachs, Lehigh University, for helpful discussions and the measurement of Raman spectra shown in the ESI.† This work was supported by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) through the cooperative research center ‘‘Structure, dynamics, and reactivity of transition metal oxide aggregates’’ (Sonderforschungsbereich 546, http://www.chemie.hu-berlin.de/sfb546).References
- F. Cavani and J. H. Teles, ChemSusChem, 2009, 2, 508 CrossRef CAS.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113 CrossRef CAS.
- R. Schlögl, Top. Catal., 2011, 54, 627 Search PubMed.
- T. Blasco and J. M. Lopez Nieto, Appl. Catal., A, 1997, 157, 117 CrossRef CAS.
- A. Khodakov, B. Olthof, A. T. Bell and E. Iglesia, J. Catal., 1999, 181, 205 CrossRef CAS.
- A. A. Lemonidou, L. Nalbandian and I. A. Vasalos, Catal. Today, 2000, 61, 333 CrossRef CAS.
- G. Martra, F. Arena, S. Coluccia, F. Frusteri and A. Parmaliana, Catal. Today, 2000, 63, 197 CrossRef CAS.
- N. Ballarini, F. Cavani, A. Cericola, C. Cortelli, M. Ferrari, F. Trifiro, G. Capannelli, A. Comite, R. Catani and U. Cornaro, Catal. Today, 2004, 91 and 92, 99 Search PubMed.
- A. Christodoulakis, M. Machli, A. A. Lemonidou and S. Boghosian, J. Catal., 2004, 222, 293 CrossRef CAS.
- E. V. Kondratenko, M. Cherian and M. Baerns, Catal. Today, 2006, 112, 60 Search PubMed.
- D. Shee, T. V. M. Rao and G. Deo, Catal. Today, 2006, 118, 288 CrossRef CAS.
- A. Dinse, B. Frank, C. Hess, D. Habel and R. Schomäcker, J. Mol. Catal. A: Chem., 2008, 289, 28 CrossRef CAS.
- H. Tian, E. I. Ross and I. E. Wachs, J. Phys. Chem. B, 2006, 110, 9593 CrossRef CAS.
- K. Chen, A. T. Bell and E. Iglesia, J. Catal., 2002, 209, 35 CrossRef CAS.
- E. V. Kondratenko, M. Cherian, M. Baerns, D. Su, R. Schlögl, X. Wang and I. E. Wachs, J. Catal., 2005, 234, 131 CrossRef CAS.
- B. Olthof, A. Khodakov, A. T. Bell and E. Iglesia, Effects of Support Composition and Pretreatment Conditions on the Structure of Vanadia Dispersed on SiO2, Al2O3, TiO2, ZrO2, and HfO2, 2000, 107, 1516 Search PubMed.
- K. Chen, E. Iglesia and A. T. Bell, J. Catal., 2000, 192, 197 CrossRef CAS.
- A. Dinse, A. Ozarowski, C. Hess, R. Schomäcker and K.-P. Dinse, J. Phys. Chem. C, 2008, 112, 17664 Search PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, Ray Photoelectron Spectroscopy for Investigation of Heterogeneous Catalytic Processes, ch. 4 in Advances in Catalysis, ed. C. G. Bruce and K. Helmut, Academic Press, 2009, p. 213 Search PubMed.
- M. Hävecker, M. Cavalleri, R. Herbert, R. Follath, A. Knop-Gericke, C. Hess, K. Hermann and R. Schlögl, Phys. Status Solidi B, 2009, 246, 1459 Search PubMed.
- F. de Groot, Chem. Rev., 2001, 101, 1779 CrossRef CAS.
- F. M. F. de Groot and A. Kotani, Core spectroscopy of solids, Taylor and Francis, New York, 2008 Search PubMed.
- H. Ikeno, F. M. F. de Groot, E. Stavitski and I. Tanaka, J. Phys.: Condens. Matter, 2009, 21, 104208 Search PubMed.
- E. Stavitski and F. M. F. de Groot, Micron, 2010, 41, 687 Search PubMed.
- K. Inumaru, T. Okuhara and M. Misono, J. Phys. Chem., 1991, 95, 4826 CrossRef CAS.
- X. Gao, S. R. Bare, J. L. G. Fierro and I. E. Wachs, J. Phys. Chem. B, 1999, 103, 618 CrossRef CAS.
- J. Keranen, C. Guimon, E. Iiskola, A. Auroux and L. Niinisto, J. Phys. Chem. B, 2003, 107, 10773 CrossRef CAS.
- M. Cozzolino, M. Di Serio, R. Tesser and E. Santacesaria, Appl. Catal., A, 2007, 325, 256 CrossRef CAS.
- B. Bonelli, M. Cozzolino, R. Tesser, M. Di Serio, M. Piumetti, E. Garrone and E. Santacesaria, J. Catal., 2007, 246, 293 CrossRef CAS.
- A. Bruneau and J. P. Gallas, in Surface Properties of Silica, ed. A. P. Legrand, Wiley, New York, 1999, p. 194 Search PubMed.
- D. E. Keller, T. Visser, F. Soulimani, D. C. Koningsberger and B. M. Weckhuysen, Vib. Spectrosc., 2007, 43, 140 CrossRef CAS.
- D. J. C. Yates, J. Phys. Chem., 1961, 65, 746 CAS.
- R. J. Davis and Z. F. Liu, Chem. Mater., 1997, 9, 2311 CrossRef CAS.
- C. d. Carteret, J. Phys. Chem. C, 2009, 113, 13300 Search PubMed.
- J.-P. Gallas, J.-M. Goupil, A. Vimont, J.-C. Lavalley, B. Gil, J.-P. Gilson and O. Miserque, Langmuir, 2009, 25, 5825 CrossRef CAS.
- E. Santacesaria, M. Cozzolino, M. Di Serio, A. M. Venezia and R. Tesser, Appl. Catal., A, 2004, 270, 177 CrossRef CAS.
- M. Graetzel and F. P. Rotzinger, Inorg. Chem., 1985, 24, 2320 CrossRef.
- T. Ohsaka, F. Izumi and Y. Fujiki, J. Raman Spectrosc., 1978, 7, 321.
- S. P. S. Porto, P. A. Fleury and T. C. Damen, Phys. Rev., 1967, 154, 522 CrossRef CAS.
- A. Fernandez, J. Leyrer, A. R. Gonzalez-Elipe, G. Munuera and H. Knözinger, J. Catal., 1988, 112, 489 CrossRef CAS.
- X. Gao and I. E. Wachs, Catal. Today, 1999, 51, 233 CrossRef CAS.
- S. Klein, B. M. Weckhuysen, J. A. Martens, W. F. Maier and P. A. Jacobs, J. Catal., 1996, 163, 489 CrossRef CAS.
- D. T. On, L. L. Noc and L. Bonneviot, Chem. Commun., 1996, 299 RSC.
- X. Gao, S. R. Bare, J. L. G. Fierro, M. A. Banares and I. E. Wachs, J. Phys. Chem. B, 1998, 102, 5653 CrossRef CAS.
- G. S. Henderson, X. Liu and M. E. Fleet, Phys. Chem. Miner., 2002, 29, 32 CrossRef CAS.
- E. Stoyanoy, F. Langenhorst and G. Steinle-Neumann, Am. Mineral., 2007, 92, 577 Search PubMed.
- P. Krueger, Phys. Rev. B, 2010, 81, 125121 Search PubMed.
- F. M. F. de Groot, M. O. Figueiredo, M. J. Basto, M. Abbate, H. Petersen and J. C. Fuggle, Phys. Chem. Miner., 1992, 19, 140 CAS.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter, 1990, 41, 928 CrossRef.
- M. Cheynet, S. Pokrant, S. Irsen and P. Krueger, Ultramicroscopy, 2010, 110, 1046 Search PubMed.
- U. Scharf, M. Schraml-Marth, A. Wokaun and A. Baiker, J. Chem. Soc., Faraday Trans., 1991, 87, 3299 RSC.
- J.-M. Jehng and I. E. Wachs, Catal. Lett., 1992, 13, 9 CrossRef CAS.
- J. Keraenen, C. Guimon, A. Auroux, E. I. Iiskola and L. Niinistoe, Phys. Chem. Chem. Phys., 2003, 5, 5333 RSC.
- H.-M. Lin, S.-T. Kao, K.-M. Lin, J.-R. Chang and S.-G. Shyu, J. Catal., 2004, 224, 156 Search PubMed.
- D. Shee and G. Deo, Catal. Lett., 2008, 124, 340 Search PubMed.
- M. Schraml-Marth, A. Wokaun, M. Pohl and H.-L. Krauss, J. Chem. Soc., Faraday Trans., 1991, 87, 2635 RSC.
- R. Monaci, E. Rombi, V. Solinas, A. Sorrentino, E. Santacesaria and G. Colon, Appl. Catal., A, 2001, 214, 203 CrossRef CAS.
- A. Sorrentino, S. Rega, D. Sannino, A. Magliano, P. Ciambelli and E. Santacesaria, Appl. Catal., A, 2001, 209, 45 CrossRef CAS.
- A. Comite, A. Sorrentino, G. Capannelli, M. Di Serio, R. Tesser and E. Santacesaria, J. Mol. Catal. A: Chem., 2003, 198, 151 CrossRef CAS.
- E. Santacesaria, A. Sorrentino, R. Tesser, M. Di Serio and A. Ruggiero, J. Mol. Catal. A: Chem., 2003, 204 and 205, 617 Search PubMed.
- K. I. Hadjiivanov and D. G. Klissurski, Chem. Soc. Rev., 1996, 25, 61 RSC.
- H. Knözinger and S. Huber, J. Chem. Soc., Faraday Trans., 1998, 94, 2047 RSC.
- T. P. Beebe, P. Gelin and J. T. Yates Jr, Surf. Sci., 1984, 148, 526 CrossRef CAS.
- X. Gao and I. E. Wachs, J. Phys. Chem. B, 2000, 104, 1261 CrossRef CAS.
- Z. Wu, H.-S. Kim, P. C. Stair, S. Rugmini and S. D. Jackson, J. Phys. Chem. B, 2005, 109, 2793 CrossRef CAS.
- C. Hess, G. Tzolova-Müller and R. Herbert, J. Phys. Chem. C, 2007, 111, 9471 CrossRef CAS.
- P. Grüne, T. Wolfram, K. Pelzer, R. Schlögl and A. Trunschke, Catal. Today, 2010, 157, 137 CrossRef CAS.
- Y.-M. Liu, Y. Cao, N. Yi, W.-L. Feng, W.-L. Dai, S.-R. Yan, H.-Y. He and K.-N. Fan, J. Catal., 2004, 224, 417 CrossRef CAS.
- A. Dinse, S. Khennache, B. Frank, C. Hess, R. Herbert, S. Wrabetz, R. Schlögl and R. Schomäcker, J. Mol. Catal. A: Chem., 2009, 307, 43 CrossRef CAS.
- A. Khodakov, J. Yang, S. Su, E. Iglesia and A. T. Bell, J. Catal., 1998, 177, 343 CrossRef CAS.
- E. Heracleous, M. Machli, A. A. Lemonidou and I. A. Vasalos, J. Mol. Catal. A: Chem., 2005, 232, 29 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00541g |
| This journal is © The Royal Society of Chemistry 2012 |