Factors driving the activity of commercial titanium dioxide powders towards gas phase photocatalytic oxidation of acetaldehyde
Sammy W.
Verbruggen†
ab,
Kasper
Masschaele†
b,
Elke
Moortgat
b,
Tamas E.
Korany
b,
Birger
Hauchecorne
a,
Johan A.
Martens
*b and
Silvia
Lenaerts
*a
aFaculty of Science, Department of Bioscience Engineering, Research unit Sustainable Energy and Air Purification, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium. E-mail: silvia.lenaerts@ua.ac.be
bFaculty of Bio-engineering, Department of Microbial and Molecular Systems, Kasteelpark 23, 3001 Heverlee (Leuven), Belgium. E-mail: Johan.Martens@biw.kuleuven.be
First published on 13th June 2012
Abstract
The photocatalytic activity of two commercial titanium dioxide powders (Cristal Global, Millennium PC500 and Evonik, P25) is compared towards acetaldehyde degradation in the gas phase. In contrast to the extensive literature available, we found a higher activity for the PC500 than for the P25 coating. Here, we present a comprehensive characterization of the bulk and surface properties of both powders. Our comparison shows that the material properties that dominate the overall photocatalytic activity in gas phase differ from those required for the photodegradation of water-borne pollutants.
1 Introduction
The atmosphere, and especially indoor air, contains many volatile organic compounds (VOCs) resulting from human respiration, environmental tobacco smoke (ETS) and the slow emission from infrastructural materials. Their concentrations in indoor air can amount to considerably higher levels than outdoors, even though some of them may be harmful to humans already at ppb levels. The discovery by Fujishima and Honda1 of photocatalytic splitting of water instigated extensive research for the use of TiO2 as a material for photocatalytic environmental purification. Titanium dioxide is an ideal photocatalyst since it is relatively inexpensive to produce, non-toxic, chemically stable and has relatively high reactivity for elimination of air and water borne pollutants. The gas phase photo-oxidation of organic substances was pioneered by McLintock and Ritchie already in the early 1960's.2 Since then, the solid-gas heterogeneous photocatalytic reactions have received much attention.3,4Recently, the large potential of gas phase photocatalysis for environmental applications has been postulated since a wide range of air contaminants including alkanes,5 alkenes,6 halocarbons,7 alcohols,8 aldehydes,9 nitrogen-containing compounds,10 and ketones11 can be partially or completely degraded with the TiO2-assisted UV irradiation. In addition, TiO2 based semiconductors have been used for the photocatalytic degradation of chlorinated organics, pesticides and herbicides, surfactants, dyes, and destruction of bacteria, viruses, and molds in water.3 In this context it is worthwhile pointing out that photocatalytic methods of decontamination are ideal since they operate at near ambient conditions using only light as the source of energy. Thus, photocatalysis has emerged as an advanced oxidation process (AOP). Although some success has been realized in the complete oxidation of several organics at the laboratory scale, photocatalysis using TiO2 still suffers from relatively low quantum efficiency.
Here, the photocatalytic degradation of acetaldehyde into CO2 and H2O is studied on two titanium dioxide catalysts. The goal of our study is to determine the required material properties to obtain an optimal photocatalytic activity. Previous reports have discussed the photocatalytic degradation of acetaldehyde in detail,12–17 but in addition it has been recognized that acetaldehyde is encountered as an intermediate in the photocatalytic degradation of ethanol.18,19
In this work, we study photocatalytic degradation of acetaldehyde in the gas phase using two commercial TiO2 nanopowders (Millennium PC500 and Degussa P25). In contrast to many earlier reports and especially water-borne applications, we observed better activity for the PC500 than for the P25 powder.20–23 The aim of this work is to get a fundamental understanding of the parameters that determine the photocatalytic activity of a photocatalyst for gas phase purification. A comprehensive characterization of both materials enables us to distinguish the important bulk and surface parameters. More specifically, the operating principles in the gas and water phase performance are of major interest.
2 Experimental
2.1 Chemical reagents
All the products were used as received without any modification or purification, unless stated otherwise. For our study we chose two different titanium dioxide powders; the universal reference P25 (Evonik) and Millennium PC500 (Cristal Global). The extensive reports of the superiority of P25 as an unmodified photocatalyst and the very high surface area of the PC500 motivated us to choose these powders for our study. According to the supplier data and as confirmed by various groups PC500 has a specific surface area of 350 m2 g−1 and a primary particle size of 10 nm, whereas P25 has a specific surface area of 50 ± 15 m2 g−1 and a particle size of approximately 21 nm. Acetaldehyde was studied as a pollutant gas. For the gas phase measurements, acetaldehyde was used in its gaseous form (Air Liquide, 1% in N2) mixed with compressed air (Air Liquide Alphagaz) in a way that the resulting concentration was 100 or 170 ppmv. No additional water was supplied to the gas stream.2.2 Gas phase photocatalytic tests
The photoactive powders were immobilized on glass beads (Assistent, No. 1401/2, 2 mm diameter) by means of suspension coating and loaded in a continuous gas flow reactor. The entire coating protocol, testing procedure and analyzing method were validated and described in our previous work.24 In short, the powders were suspended in ethanol and ultrasonically stirred for 30 min, after which a total amount of 108 g of cleaned glass beads was coated with the suspension and dried overnight at 338 K. In this way 50 mg of catalyst was deposited on the beads, with a minimal thickness of 100 nm assuming a dense and uniform coating. The gas phase photoreactor consists of a borosilicate glass cylinder (400 mm long, 22 mm wide) surrounding a Philips Cleo 25 W UVA lamp (500 mm long, 16 mm wide and peak intensity at 365 nm) that is placed coaxially. The emitted light intensity was measured at the lamp wall using an Avantes Avespec-3648 spectrometer, and determined to be approximately 205 μW cm−2 at the peak wavelength of 365 nm. The emitted intensity was observed to decrease slightly towards the edges. The reactor dimensions are smaller than those of the lamp, thus these small deviations are neglectable. The coated beads were then loaded in the annular space (3 mm wide) between two coaxial cylinders formed by the lamp on the inner side and the reactor wall on the outer side, resulting in a remaining void fraction of 47% leading to a gas residence time of approximately 1.0 s in the reactor. A continuous gas flow spiked with 100 or 170 ppmv acetaldehyde in air was passed over the reactor bed by means of mass flow controllers (Brooks 5850E) at a total rate of 2000 cm3 min−1. Taking into account the catalyst loading of 50 mg, this results in a contact time of 100 g h mol−1 for the 100 ppmv and 59 g h mol−1 for the 170 ppmv inlet concentration respectively. For the first 10 minutes of the measurement, the polluted gas flow is bypassed around the catalyst bed, in order to establish a reference level for 100% of the acetaldehyde inlet concentration reaching the detector. For the final 10 minutes of the measurement, the flow is bypassed again for verification. After 20 min flow through the reactor in the dark to establish adsorption equilibrium, the UVA lamp was switched on for 60 min in order to observe photocatalytic degradation of acetaldehyde and formation of reaction products. Afterwards again 20 min flow in the dark was administered in order to return to the reference concentration of acetaldehyde. The difference between the saturated level after adsorption/desorption and the level during illumination is a measure for the amount of reagent/product degraded/formed.The concentration of the reagents and products was monitored in time by on-line FTIR spectroscopy of the outlet gas flow. The absorbance peak heights of vibrations of molecules of interest were plotted as a function of time by recording four to five FTIR spectra per minute and analyzing the peaks with the MacrosBasic software (Thermo Fisher). In particular, for acetaldehyde, CO2 and CO the peak heights were recorded at wavenumbers of 2728 cm−1, 2360 cm−1 and 2179 cm−1 respectively. These positions were selected because they do not interfere with other vibrations in the spectrum. The automation of the test set-up is described in more detail by Tytgat et al.25
2.3 Aqueous photocurrent measurements
A three-electrode cell was used to perform photo-electrochemical experiments in a 1 M NaOH aqueous electrolyte solution. The reference was an Ag/AgCl electrode (radiometer, 3 M KCl–AgCl saturated) and the counter electrode consisted of a platinum ring. The work electrode consists of a 1 cm2 ITO coated glass, connected with a copper wire by conducting silver paste. The connection was sealed with an epoxy-resin. The work-electrode acted as an anode and was confined in a cell made of quartz glass. The TiO2 nanopowder coating was applied by spincoating. First, dry powder was dispersed in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of ethanol and water and ultrasonicated for 10 minutes. After deposition, samples were dried at room temperature for 24 h and finally calcined at 400 °C for 3 hours in air. The PC500 properties were not affected by the heat treatment as confirmed by XRD and BET measurements.26 The counter electrode (cathode) and reference electrode were confined in a cell made of boroslicate glass. Dark current experiments were performed under a bias of 0.2 V. Photomeasurements were conducted using a 500 W Xe/Hg(Xe) light source (Oriel, Newport) with an infrared filter. The incident light had an intensity of 100 mW cm−2. A potentiostat VersaStat4 (Princeton Applied Research, Tennessee) was used to probe photocurrents and potentials. The photocurrents were measured by chopped illumination with a frequency of 0.01 s−1 at an intensity of 100 mW.
:50 mixture of ethanol and water and ultrasonicated for 10 minutes. After deposition, samples were dried at room temperature for 24 h and finally calcined at 400 °C for 3 hours in air. The PC500 properties were not affected by the heat treatment as confirmed by XRD and BET measurements.26 The counter electrode (cathode) and reference electrode were confined in a cell made of boroslicate glass. Dark current experiments were performed under a bias of 0.2 V. Photomeasurements were conducted using a 500 W Xe/Hg(Xe) light source (Oriel, Newport) with an infrared filter. The incident light had an intensity of 100 mW cm−2. A potentiostat VersaStat4 (Princeton Applied Research, Tennessee) was used to probe photocurrents and potentials. The photocurrents were measured by chopped illumination with a frequency of 0.01 s−1 at an intensity of 100 mW.
2.4 FTIR in situ adsorption study
The adsorption of acetaldehyde in gas phase on both PC500 and P25 was studied in an FTIR in situ reactor as described by Hauchecorne et al.17 P25 or PC500 was pressed into FTIR-transparent pellets and placed in a closed reactor focusing the IR beam at the center of the pellet. After applying a gas flow of 5000 ppmv acetaldehyde for 15 min, the reactor was placed in a bath and was left to stabilize for 1 h. In the meantime, spectra of the FTIR transparent catalyst pellet were recorded every 20 s, which provided information of the physico–chemical interactions determining the adsorption processes. Gas phase corrections are performed to ensure the absence of interference and to obtain solely the absorption bands of adsorbed molecules.2.5 Instrumentation
On-line FTIR spectroscopy of the gas phase reactor outlet was performed using a NicoletTM 380 FTIR spectrometer (Thermo Fisher Scientific) with ZnSe windows and a 2 m heated gas cell. Spectra were recorded in a range of 4000–400 cm−1 at a resolution of 1 cm−1 and an absorbance detection limit of 0.001 a.u., corresponding to 4.5 ppmv acetaldehyde, as was validated by constructing a calibration curve (R = 0.98) using a Dräger Polytron Organic Vapor Sensor. Determination of the Brunauer–Emmett–Teller (BET) surface area was performed with a Micrometrics Tristar Surface Area and Porosity Analyzer. The samples were degassed at 573 K for 3 h. X-ray powder diffraction (XRD) was carried out using a STOE StadiP apparatus with Cu Kα radiation and an image plate detector.UV-VIS absorption spectra were collected in the range of 300–700 nm using a Shimadzu UV-2501PC double beam spectrophotometer equipped with a 60 mm BaSO4 coated integrating sphere and a Photomultiplier R-446U detector.
3 Results and discussion
3.1 Photo-induced degradation of acetaldehyde
In a typical gas phase degradation experiment, the photoreactor was loaded with glass spheres coated with P25 or PC500 nanopowder. A gas stream containing 100 or 170 ppmv acetaldehyde was then introduced into the photoreactor. The concentration profiles of acetaldehyde and the degradation product CO2 in the outlet gas stream were monitored by FTIR peak heights. An example for a stream containing 100 ppmv is shown in Fig. 1. In the first step (I) the inlet gas-stream is by-passed directly to the outlet and the thus obtained FTIR signal defines the reference level. The flow is allowed to enter the reactor in the second step (II) under dark conditions. The drop in acetaldehyde concentration in step II is attributed to adsorption of the acetaldehyde molecules on the catalyst surface, reactor walls or any interaction with chemical species present (adsorbents, water, etc.). Blank tests with an empty reactor or loaded with uncoated glass beads show no such drop, providing evidence that adsorption on the catalyst surface is prevailing. The transient behavior in step II shows that more acetaldehyde can be adsorbed on the PC500 catalyst compared to the P25 system.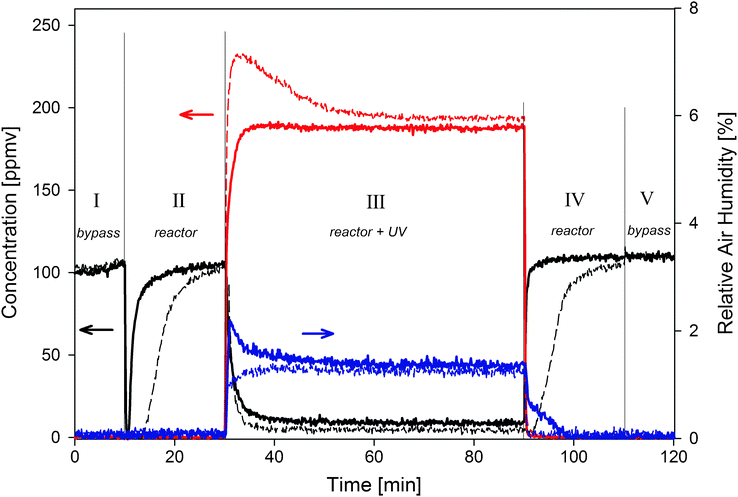 | ||
| Fig. 1 Concentration profiles of acetaldehyde and CO2 over P25 (solid line) and PC500 (dashed line) in the outlet gas stream during one experiment. The acetaldehyde inlet concentration is 100 ppmv in air at a total flow rate of 2000 cm3 min−1. Red: CO2, blue: H2O, black: acetaldehyde. | ||
When UV illumination is switched on in step III, the FTIR signal reveals an immediate decrease in the acetaldehyde concentration in the outlet gas-stream, and additionally CO2 is formed. The decreased acetaldehyde concentration in combination with the formation of CO2 indicates photo-induced degradation of acetaldehyde. When the reactor was loaded with uncoated glass beads no acetaldehyde conversion was found, indicating the absence of photolysis.
Probing the acetaldehyde concentration in the outlet gas-stream it is suggested that for the P25, an equilibrium is established after approximately 10 minutes and a steady-state conversion of 93 ± 1% is observed. The PC500 material shows steady state behavior after more than 30 minutes and shows nearly 100% conversion. The steady-state concentrations of CO2 monitored by FTIR peak height are consistent with the trend observed from the acetaldehyde levels during degradation, with more acetaldehyde degradation at PC500 coated beads compared to P25 coated ones. Initially, the excessive conversion on PC500 is significant compared to the P25 powder, as can be verified from Fig. 1, step III. The observed overshoot in the produced CO2 concentration for PC500 is reproducible (data not shown) and the height of the overshoot is determined by the acetaldehyde loading concentration. For an increasing inlet concentration of acetaldehyde the overshoot gradually decreased and finally disappeared completely. The P25 samples on the other hand succeeded in a degradation of 76% of the introduced acetaldehyde in a 170 ppmv flow, in contrast to a conversion of 99% for the PC500 coated beads (data not shown). For a 100 ppmv inlet stream the turnover frequency (TOF) of acetaldehyde on PC500 and P25 equals 0.7 mol per mol TiO2 per hour, and 0.54 mol per mol TiO2 per hour, respectively. Furthermore, for the P25 the absolute amount of acetaldehyde degraded was independent of the inlet concentration (for concentrations exceeding 170 ppmv). This suggests that the degradation rate is kinetically determined (light absorption or charge transfer) and mass transfer issues can be neglected in this case.
The steady state conversion remains constant for long times which convinces us that adsorbed intermediates do not affect the activity of the photocatalyst, nor the absence of additional humidity becomes the rate limiting step. Fouling or poisoning of the catalyst would result in a gradual decrease in activity because all active sites are occupied and no further conversion is observed. Absence of water near the surface could result in a lower rate of OH radical formation during illumination. In any case, initially the photocatalysts contain an appreciable amount of physically and chemically adsorbed water. Furthermore, water is formed during the photocatalytic degradation of acetaldehyde as shown in Fig. 1 and previously reported by us.17 Long term measurements for 30 hours showed that there was no observable decrease in acetaldehyde degradation or CO2 formation (data not shown).
In the following we characterize the PC500 and P25 powders and distinguish the important material properties:
1. Surface properties
2. Bulk properties
3. Morphology
The bulk properties will determine the photon-to-electrical conversion in the form of electron–hole pairs (quantum efficiency). Ideally the bulk properties support charge separation of the photo-generated holes and electrons, resulting in good apparent quantum efficiency. Secondly, the surface properties will determine to a great extent the activity of the catalyst. A high concentration of reactive species and optimal adsorption behavior are preferred. Finally, the morphology is important because of possible diffusion limitations and effective available surface area. Therefore, the thickness, porosity and crystal structure of the coatings need to be optimized.27
3.2 Bulk versus surface recombination
The bulk properties, related to the photon absorption, together with the surface properties determine the overall quantum efficiency of the photocatalyst materials. The bulk properties depend on the crystallinity of the semiconductor, the primary particle size and the morphology of the coating. The UV-VIS absorption spectra of P25 and PC500 are shown in Fig. 2(a).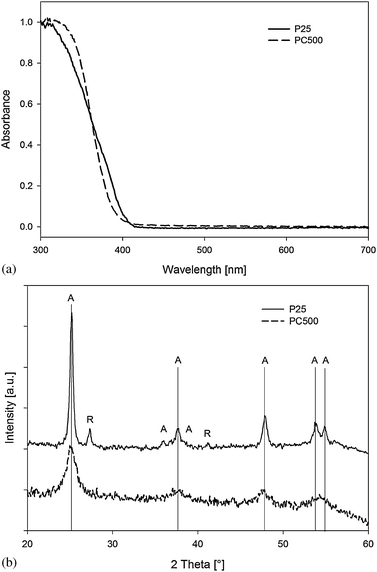 | ||
| Fig. 2 (a) DRS UV-VIS and XRD (b) scans for titanium dioxide powders P25 and PC500. Anatase: A, rutile: R. | ||
Using the Kubelka–Munk function, the bandgap energies of P25 and PC500 are calculated to be 3.08 and 3.20 eV respectively. The bandgap of PC500 corresponds to that of pure TiO2 anatase, whereas the lower bandgap of P25 can be explained by the presence of 20% rutile. Although, the absorption spectra reveal the photoresponse, the photocatalytic behavior is determined by measuring the generated photocurrents upon illumination in different electrolyte solutions.28 To this extent, transparent electrodes of P25 and PC500 were coated on ITO glass slides and subsequently illuminated under a bias of +0.2 V vs. Ag/AgCl. When an electrolyte containing 1 M NaOH was used, the photocurrent generated on the P25 electrodes was found to be much higher than for the PC500 electrode, as shown in Fig. 3. However, when methanol (15 vol%) was added to the electrolyte the P25 electrode gave higher photocurrents compared to the pure water system (an increase from 950 to 1900 μA), but the PC500 electrode showed an increase in the photocurrent by a factor of 200 (from 4 to 730 μA).
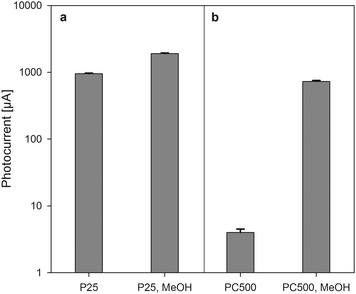 | ||
| Fig. 3 Average photocurrents measured over P25 (a) and PC500 (b) electrodes. The first bar is the average photocurrent measured in water only, the second bar is the average photocurrent measured in water with methanol added. | ||
These results enable a direct distinction between pure bulk and surface behavior. In a pristine 1 M NaOH electrolyte the generated photoholes are consumed by OH-groups, while dissolved oxygen is reduced by the photo-electrons. When methanol is added, on the other hand, methanol oxidation is preferred over water splitting. It has been shown that the TiO2 hole transfer to methanol is an ultrafast process and can proceed with a time constant of ≈300 ps, while for water it can be as high as 2 ms.29 By overcoming the limitations of the hole transfer step with sacrificial donors (methanol), it is possible to boost the lifetime of electrons.30 Use of alcohols such as methanol31 as sacrificial donors has also been found to be favorable in doubling the photocurrent generation in photoelectrochemical cells since the alkoxy radicals formed upon hole oxidation are capable of delivering electrons to the semiconductor electrode.32 By adding methanol to the electrolyte we have eliminated to a certain extent the effect of the surface properties that enables us to conclude that more bulk recombination occurs in the PC500 catalyst compared to the P25 (approximately a factor of 2–3). For two materials having a similar surface concentration of surface hydroxyls, but different quantum efficiency (photon-to-exciton conversion), the material with the highest quantum efficiency will yield a higher number of hydroxyl radicals per unit surface area.
The improved quantum efficiency of P25 is attributed to the crystalline structure with crystallites of 30 nm and an anatase/rutile ratio of approximately 4/1, as can be seen in Fig. 2(b). Although the mechanism remains speculative, it is expected that the interface between the rutile and anatase crystals favors charge separation for longer time periods.33 The PC500 on the other hand was found to be 75% anatase with a primary particle size of approximately 5–10 nanometers and 25% amorphous titanium dioxide which favors bulk recombination resulting in a lower overall conversion of photons in photo-generated charge carriers. Although the photocurrent measurements give direct evidence for a lower photon conversion per unit of surface area on the PC500, some additional sources for recombination are introduced in the latter experiments. The photogenerated electrons need to travel towards the ITO substrate. The amorphous nature of PC500 and the small primary particle size create many possible trapping sites and thus an enhanced recombination of photo-generated charge carriers, lowering the observed photocurrents. In the original gasphase experiments the electrons have to diffuse only to small distances before they reach the electrolyte and reduce the adsorbed oxygen.
3.3 Reaction pathway
The lower quantum efficiency of the PC500 may also affect the exact reaction pathway.27 It is known that the oxidation of acetaldehyde depends on the concentration of available photoholes. For a high concentration of holes, acetaldehyde is oxidized readily into CO2 and H2O, where for lower photohole concentration acetic acid is found as an intermediate, as given in eqn (1) and (2).| CH3CHO + 3H2O + 10h+ → 2CO2 + 10H+ | (1) |
| CH3CHO + H2O + 2h+ → CH3COOH + 2H+ | (2) |
3.4 Surface properties
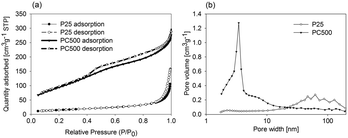 | ||
| Fig. 4 (a) Adsorption/desorption isotherms of P25 and PC500 and (b) pore volume distribution plot of P25 and PC500. A logarithmic scale for the pore width was used for clarity purposes. | ||
The effective surface area available for acetaldehyde adsorption on PC-500 was found to be 3.5 times higher compared to the P25, as determined from Fig. 5. Integration of the area in zone I, and in zones I + II, provides estimates of the amount of adsorbed acetaldehyde on the P25 and PC500, respectively. The total surface area of PC500, as determined from BET, was six times larger than that of P25, but the effective surface area for acetaldehyde adsorption is found to be much smaller. A study of the surface chemistry of both powders should indicate the origin of this difference.
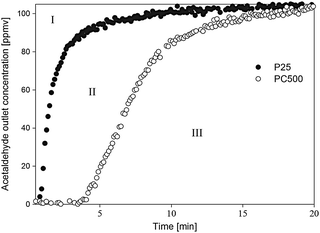 | ||
| Fig. 5 Outlet concentration of acetaldehyde after adsorption on P25 and PC500 in function of time. | ||
1. the surface hydroxyl concentration
2. the surface cation concentration
3. the presence of adsorbed intermediates, oxygen, surface groups. It is commonly acknowledged that hydroxyl groups play a major role in the photo-oxidation of organic compounds. Here, we will investigate the total number of surface hydroxyls and the surface density of hydroxyls on both materials. Secondly, Fourier Transformed Infrared spectra enable us to study the interactions between acetaldehyde molecules and the surface hydroxyl groups. A first estimate of the physically and chemically bonded water or hydroxyls is given by thermogravimetric measurements (TGA). Most of the physically bound water is expected to evaporate at temperatures around 170–200 °C. At higher temperatures, the chemically bound hydroxyls are released, and at temperatures exceeding 400 °C phase transformation can be expected. For the P25, the total mass loss at 400 °C is approximately 2.5%. The total mass of the PC500 decreases rapidly up to temperatures of 400 °C, and then levels off. At a steady temperature increase of 10 °C min−1 almost 17% mass loss was found for the PC500 at 400 °C. If only the mass loss between 170 °C and 400 °C is considered, a simple estimation yields values of 2.7 OH per nm2 for PC500 and 5 OH per nm2 for P25, which is in good agreement with suppliers info. FTIR analysis of the OH stretching region (4000–3500 cm−1) on both materials was in agreement with the spectrum earlier reported in the literature.34
The FTIR spectra of P25 and PC500 when brought into contact with acetaldehyde discern differences in acetaldehyde adsorption. Fig. 6 shows the spectra after equilibrating the reactor for 10 min with acetaldehyde in air. The FTIR spectrum of acetaldehyde in the gas phase is shown in Fig. 6(a), and the typical peaks for the carbonyl group are present at 1761, 1746, and 1734 cm−1. When the acetaldehyde is adsorbed, the carbonyl bands are redshifted towards lower wavenumbers, indicating a significant interaction between the acetaldehyde molecules and the TiO2 surface. In the case of P25 (see Fig. 6(b)), it is found that acetaldehyde adsorbs in two different ways on the TiO2 surface. A H-bridge can be formed with a surface hydroxyl group (CH3CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯HO–Ti), resulting in a redshift of the IR band to 1714 cm−1. The second mode results in an IR band at 1693 cm−1 and is assigned to the carbonyl bonding with a cation Ti4+-surface group (CH3CHO⋯Ti4+).35 For the adsorption on PC500 (see Fig. 6(c)) on the other hand, only at 1714 cm−1 an IR absorption peak was found, indicating that the acetaldehyde dominantly binds with a surface hydroxyl of the PC500 surface. The presence of high amounts of water that preferentially bind to the cations might be a possible explanation. The peak absorbed at 1633 cm−1 is attributed to Ti4+ coordinated water, and that at 1621 cm−1 to water coordinated through hydrogen bonds.36 Overall, bands near 1630 cm−1 are assigned to adsorbed water.
O⋯HO–Ti), resulting in a redshift of the IR band to 1714 cm−1. The second mode results in an IR band at 1693 cm−1 and is assigned to the carbonyl bonding with a cation Ti4+-surface group (CH3CHO⋯Ti4+).35 For the adsorption on PC500 (see Fig. 6(c)) on the other hand, only at 1714 cm−1 an IR absorption peak was found, indicating that the acetaldehyde dominantly binds with a surface hydroxyl of the PC500 surface. The presence of high amounts of water that preferentially bind to the cations might be a possible explanation. The peak absorbed at 1633 cm−1 is attributed to Ti4+ coordinated water, and that at 1621 cm−1 to water coordinated through hydrogen bonds.36 Overall, bands near 1630 cm−1 are assigned to adsorbed water.
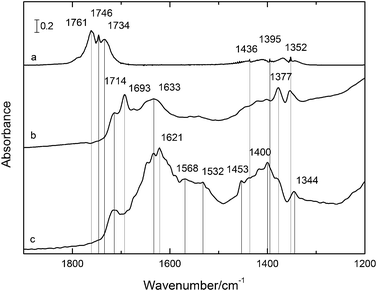 | ||
| Fig. 6 IR spectra at RT of (a) acetaldehyde in the gas phase; (b) acetaldehyde after 10 min adsorption on P25; (c) acetaldehyde after 10 min adsorption on PC500. Wavenumbers are summarized in Table 1. | ||
| Species [1] | Mode [1] | Gas | P25 | PC500 |
|---|---|---|---|---|
| CH3CHOg |
ν(CO) |
1761 | ||
| CH3CHOg |
ν(CO) |
1746 | ||
| CH3CHOg |
ν(CO) |
1734 | ||
| CH3CHOad |
ν(CO) |
1714 | 1714 | |
| CH3CHOad |
ν(CO) |
1693 | ||
| Ti–OH2 | δ(OH) | 1633 | ||
| Ti–OH2 | δ(OH) | 1621 | ||
| CH3COOad− | ν as(COO) | 1568 | ||
| CH3COOad− | ν as(COO) | 1532 | ||
| CH3COOad− | ν s(COO) | 1453 | ||
| CH3CHOg | δ as(CH3) | 1436 | ||
| CH3CHOad | δ(CH3) | 1400 | ||
| CH3CHOg | δ s(CH3) | 1395 | ||
| CH3CHOad | δ(CH3) | 1377 | ||
| CH3CHOg | δ(CH) | 1352 | ||
| CH3CHOad | δ(CH) | 1353 | 1344 |
Broad bands near 1550 cm−1 and 1444 and 1400 cm−1 were observed only for the PC500, which are attributed to acetic acid and acetate νas and νs vibrations respectively.37 Although acetic acid adsorbed on titanium dioxide is expected to induce absorption peaks at 1415 cm−1 and 1540 cm−1, the peak at 1400 cm−1 is intriguing. The coordination of adsorbed acetic acid or acetate by adsorbed or dissociated water may reduce possible vibration modes. The band at 1540 and 1400 cm−1 persisted upon heating of the PC500 sample, where the 1541 and 1414 cm−1 peaks remained even up to 400 °C, which may indicate strong adsorption of the acetic acid on the TiO2 surface.37
In Table 2 a brief overview of the studied parameters is given. The high surface area of PC500 is found to be an enormous advantage for the gas phase degradation of acetaldehyde. The adsorbed amount of acetaldehyde agrees with the total number of OH groups on the PC500 surface, and thus exceeds the adsorbed quantity on P25 by a factor of 3.5 (ratio equals 0.28). If acetaldehyde adsorption would only occur through interaction with surface hydroxyls, a factor of 4 would be expected, but from FTIR analysis it was found that on P25 adsorption occurs also on Ti4+ cations. The improved activity (TOF) of PC500 in comparison with P25 is thus rationalized by considering the high surface area, absolute amount of surface hydroxyls, adsorption and the possible reaction paths which can be determined by the photo-absorption efficiency, as discussed in Section 3.3. Considering surface properties alone, the TOF on P25 would be approximately 0.25 of that on PC500. In our experiments, however, the TOF on P25 is 0.77 of that on PC500, which is believed to be attributed to the bulk properties. Indeed the high crystallinity of P25 compared to PC500 is confirmed by XRD in Fig. 2 and the larger bandgap for PC500 implies a lower quantum efficiency for PC500 compared to P25. Unfortunately, current data do not provide enough information to quantitatively determine the major losses. Recombination losses due to the high fraction of amorphous titanium dioxide are believed to further decrease the quantum efficiency on PC500, eventually resulting in a ratio of the TOF of 0.77.
| Property | Ratio P25/PC500 |
|---|---|
| TOF | 0.77 |
| Total surface area | 0.15 |
| OH surface density | 2 |
| OH total amount | 0.25 |
| Acetaldehyde adsorbed | 0.28 |
In summary, in the specific case of acetaldehyde oxidation in the gasphase the high surface area of PC500 can be fully exploited. Although the low hydroxyl concentration per unit area and the inferior quantum efficiency will limit the available hydroxyl radicals during illumination, the PC500 material is found to have a higher TOF than P25.
Our results confirm that for photocatalytic degradation of VOC in the gas phase a high surface area is beneficial. Earlier reports, however, indicate that the photodegradation of water-borne organic dyes or organic pollutants occurs better on P25 compared to PC500. The reason is believed to be two-fold. First, the experiments are performed at slightly acidic pH, due to dissolved CO2 in DI or RO water. The isoelectric points (IEP) of P25 and PC500 are 7.0 and 6.2, respectively,38 meaning that a P25 surface will have a net positive charge, while the PC500 is slightly negative, in slightly acidic media. Most targeted pollutants in water purification applications, like (azo-)dyes, have a negative charge, due to a relatively low pKa value.39 Electrostatic repulsions between pollutants and the PC500 catalyst surface inhibit adsorption and prevent facile charge transfer. The positive Ti4+ groups on the P25 surface on the other hand attract the anionic pollutants and promote subsequent adsorption. For very acid solutions, where the PC500 surface also carries a net positive charge, it has been shown that PC500 shows a higher activity compared to P25.3 Secondly, the effective available surface for PC500 is expected to be much smaller due to the complex structure of the pores, and the full potential of the PC500 surface cannot be exploited. The adsorbed and bulk water in the nanopores is believed to hinder an easy diffusion of molecules. On the other hand, the absence of bulk water in gas phase experiments is expected to complicate desorption of intermediates.40 The accumulation of intermediates on the catalysts surface is expected to result in a gradual decrease in activity, which was not observed in the experiments presented here.
Finally, based on our work we conclude with suggestions for an optimized photocatalyst. A first possible route is the improvement of the degree of crystallinity of PC500, while maintaining the high surface area, by decoupling morphological and functional effects of annealed nanostructured porous coatings. Prior to a 550 °C annealing, the PC500 coating is encapsulated by a SiO2 scaffold to convert all amorphous titanium dioxide into highly crystalline anatase.41 Subsequently, the silica is dissolved in a 2 M NaOH solution. In this way, the high surface area is retained, and the degree of crystallinity increases. Secondly, the surface density of the hydroxyls can be increased by a hydrothermal treatment,42 enhancing the activity.
4 Conclusion
In this work we studied the photocatalytic degradation of acetaldehyde in the gas phase on Millennium PC500 and P25. The significant differences in bulk and surface properties between both materials allow a fundamental study of the material characteristics that are important in VOC gas phase purification. Although the quantum efficiency of P25 is found to be superior to PC500, as demonstrated by photocurrent measurements, the TOF of acetaldehyde on PC500 exceeds that on P25, mainly due to the higher available surface area that is the prevailing factor when working in the gas phase. Additional parameters that are in favor of the high TOF on PC500 are the high absolute amount of surface hydroxyl groups and the existence of an alternative reaction pathway for photocatalysts that allows conversion when a lower amount of photoholes is available.Acknowledgements
S.W.V. acknowledges the Research Foundation of Flanders (FWO) for the financial support. J.A.M acknowledges long term funding (Methusalem).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- Is. Mclintoc and M. Ritchie, Trans. Faraday Soc., 1965, 61, 1007 RSC.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- W. E. Jardim and R. M. Alberici, Appl. Catal., B, 1997, 14, 55–68 CrossRef.
- J. F. Rontani and P. J. P. Giral, Int. J. Environ. Anal. Chem., 1990, 42, 61–68 CrossRef CAS.
- M. A. Anderson, M. E. Zorn, D. T. Tompkins and W. A. Zeltner, Environ. Sci. Technol., 2000, 34, 5206–5210 CrossRef.
- K. Urashima and J. S. Chang, IEEE Trans. Dielectr. Electr. Insul., 2000, 7, 602–614 CrossRef CAS.
- N. R. Blake and G. L. Griffin, J. Phys. Chem., 1988, 92, 5697–5701 CrossRef CAS.
- L. Stevens, J. A. Lanning, L. G. Anderson, W. A. Jacoby and N. Chornet, J. Air Waste Manage. Assoc., 1998, 48, 979–984 CAS.
- E. Pelizzetti, C. Minero, P. Piccinini and M. Vincenti, Coord. Chem. Rev., 1993, 125, 183–193 CrossRef CAS.
- J. M. Coronado, M. E. Zorn, I. Tejedor-Tejedor and M. A. Anderson, Appl. Catal., B, 2003, 43, 329–344 CrossRef CAS.
- I. Sopyan, M. Watanabe, S. Murasawa, K. Hashimoto and A. Fujishima, J. Photochem. Photobiol., A, 1996, 98, 79–86 CrossRef CAS.
- H. Hu, W. J. Xiao, J. Yuan, J. W. Shi, M. X. Chen and G. W. F. Shang, J. Environ. Sci. (Beijing, China), 2007, 19, 80–85 CAS.
- Z. Y. Liu, X. T. Zhang, S. Nishimoto, T. Murakami and A. Fujishima, Environ. Sci. Technol., 2008, 42, 8547–8551 CrossRef CAS.
- M. Sung, S. Kato, F. Kawanami and M. Sudo, Build. Environ., 2010, 45, 2002–2007 CrossRef.
- D. Meroni, S. Ardizzone, G. Cappelletti, C. Oliva, M. Ceotto, D. Poelman and H. Poelman, Catal. Today, 2011, 161, 169–174 CrossRef CAS.
- B. Hauchecorne, D. Terrens, S. Verbruggen, J. A. Martens, H. Van Langenhove, K. Demeestere and S. Lenaerts, Appl. Catal., B, 2011, 106, 630–638 CrossRef CAS.
- M. L. Sauer and D. F. Ollis, J. Catal., 1996, 158, 570–582 CrossRef CAS.
- D. S. Muggli, K. H. Lowery and J. L. Falconer, J. Catal., 1998, 180, 111–122 CrossRef CAS.
- C. Guillard, J. Disdier, J. M. Herrmann, C. Lehaut, T. Chopin, S. Malato and J. Blanco, Catal. Today, 1999, 54, 217–228 CrossRef CAS.
- D. Mantzavinos, D. E. Kritikos, N. P. Xekoukoulotakis and E. Psillakis, Water Res., 2007, 41, 2236–2246 CrossRef.
- S. Brosillon, L. Lhomme, D. Wolbert and J. Dussaud, Appl. Catal., B, 2005, 61, 227–235 CrossRef.
- M. Saquib, M. Abu Tariq, M. M. Haque and M. Muneer, J. Environ. Manage., 2008, 88, 300–306 CrossRef CAS.
- S. W. Verbruggen, S. Ribbens, T. Tytgat, B. Hauchecorne, M. Smits, V. Meynen, P. Cool, J. A. Martens and S. Lenaerts, Chem. Eng. J. (Amsterdam, Neth.), 2011, 174, 318–325 CAS.
- T. Tytgat, B. Hauchecorne, S. W. Verbruggen, M. Smits and S. Lenaerts, J. Assoc. Lab. Autom., 2012, 17, 134–143 Search PubMed.
- V. M. Gun'ko, J. P. Blitz, V. I. Zarko, V. V. Turov, E. M. Pakhlov, O. I. Oranska, E. V. Goncharuk, Y. I. Gornikov, V. S. Sergeev, T. V. Kulik, B. B. Palyanytsya and R. K. Samala, J. Colloid Interface Sci., 2009, 330, 125–137 CrossRef CAS.
- A. Fujishima, Z. Y. Liu, X. T. Zhang, S. Nishimoto and T. Murakami, Environ. Sci. Technol., 2008, 42, 8547–8551 CrossRef.
- H. Dotan, K. Sivula, M. Gratzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 CAS.
- A. Furube, Y. Tamaki, M. Murai, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2006, 128, 416–417 CrossRef.
- P. V. Kamat, I. Bedja and S. Hotchandani, J. Phys. Chem., 1994, 98, 9137–9142 CrossRef CAS.
- T. Kawai and T. Sakata, J. Chem. Soc., Chem. Commun., 1980, 694–695 RSC.
- A. E. Regazzoni, P. A. Mandelbaum, M. A. Blesa and S. A. Bilmes, J. Phys. Chem. B, 1999, 103, 5505–5511 CrossRef.
- J. P. Blitz, V. M. Gun'ko, V. I. Zarko, V. V. Turov, E. M. Pakhlov, O. I. Oranska, E. V. Goncharuk, Y. I. Gornikov, V. S. Sergeev, T. V. Kulik, B. B. Palyanytsya and R. K. Samala, J. Colloid Interface Sci., 2009, 330, 125–137 CrossRef.
- J. T. Yates and D. A. Panayotov, Chem. Phys. Lett., 2005, 410, 11–17 CrossRef.
- D. K. Paul, M. Singh, N. Zhou and K. J. Klabunde, J. Catal., 2008, 260, 371–379 CrossRef.
- G. Munuera and F. S. Stone, Discuss. Faraday Soc., 1971, 205 RSC.
- J. Arana, J. M. D. Rodriguez, O. G. Diaz, J. A. H. Melian, C. F. Rodriguez and J. P. Pena, Appl. Catal., A, 2006, 299, 274–284 CrossRef CAS.
- J. Kiwi, D. Gumy, C. Morais, P. Bowen, C. Pulgarin, S. Giraldo and R. Hajdu, Appl. Catal., B, 2006, 63, 76–84 CrossRef.
- M. Kurtoglu, N. Birbicer, U. Kimyonsen and S. Serin, Dyes Pigm., 1999, 41, 143–147 CrossRef CAS.
- W. F. Shangguan, H. Hu, W. J. Xiao, J. Yuan, J. W. Shi and D. N. He, J. Sol-Gel Sci. Technol., 2008, 45, 1–8 CrossRef.
- K. Sivula, J. Brillet and M. Gratzel, Nano Lett., 2010, 10, 4155–4160 CrossRef.
- J. G. Yu, H. G. Yu, B. Cheng, M. H. Zhou and X. J. Zhao, J. Mol. Catal. A: Chem., 2006, 253, 112–118 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |