Stereoselective synthesis of bulky 1,2-diols with alcohol dehydrogenases†
Justyna
Kulig
a,
Robert C.
Simon‡
ab,
Christopher A.
Rose
c,
Syed Masood
Husain
d,
Matthias
Häckh
d,
Steffen
Lüdeke
d,
Kirsten
Zeitler
c,
Wolfgang
Kroutil
b,
Martina
Pohl
a and
Dörte
Rother
*a
aInstitute of Bio- and Geosciences, IBG-1: Biotechnology, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: do.rother@fz-juelich.de; Fax: +49 2461-613870; Tel: +49 2461-616772
bInstitute of Chemistry, Organic and Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria
cInstitute of Organic Chemistry, University of Regensburg, Universitätsstrasse 31, 93053 Regensburg, Germany
dInstitute of Pharmaceutical Sciences, University of Freiburg, Albertstrasse 25, 79104 Freiburg, Germany
First published on 27th March 2012
Abstract
Although biotransformations implementing alcohol dehydrogenases (ADHs) are widespread, enzymes which catalyse the reduction and oxidation of sterically demanding substrates, especially 2-hydroxy ketones, are still rare. To fill this gap eight ADHs were investigated concerning their potential to reduce bulky 2-hydroxy ketones. All of these enzymes showed good activities along with excellent enantio- (ee > 99%) and diastereoselectivities (de > 99%). Due to their differences in substrate preferences and stereoselectivity a broad range of diastereomerically pure 1,2-diols is now accessible via biotransformation. Best results were obtained using the alcohol dehydrogenase from Ralstonia sp. (Cupriavidus sp.) (RADH), which showed a broad substrate range, especially for sterically demanding compounds. Araliphatic 2-hydroxy ketones, like (R)-2-hydroxy-1-phenylpropan-1-one ((R)-2-HPP), were reduced much faster than aliphatic or aromatic aldehydes (e.g. benzaldehyde) under the applied conditions. Additionally (R)- as well as (S)-2-hydroxy ketones were converted with high diastereoselectivities (de > 99%). RADH, which was up to now only studied as a whole cell biocatalyst overexpressed in E. coli, was purified and thoroughly characterised concerning its catalytic properties.
1. Introduction
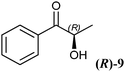
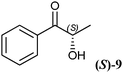
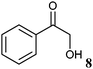
A key interest in organic chemistry is the synthesis of chiral building blocks.1 Among these, sterically demanding chiral 1,2-diols with two chiral centres are of high interest since they find versatile application as synthons for chemical catalysts, agrochemicals and pharmaceuticals.2–4 However, the development of methods covering the synthesis of all forms of the stereoisomers is still challenging. The production of enantiomerically pure 1,2-diols has been described using chemical and enzymatic methods. Since four stereoisomers can occur, the chemical synthesis of chiral 1,2-diols is not a trivial task. An approved method is the synthesis of syn-1,2-diols applying Sharpless dihydroxylation of (E)-olefins. It results in high stereoselectivities, while the synthesis of anti-1,2-diols is limited by the availability of (Z)-olefins.5 Moreover, the Sharpless asymmetric dihydroxylation requires the application of potassium osmate(VI) dihydrate (K2OsO4·2H2O), which is a dangerous and toxic catalyst. Therefore, the enzymatic approach is an attractive alternative. Here the synthesis takes place under mild conditions yielding chiral diols with high chemo-, regio- and stereoselectivity. Different biocatalytic routes to chiral alcohols have been described using oxidoreductases, hydrolases and lyases, either with isolated enzymes or whole cells.6One powerful approach toward chiral 1,2-diols is the reduction of the prochiral keto group of 2-hydroxy ketones via NAD(P)H-dependent oxidoreductases. The reduction of small and small-bulky 2-hydroxy ketones, such as (R)- and (S)-2-hydroxy-1-phenylpropan-1-one (2-HPP, 9), using alcohol dehydrogenases from Lactobacillus brevis (LBADH) and Thermoanaerobacter sp. (ADHT) has already been described earlier.7,8 However, there is still a gap of enzymes accepting bulky-bulky substrates which has to be filled. Bulky-bulky3 2-hydroxy ketones are defined as 2-hydroxy ketones possessing two substituents which are larger than ethyl, azido-, cyano-, or halomethyl groups. Recently, the biocatalytic reduction of bulky-bulky 2-hydroxy ketones such as methoxy-substituted (R)-2-HPP and benzoin derivatives was described using whole cells of Pichia glucozyma.9 Unfortunately, the respective enzyme(s) responsible for this reaction is/are not yet known.
An alcohol dehydrogenase identified from Ralstonia sp. DSM 6428 (also known as Cupriavidus sp. DSM 6428) has earlier been described by Kroutil's group as a suitable enzyme for the reduction of bulky-bulky ketones by whole cell biotransformations.3 Motivated by these results, we started a broader screening of available ADHs concerning their activity towards various 2-hydroxy ketones, which was initially performed using crude extracts of recombinant E. coli cells overexpressing the respective enzymes: alcohol dehydrogenases from Thermoanaerobacter sp. (ADHT), Flavobacterium frigidimaris (FADH), horse liver (HLADH), Lactobacillus brevis (LBADH), Ralstonia sp. (RADH), Sphingobium yanoikuyae (SADH), Thermus sp. (TADH), and carbonyl reductase from Candida parapsilosis (CPCR). Among these ADHT, LBADH and RADH showed promising activity towards 2-hydroxy ketones, with RADH being most active towards bulky-bulky 2-hydroxy ketones. Therefore its high potential for the synthesis of chiral diols was studied in more detail.
2. Material and methods
2.1. Origin of chemicals
All chemicals for chemical syntheses were of high analytical grade and purchased from Sigma (Steinheim, Germany), Aldrich (Steinheim, Germany), Fluka (Steinheim, Germany), Merck (Darmstadt, Germany) and Roth (Karlsruhe, Germany). NADPH and NADH were purchased from Biomol (Hamburg, Germany).2.2. Chemical and biochemical synthesis of 2-hydroxy ketones
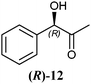
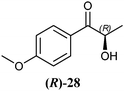
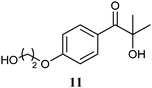
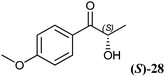
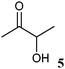
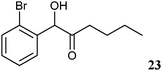
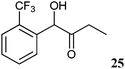
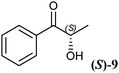
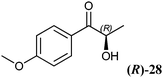
The 2-hydroxy ketones 5–8, 10–11 and 13–14 were bought from one of the above-mentioned companies. Phenylacetylcarbinol, PAC ((R)-12), was received from BASF AG and purified as described below. The 2-hydroxy ketones 18–27 and 32–34 were obtained via carbene-catalysed regioselective chemical syntheses as recently described.10,11 2-Hydroxy ketones rac-9, (R)-9, (S)-9, (R)-28 and (S)-28 were chemically or biochemically synthesised as described in the following.The biocatalytic syntheses of 2-hydroxy ketones 15–17, 29–30 and 31 were conducted as previously described.19–22
2.3. Cloning, expression and purification of recombinant ADHs
Cloning, expression and purification procedures of recombinant ADHs can be found in the ESI.†2.4. Enzyme activity assays
Activity measurements of ADHs were performed spectrophotometrically at 340 nm by determining the consumption of NAD(P)H at 30 °C in a half-micro cuvette (total volume: 1 mL) for 60 seconds. One unit (U) of activity is defined as the amount of enzyme which catalyses the consumption of 1 μmol of NAD(P)H per minute in an appropriate buffer system (see Table 2) at 30 °C. The standard activity tests were conducted using benzaldehyde (10 mM) (ADHT and RADH) or acetophenone (10 mM) (LBADH) as substrate. All activity measurements were performed in triplicate.| Substrate | Products | ||||
|---|---|---|---|---|---|

|

|

|

|
|

|

|
|
|
|||

|
|

|
|||
|

|

|
|||

|

|
|

|
||
|

|
|

|
||

|

|

|
|||
| Substrate | c/mM | Specific activity/U mg−1 | ||
|---|---|---|---|---|
| ADHT | LBADH | RADH | ||
| Aldehydes | ||||

|
10 | 6.0 ± 0.3 | 109.8 ± 3.1 | n.a. |
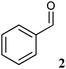
|
10 | 5.9 ± 0.2 | 8.0 ± 0.1 | 3.0 ± 0.1 |

|
10 | 7.7 ± 0.2 | 4.2 ± 0.0 | 4.1 ± 0.0 |
| Ketones | ||||

|
10 | 7.7 ± 0.2 | 89.6 ± 1.0 | 2.4 ± 0.0 |
| 2-Hydroxy ketones: aliphatic | ||||
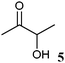
|
10 | 18.1 ± 0.1 | 127.8 ± 3.7 | 0.5 ± 0.0 |

|
10 | 13.3 ± 0.3 | 36.7 ± 0.4 | 6.6 ± 0.1 |
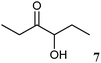
|
10 | 6.4 ± 0.3 | 66.1 ± 0.9 | 4.8 ± 0.1 |
| 2-Hydroxy ketones: 2-HPP and derivatives thereof | ||||

|
10 | 4.2 ± 0.1 | 28.7 ± 0.2 | 5.6 ± 0.1 |

|
10 | 0.8 ± 0.0 | 3.5 ± 0.0 | 112.2 ± 7.2 |
|
10 | 2.2 ± 0.2 | 3.0 ± 0.0 | 239.9 ± 7.6 |

|
10 | 2.9 ± 0.0 | 3.2 ± 0.1 | 10.0 ± 0.1 |

|
10 | 4.4 ± 0.5 | 0.3 ± 0.0 | 21.5 ± 0.4 |
|
5 | 0.7 ± 0.2 | 0.0 ± 0.0 | 4.1 ± 0.1 |
| 2-Hydroxy ketones: PAC | ||||

|
10 | 3.5 ± 0.0 | 20.4 ± 0.2 | 36.3 ± 1.8 |
| 2-Hydroxy ketones: benzoin and α-pyridoin | ||||
|
10 | 0.1 ± 0.0 | n.d. | n.a. |

|
10 | 0.5 ± 0.0 | 0.1 ± 0.0 | 0.6 ± 0.0 |
| Substrate | Oxidoreductase | ||
|---|---|---|---|
| ADHT | LBADH | RADH | |
| ee—enantiomeric excess, de—diastereomeric excess, dr—diastereomeric ratio, n.d.—not determined.a Employing crude cell extracts with overexpressed ADHs.b Employing purified RADH.c de determined by 1H NMR measurement.d Absolute configuration confirmed by VCD studies. | |||

|
(R) | (S) | (R) |
| dr (syn/anti) | dr (syn/anti) | dr (syn/anti) | |
23.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1a :1a |
1:1a |
2:1a |
|
| 4:1b |
|||

|
(R) | (S) | (R) |
| de > 99% | de > 99% | de > 99% | |
| (syn)a | (anti)a | (syn)a,b | |
|
(R) | (S) | (R) |
| de > 99% | de > 99% | de > 99% | |
| (anti)a | (syn)a | (anti)a,b | |

|
(R) | (S) | (R) |
| ee > 99%a | ee > 99%a | ee > 99%a,b | |

|
(S) | (R) | (S) |
| de > 99% | de 89% | de > 99% | |
| (anti)a | (syn)a | (anti)a,b | |

|
n.d. | n.d. | (R) |
| de > 99% | |||
| (syn)b,c,d | |||
|
n.d. | n.d. | (R) |
| de 87% | |||
| (anti)b,c | |||
| Substrate | c/mM | Specific activity/U mg−1 |
|---|---|---|
| a Absorption limitations occurred already at 1 mM of substrate concentration. b Saturated substrate solution, diluted two-fold because of absorption limitations. | ||
| Acyloins | ||
|
10 | 1.1 ± 0.0 |

|
10 | 9.8 ± 0.0 |

|
10 | 5.1 ± 0.1 |

|
10 | 0.8 ± 0.1 |
| PAC and derivatives thereof | ||

|
10 | 49.3 ± 1.1 |

|
10 | 61.9 ± 1.9 |

|
10 | 19.0 ± 0.5 |

|
10 | 60.0 ± 2.4 |

|
Saturated | 0.7 ± 0.1 |

|
Saturated | 0.9 ± 0.0 |

|
Saturated | 0.1 ± 0.1 |

|
1 | 0.2 ± 0.0 |

|
5 | 0.2 ± 0.0 |
|
1 | 0.2 ± 0.1 |

|
5 | 0.5 ± 0.0 |
|
5 | 1.1 ± 0.1 |

|
1 | 0.3 ± 0.0 |

|
Saturated | 0.6 ± 0.2 |
| 2-HPP and derivatives thereof | ||
|
10 | 4.3 ± 0.1 |

|
10 | 259.2 ± 3.5 |

|
10 | 362.6 ± 1.9 |
|
10 | 17.1 ± 0.3 |

|
10 | 44.4 ± 0.5 |
|
10 | 138.7 ± 1.8 |

|
10 | 17.4 ± 0.2 |
|
10 | 0.6 ± 0.0 |

|
10 | 0.2 ± 0.1 |

|
10a | Absorption limitation |

|
5 | 6.2 ± 0.3 |
| Other substrates | ||

|
Saturated | 0.1 ± 0.0 |

|
Saturated | 0.00 |
|
Saturated | 0.00 |

|
Saturatedb | 0.4 ± 0.0 |
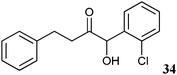
|
Saturated | 0.3 ± 0.0 |
2.5. Chemical syntheses of reference compounds
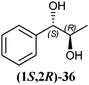
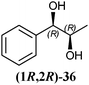
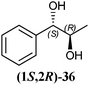
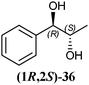
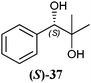
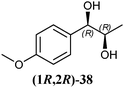
In Table 1 all possible products of the enzymatic reduction of rac-9, (R)-9, (S)-9, 10, (R)-12, (R)-28 and (S)-28 are summarised. Reduction of the prochiral carbonyl group of 10 can only yield the (R)- or (S)-diol 37 (Table 1). The reduction of rac-9, (R)-9, (S)-9 and (R)-12 may yield four stereoisomers of 1-phenylpropane-1,2-diol (36), whereas the reduction of (R)-28 and (S)-28 can theoretically give up to four stereoisomers of the 1-(4-methoxyphenyl)-propane-1,2-diol (38) depending on the employed enzyme. As will be seen in the discussion, neither a racemisation nor a tautomerisation occurs under the applied conditions for the enzymatic reductions.In order to assign the absolute configuration of the enzymatically obtained 1,2-diols, compounds 36–38 were chemically synthesised as described below.
1H-NMR and 13C-NMR spectra were recorded on a Bruker Advance-DRX 600 spectrometer in CDCl3 with TMS (Me4Si) as the internal standard. Chemical shifts are given in ppm relative to the Me4Si (1H, Me4Si = 0 ppm) or relative to the resonance of the solvent (13C, CDCl3 = 77.2 ppm). GC-MS analysis was performed on a Varian CP-3800/Saturn 2000 instrument equipped with HP-5 column (30 m × 0.32 mm × 0.25 μm). The sample analysis (1 μL) was carried out in following program: injection temperature: 60 °C and kept constant for 3 min, then in a slope 20.0 °C min−1 temperature was increased to 180 °C and subsequently kept constant for 5 min. Optical rotation was measured at 20 °C on a Jasco P-2000 polarimeter using the sodium D-line.
syn:29,301H NMR (600 MHz, CDCl3): 1.03 (d, 3JMe,2 = 6.3 Hz, 3 H, CH3), 2.57 (brd, 3JOH,2 = 3.1 Hz, 1 H, OH), 2.63 (brd, 3JOH,1 = 3.1 Hz, 1 H, OH), 3.80 (s, 3 H, OMe), 3.82 (ddq, 3J2,1 = 7.2 Hz, 3J2,Me = 6.2 Hz, 3J2,OH = 3.0 Hz, 1H, 2-H), 4.31 (d, 3J1,2 = 7.6 Hz, 1H, 1-H), 6.88 (d, 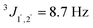 , 2H, arom.-H), 7.25 (d,
, 2H, arom.-H), 7.25 (d, 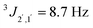 , 2 H, arom.-H) ppm. 13C-NMR (151 MHz, CDCl3): 18.8 (C-3), 55.3 (OMe), 72.3 (C-2), 79.2 (C-1), 113.9 (arom.-CH), 128.0 (arom.-CH), 133.2 (arom.-Cipso), 159.5 (arom.-Cipso) ppm.
, 2 H, arom.-H) ppm. 13C-NMR (151 MHz, CDCl3): 18.8 (C-3), 55.3 (OMe), 72.3 (C-2), 79.2 (C-1), 113.9 (arom.-CH), 128.0 (arom.-CH), 133.2 (arom.-Cipso), 159.5 (arom.-Cipso) ppm.
anti:311H NMR (600 MHz, CDCl3): 1.09 (d, 3JMe,2 = 6.4 Hz, 3 H, CH3), 2.50 (brs, 1 H, OH), 2.77 (brs, 1 H, OH), 3.80 (s, 3 H, OMe), 3.97 (ddq, 3J2,1 = 4.4 Hz, 3J2,Me = 6.4 Hz, 3J2,OH = 10.8 Hz, 1 H, 2-H), 4.59 (d, 3J1,2 = 4.5 Hz, 1 H, 1-H), 6.89 (d, 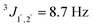 , 1 H, arom.-H), 7.28 (d,
, 1 H, arom.-H), 7.28 (d, 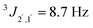 , 1 H, arom.-H) ppm. 13C-NMR (151 MHz, CDCl3): 17.5 (C-3), 55.3 (OMe), 71.3 (C-2), 77.3 (C-1), 113.8 (arom.-CH), 127.9 (arom.-CH), 132.5 (arom.-Cipso), 159.3 (arom.-Cipso) ppm.
, 1 H, arom.-H) ppm. 13C-NMR (151 MHz, CDCl3): 17.5 (C-3), 55.3 (OMe), 71.3 (C-2), 77.3 (C-1), 113.8 (arom.-CH), 127.9 (arom.-CH), 132.5 (arom.-Cipso), 159.3 (arom.-Cipso) ppm.
2.6. Set-up of biotransformations
The stereoselectivity of all different ADH-catalysed reductions was determined using racemic or enantiomerically pure 2-hydroxy ketones (Table 3). Asymmetric biotransformations were conducted in 1.5 mL Eppendorf vials at 20 °C under constant shaking (500 rpm) for 24 hours in a total volume of 1 mL. The following conditions were used: TEA-HCl buffer (50 mM), pH 7.0, if not otherwise stated (Table S2, ESI†), 2-hydroxy ketone (10 mM), NADPH (1 mM), crude cell extracts containing overexpressed ADHs (2 mg mL−1) or isolated RADH (0.2 mg mL−1), respectively.2.7. Chiral analysis of biotransformation products
Samples (1 μL) were analysed by gas chromatography on a chiral CP-chirasil-DEX CB column (25 m × 0.25 mm × 0.25 μm, Varian, Germany) with a flame ionisation detector (FID) and hydrogen as the carrier gas. Separation and determination of the enantiomeric excess of the non-derivatised stereoisomers of 1-phenylpropane-1,2-diol (36) was carried out using the following program: injection temperature: 140 °C, isotherm run for 30 min. The retention times of the stereoisomers (36) were as follows: Rt,(1S,2S)-3636 = 24.1 min, Rt,(1R,2R)-3636 = 25.8 min, Rt,(1S,2R)-3636 = 27.4 min, Rt,(1R,2S)-373737 = 28.5 min. The retention times of anti-diols (36) were assigned according to the literature.7 For separation and determination of the enantiomeric excess of 2-methyl-1-phenylpropane-1,2-diol (37) the following program was applied: injection temperature: 110 °C, then in a linear gradient of 2.5 °C min−1 the temperature was increased to 160 °C. The retention times of the enantiomers of 2-methyl-1-phenylpropane-1,2-diol (37) were: Rt,(S)-373737 = 21.9 min, Rt,(R)-373737 = 22.5 min. The determination of the diastereomeric excess of the stereoisomers of 1-(4-methoxyphenyl)propane-1,2-diol (38) was conducted by nuclear magnetic resonance (1H NMR).3. Results and discussion
3.1. Activity-screening of ADHs
To study the potential of the eight well established oxidoreductases for the reduction of different 2-hydroxy ketones to 1,2-diols, initial tests were conducted with crude cell extracts of the recombinant strains. Additionally, some aldehydes and ketones were tested to complete the substrate range for small compounds. Among the eight enzymes only ADHT, LBADH and RADH showed significant activity towards the tested 2-hydroxy ketones under the applied conditions (Table S2 and Table S3, ESI†).The first tests using crude cell extracts demonstrated the high potential of the three selected enzymes for the reduction of a broad range of 2-hydroxy ketones.
Almost every substrate could be reduced by at least one of the enzymes. ADHT and LBADH showed higher activities towards aliphatic substrates, whereas RADH clearly preferred sterically demanding mixed aromatic–aliphatic 2-hydroxy ketones. Also the sterically hindered substrate 10 was reduced by RADH effectively. Since the kinetic parameters for the reduction of the 2-hydroxy ketones are not yet known, the activities cannot be compared in terms of maximal velocities; they could be significantly higher at different substrate concentrations.
Conclusively, these initial results suggested that a broad range of chiral 1,2-diols was accessible by these three enzymes. In the following their stereoselectivity was studied for selected reactions.
3.2. Stereoselectivity studies
The three selected enzymes were investigated towards their stereoselectivity for the reduction reaction of seven different 2-hydroxy ketones (Table 3). The stereochemistry of the reduction products was determined by referring to the absolute configuration of the respective syn-diols (1R,2R)-36, (1S,2S)-36, (1R,2R)-38, and (1S,2S)-38 from asymmetric Sharpless synthesis (Table 1). This allowed for assignment of retention times from chiral solid phase gas chromatography to a particular configuration of both syn-diols and the corresponding anti-diols. The chiroptical data for syn-36, (R)-37 and (S)-37 were in agreement with the literature values for the expected configurations.7,28 Absolute configuration of the syn-38 enantiomers was confirmed by vibrational circular dichroism (VCD) analysis (see ESI† for spectra and details).The only achiral substrate 10 was reduced by all three oxidoreductases with high stereoselectivity. Thereby LBADH yielded the (S)-enantiomer, while the other two gave the (R)-enantiomer with an ee of >99%.
The other tested substrates in Table 3 already contain one chiral centre ((R)-9, (S)-9, (R)-12, (R)-28 and (S)-28). This chiral centre is maintained during biotransformation in all cases and has no visible chiral induction on the formation of the second chiral centre, except substrate (S)-28, in which the diastereomeric excess drops to 87%. Under the applied reaction conditions we could not observe any racemisation or tautomerisation of the 2-hydroxy ketones. This is a clear advantage of the enzymatic approach. Almost all of the tested substrates were reduced with very high stereoselectivity (ee and de > 99%).
Conclusively, syn- and anti-1,2-diols with high diastereoselectivity were obtained by the appropriate choice of the biocatalyst. The diastereomeric ratio of the syn/anti-diols provided information about the enzyme's selectivity towards the two respective enantiomers of the substrate. This was specifically tested with both enantiomers of 2-HPP (9), which were accepted by all tested enzymes and reduced selectively. Whereas LBADH reduced both enantiomers with the same velocity under the applied conditions, RADH and ADHT preferred the (R)-enantiomer of 9 yielding diastereomeric ratios (syn/anti) of 2:1 (RADH) and 23.6:1 (ADHT), respectively.
LBADH showed inverse stereoselectivity compared to the other tested oxidoreductases. Interestingly for all of these enzymes, the location of the already existing chiral centre in two similar 2-hydroxy ketones resulted in the formation of inverse product stereoselectivities, e.g. (R)-PAC ((R)-12) and (R)-HPP ((R)-9) were reduced to the respective anti- and syn-diols. With ADHT and RADH (R)-2-HPP ((R)-9) was reduced (R)-selectively to the syn-product (1R,2R)-36, while (R)-PAC ((R)-12) was reduced (S)-selectively yielding the anti-product (1R,2S)-36. Besides, LBADH catalysed also the formation of the syn-diol (1R,2R)-36 by (R)-selective reduction of (R)-PAC ((R)-12) (Table 3). This was caused by the orientation of the substrate in the enzymes' active sites, where the cofactor's hydride ion attack could occur either from the si-face of the reduced molecule or from the re-face, depending on the enzyme.32,33
These data demonstrate that at least all four different stereoisomers of the methyl-phenylpropanediols are now accessible with the three tested ADHs, depending on the chirality of the starting compound and the stereoselectivity of the enzyme.
3.3. Detailed characterisation of RADH's substrate range
The high potential of RADH to reduce bulky-bulky 2-hydroxy ketones (Table 2) motivated us to characterise the catalytic potential of this enzyme in more detail. Therefore, a purification procedure was established to get access to the pure biocatalyst by avoiding potential side effects of the crude cell extract, and to allow quantitative measurements. Detailed characterisation of the enzyme, which will be published elsewhere (manuscript in preparation) revealed the addition of CaCl2 as essential to maintain the stability of the purified enzyme. To analyse the substrate range of RADH in more detail, its initial rate activities towards the reduction of 34 different 2-hydroxy ketones were analysed.A screening of 2-hydroxy ketones with isolated RADH confirmed the trend already observed with crude cell extracts, demonstrating that this enzyme was extremely active towards the reduction of bulky-bulky compounds. The highest activity among the tested substrates was found with (R)-2-HPP ((R)-9), which was reduced to the respective vicinal 1,2-diol with about 360 U mg−1, followed by racemic 2-HPP (rac-9), with a specific activity of 260 U mg−1, and (R)-p-methoxy-2-HPP ((R)-28) with about 140 U mg−1. However, these activities could not be compared in absolute numbers as they do not necessarily represent the maximal velocity.
Regarding two very similar molecules such as (R)-2-HPP ((R)-9) and (R)-PAC ((R)-12), the relative position of the hydroxyl- and the keto group has significant impact on RADH's activity, since (R)-2-HPP ((R)-9) is about 6-fold faster converted to the respective 1,2-diol than (R)-PAC ((R)-12). This goes along with data obtained with the crude cell extract where a similar difference (6.5-fold) was observed (Table 2).
Our results demonstrate that RADH prefers 2-HPP-derivatives as excellent substrates for reductions. Although PAC and its derivatives are reduced with good activities as well, the reduction of the keto function next to the aromatic ring seems to be preferred over the reduction of the keto group next to the aliphatic side chain. Additionally, although the activity towards the sterically extremely demanding bulky-bulky substrates 14 and 34 is rather low, it is worth pointing out that they are converted at all.
As already visible with the crude cell extract, the chiral centre of chiral substrates has a strong influence on the specific activity of the RADH. For example (S)-p-methoxy-2-HPP ((S)-28) is reduced with a specific activity of 17 U mg−1 while the (R)-enantiomer ((R)-28) is reduced 8-fold faster (139 U mg−1). The same holds for (R)-2-HPP ((R)-9) and (S)-2-HPP ((S)-9), supporting the results obtained with crude cell extracts (Table 2). An inverse situation was observed in the case of 1-hydroxy-1-phenylbutan-2-one ((R)-17, (S)-17), where the (S)-enantiomer ((S)-17) is reduced 3-fold faster than the (R)-enantiomer ((R)-17).
The enzyme exhibits severe activity differences towards substrates not only with respect to the position of the carbonyl group but also regarding the configuration of the adjacent asymmetric chiral carbon atom.
Conclusively not only the position of the hydroxyl group to be reduced has a strong influence on the enzyme's activity but also the configuration of the asymmetric carbon atom is decisive.
Therewith the results obtained with the purified enzyme confirm the data obtained with crude cell extracts (Table 3). The only difference was a shift of the ratio of syn and anti diol formation from racemic 2-HPP from 2:1 (crude cell extract) to 4:1 (purified RADH).
4. Conclusions and outlook
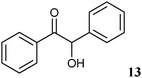
We demonstrated the high potential of biocatalysis for the stereoselective synthesis of a broad range of 1,2-diols starting from chiral 2-hydroxy ketones. Among the enzymes tested, ADHT, LBADH and especially RADH showed high catalytic activities and stereoselectivities and enable the excess to all stereoisomers of a respective 1,2-diol, particularly to anti-diols.RADH was isolated for the first time and a protocol to keep the enzyme stable in buffer was established. The reduction activity of the isolated enzyme was studied with 34 different 2-hydroxy ketones (Table 4), where only two (benzoin 13 and 2-(4-bromophenyl)-1-cyclohexyl-2-hydroxyethanone 33) did not show any activity at all, which might be due to the low solubility of these compounds. Highest activities were observed with (R)-2-HPP ((R)-9) and its 4-methoxy derivative ((R)-28) with a methyl group next to the prochiral carbonyl group.
In our ongoing studies RADH is evaluated in biotransformations in order to elucidate the stability of the enzyme in a purified form and to identify optimal reaction parameters.
Acknowledgements
This work was financed by the Marie Curie Initial Training Network “BIOTRAINS—a European biotechnology training network for the support of chemical manufacturing”, grant agreement no. 238531, and partially financed by the DFG (German Research Foundation) in frame of the Research Group FOR 1296.Notes and references
- M. Hall and A. S. Bommarius, Chem. Rev., 2011, 111, 4088–4110 CrossRef CAS.
- T. Daußmann, H. G. Hennemann, T. C. Rosen and P. Dünkelmann, Chem. Eng. Technol., 2006, 78, 249–255 Search PubMed.
- I. Lavandera, A. Kern, B. Ferreira-Silva, A. Glieder, S. de Wildeman and W. Kroutil, J. Org. Chem., 2008, 73, 6003–6005 CrossRef CAS.
- R. N. Patel, Coord. Chem. Rev., 2008, 252, 659–701 CrossRef CAS.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- K. Goldberg, K. Schroer, S. Lütz and A. Liese, Appl. Microbiol. Biotechnol., 2007, 76, 237–248 CrossRef CAS.
- D. Kihumbu, T. Stillger, W. Hummel and A. Liese, Tetrahedron: Asymmetry, 2002, 13, 1069–1072 CrossRef CAS.
- S. Shanmuganathan, D. Natalia, L. Greiner and P. Dominguez de Maria, Green Chem., 2012, 14, 94–97 RSC.
- S. M. Husain, T. Stillger, P. Dünkelmann, M. Lodige, L. Walter, E. Breitling, M. Pohl, M. Burchner, I. Krossing, M. Müller, D. Romano and F. Molinari, Adv. Synth. Catal., 2011, 353, 2359–2362 CrossRef CAS.
- S. E. O'Toole, C. A. Rose, S. Gundala, K. Zeitler and S. J. Connon, J. Org. Chem., 2011, 76, 347–357 CrossRef CAS.
- C. A. Rose, S. Gundala, S. J. Connon and K. Zeitler, Synthesis, 2011, 190–198 CAS.
- C. Q. Chen, X. H. Feng, G. Z. Zhang, Q. Zhao and G. S. Huang, Synthesis, 2008, 3205–3208 Search PubMed.
- S. B. Sopaci, I. Simsek, B. Tural, M. Volkan and A. S. Demir, Org. Biomol. Chem., 2009, 7, 1658–1664 CAS.
- P. Dominguez de Maria, T. Stillger, M. Pohl, S. Wallert, K. Drauz, H. Gröger, H. Trauthwein and A. Liese, J. Mol. Catal. B: Enzym., 2006, 38, 43–47 CrossRef CAS.
- A. S. Demir, M. Pohl, E. Janzen and M. Müller, J. Chem. Soc., Perkin Trans. 1, 2001, 633–635 RSC.
- T. Dünnwald, A. S. Demir, P. Siegert, M. Pohl and M. Müller, Eur. J. Org. Chem., 2000, 2161–2170 CrossRef.
- N. Kurlemann, M. Lara, M. Pohl, W. Kroutil and A. Liese, J. Mol. Catal. B: Enzym., 2009, 61, 111–116 CrossRef CAS.
- A. S. Demir, O. Sesenoglu, E. Eren, B. Hosrik, M. Pohl, E. Janzen, D. Kolter, R. Feldmann, P. Dünkelmann and M. Müller, Adv. Synth. Catal., 2002, 344, 96–103 CrossRef CAS.
- D. Rother, G. Kolter, T. Gerhards, C. L. Berthold, E. Gauchenova, M. Knoll, J. Pleiss, M. Müller, G. Schneider and M. Pohl, ChemCatChem, 2011, 3, 1587–1596 CrossRef CAS.
- P. Dominguez de Maria, M. Pohl, D. Gocke, H. Gröger, H. Trauthwein, T. Stillger, L. Walter and M. Müller, Eur. J. Org. Chem., 2007, 2940–2944 CrossRef CAS.
- P. Lehwald, M. Richter, C. Rohr, H. W. Liu and M. Müller, Angew. Chem., Int. Ed., 2010, 49, 2389–2392 CrossRef CAS.
- A. S. Demir, O. Sesenoglu, P. Dünkelmann and M. Müller, Org. Lett., 2003, 5, 2047–2050 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- M. Klawonn, M. K. Tse, S. Bhor, C. Dobler and M. Beller, J. Mol. Catal. A: Chem., 2004, 218, 13–19 CrossRef CAS.
- Z. Wang, Y. T. Cui, Z. B. Xu and J. Qu, J. Org. Chem., 2008, 73, 2270–2274 CrossRef CAS.
- N. Schöne, Doctoral thesis, Universität Stuttgart, 2007 Search PubMed.
- K. B. Sharpless, W. Amberg, Y. L. Bennani, G. A. Crispino, J. Hartung, K. S. Jeong, H. L. Kwong, K. Morikawa, Z. M. Wang, D. Q. Xu and X. L. Zhang, J. Org. Chem., 1992, 57, 2768–2771 CrossRef CAS.
- S. Negishi, H. Ishibashi and J. Matsuo, Org. Lett., 2010, 12, 4984–4987 CrossRef CAS.
- S. Trudeau, J. B. Morgan, M. Shrestha and J. P. Morken, J. Org. Chem., 2005, 70, 9538–9544 CrossRef CAS.
- J. B. Morgan, S. P. Miller and J. P. Morken, J. Am. Chem. Soc., 2003, 125, 8702–8703 CrossRef CAS.
- D. Acetti, E. Brenna and C. Fuganti, Tetrahedron: Asymmetry, 2007, 18, 488–492 CrossRef CAS.
- T. Matsuda, R. Yamanaka and K. Nakamura, Tetrahedron: Asymmetry, 2009, 20, 513–557 CrossRef CAS.
- K. Nakamura, R. Yamanaka, T. Matsuda and T. Harada, Tetrahedron: Asymmetry, 2003, 14, 2659–2681 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The vector maps of RADH and SADH, cloning, expression and purification procedures of recombinant ADHs, tables with all crude extract data as well as infrared and vibrational circular dichroism spectra are presented. See DOI: 10.1039/c2cy20120h |
| ‡ Present address: Institute of Chemistry, Organic and Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria |
| This journal is © The Royal Society of Chemistry 2012 |