Transesterifications using a hydrocalumite synthesized from waste slag: an economical and ecological route for biofuel production†
Yasutaka
Kuwahara
a,
Keita
Tsuji
b,
Tetsutaro
Ohmichi
a,
Takashi
Kamegawa
a,
Kohsuke
Mori
a and
Hiromi
Yamashita
*a
aDivision of Materials and Manufacturing Science, Graduate School of Engineering, Osaka University, 2-1 Yamada-oka, Suita, Osaka, Japan. E-mail: yamashita@mat.eng.osaka-u.ac.jp; Fax: +81 6-6879-7457; Tel: +81 6-6879-7457
bBEL Japan, Inc., 1-9-1 Haradanaka, Toyonaka, Osaka, Japan
First published on 10th April 2012
Abstract
Blast furnace slag (BFS), a high volume byproduct resulting from iron-making processes, was used as a low-cost and abundant precursor for preparing a hydrocalumite, and the thus prepared slag-made hydrocalumite and its derivatives were applied for transesterifications of esters including triglycerides. In the transesterification of n-ethyl butyrate, calcined samples provided higher catalytic activities than those of the as-synthesized hydrocalumite due to the interfusion of slag-derived impurity elements, such as Fe and Mn, which act as catalyst promoters. Furthermore, the catalyst calcined at 800 °C in air worked as an efficient catalyst for the transesterification of soybean oil with methanol, affording up to 97% yield of fatty acid methyl esters (FAME) after 6 h under relatively moderate reaction conditions (i.e., methanol/soybean oil = 12, reaction temperature = 60 °C, use of 1 wt% catalyst), and its catalytic performance was reproduced even after air-exposure for 1 day. It is believed that the slag-made hydrocalumite can replace existing solid base catalysts as a low-cost alternative for biodiesel production and potentially contribute to the sustainable chemical processes in an economical and ecological way.
Introduction
The world is currently facing the worst energy crisis which is derived from an increasing energy demand and limitations of fossil fuel and natural resource reserves.1,2 Finding a way to ease our dependence on fossil fuel and to minimize the consumption of natural resources is one of the paramount challenges of our age. Motivated by such a situation, recent energy research has been directed towards alternative renewable sources and advanced material recycling processes.3One of promising approaches to alleviate the energy problems is use of biomass energies, such as bioethanol and biodiesel, as abundant and environmentally-friendly energy sources.4 Particularly, biodiesel fuels produced from vegetable oils and animal fats are relatively clean-burning, non-toxic, bio-degradable and renewable fuels.5,6 They can replace the petroleum-derived diesel in many applications with a few special modifications, which potentially leads to the reduction of the emission of global-warming and harmful gases (CO, SOx, etc.) and particulate matter.7 Normally, most biodiesel fuels, exemplified by fatty acid methyl esters (FAME) and fatty acid ethyl esters (FAEE), are produced by transesterification of triglycerides such as vegetable oils and animal fats with alcohols using homogeneous basic catalysts (e.g., sodium hydroxide, potassium hydroxide and alkali metal alkoxides), which can provide high biodiesel yields within a short reaction period.8 However, such homogeneous base catalysts are unrecyclable after the reaction and suffer from separation problems; the current practice of aqueous quenching with acid results in saponification as well as emulsion formation, thereby making separation steps difficult. Furthermore, it also causes an alkaline wastewater stream, which leads to corrosion of equipment and a high cost of operation. Therefore, development of heterogeneous catalysts that are easily recoverable and recyclable with low energy consumption and cheaper, safer and cleaner operations is industrially and environmentally required.
A number of studies have, so far, made use of basic alumina,9 alkaline earth metal oxides (MgO, CaO, SrO, etc.)10 and transition metal oxides (ZnO, PbOx, etc.)11 as solid base catalysts, among which calcium oxide has attracted much attention in biodiesel production due to its high basicity, low solubility and low cost of preparation. However, it requires thermal activation at temperatures higher than 700 °C under an inert gas atmosphere to remove the surface carbonate species and hydroxyl groups prior to use, limiting its extensive use at scalable and commercializable levels.12,13 A worthwhile challenge to this issue is to develop an alternative solid base catalyst which will meet all the above technological requirements. Basic metal oxides derived from layered double hydroxides (LDH) have been extensively studied as solid base catalysts or catalyst precursors as well due to their high basicities and unique structural features.14 For instance, Cantrell et al.15 and Xie et al.16 demonstrated that calcined Mg–Al hydrotalcites are effective catalysts in the methanolysis of triglycerides and that their activities are strongly influenced by both Mg/Al ratio and calcination temperature. Di Serio et al. reported that the catalytic activities of calcined Mg–Al hydrotalcites in the soybean oil transesterification are correlated with their basicities, which are dependent on both the nature of precursors and preparation methods.17 Recently, Campos-Molina et al. applied the Ca–Al hydrocalumite (Ca2Al(OH)6Cl·2H2O), a structurally close analogue to Mg–Al hydrotalcite, to the transesterification of soybean oil with methanol, but a catalyst pretreatment under a flow of inert gas is still essential for achieving a high FAME yield.18
Regarding the synthesis of LDH material, we recently presented a facile synthetic route for Ca–Al hydrocalumite from blast furnace slag (BFS).19 BFS is an important high volume byproduct resulting from iron-making processes (world production in 2008: 230–280 million tons).20 Its current disposal route is recycling as hydraulic cement, concrete aggregate and pavement materials in civil engineering work, however, the urgent problems such as increasing production, shortage of storage sites and severe environmental regulations have required continuous development of advanced recycling processes of BFS.21 This conversion process was achieved by simple two-step acid-dissolution and coprecipitation procedures using HCl and NaOH, respectively, and the synthesized hydrocalumite contains slag-derived impurity elements, such as Mg, Fe, Ti and Mn. Such studies on material recycling processes which utilize industrial wastes as substitutes for chemical components of active materials have been attracting much attention in the past few decades due to both economical and ecological considerations.3,22–25 The predominant interest in waste material recycling in such a way does not lie so much in the provision of solutions to disposal problems, but lies in the availability of low cost alternatives to existing materials or as precursors in their preparation. Accordingly, the successful applications of the synthesized materials will be a key to promote their practical uses.26
In this study, we examined the catalytic activities of the slag-made hydrocalumite (slagHC) and its derivatives by the transesterifications of esters including triglycerides (Scheme 1). Synthesis of a solid catalyst practically available for biodiesel production from BFS will, by definition, lead to the reduction of manufacturing cost, energy, use and generation of hazardous substances, and such an approach would meet the strong demand of effective utilization of waste slag in the metallurgical industry. Furthermore, the thus synthesized hydrocalumite may confer new and unexpected properties that would allow efficient catalysis in transesterification of triglycerides.
 | ||
| Scheme 1 Recycling of blast furnace slag (BFS) as a solid base catalyst and its applications to biodiesel production. | ||
For catalytic applications of waste materials, consideration especially needs to be given to the potential influence of impurity elements on catalyst structure and catalytic performance. Structures were verified by using XRD, N2 adsorption and thermogravimetric analysis in detail. To evaluate the fundamental catalytic properties of slagHC, we preliminarily examined the catalytic activity of slagHC by the transesterification of aliphatic ester, n-ethyl butyrate, as a model reaction, and surface basicity that substantially affects catalytic activities was investigated by means of CO2-TPD. Furthermore, slagHC was applied to the soybean oil transesterification under relatively mild reaction conditions.
Results and discussion
Structural analyses
The structures of the synthesized materials were identified by means of XRD, N2 adsorption and thermogravimetric analyses. Fig. 1 shows the XRD patterns of the slag-made hydrocalumite and the corresponding mixed oxides obtained by calcination in air, together with that of the pure Ca–Al–Cl hydrocalumite as a reference. The XRD patterns of as-synthesized slagHC showed sharp and symmetric peaks for (003), (006) and (110) planes all attributable to the Ca–Al–Cl hydrocalumite, and no other dominant crystalline phases were observed (Fig. 1(a) and (b)). This XRD pattern can be indexed in a hexagonal lattice with an R-3 rhombohedral symmetry where layers of Ca(OH)6·H2O and Al(OH)6 are alternated and the Cl− ions are placed between interlayers.27–29 Based on the XRD data, the basal spacings and the lattice parameters, which represent the interlayer distance and the average cation–cation distance within the hydroxide layers, respectively,30 were calculated to be 7.86 Å and 5.747 Å, being in good agreement with published data for pure Ca–Al–Cl hydrocalumite, irrespective of its considerable impurity levels.27,28 | ||
| Fig. 1 X-ray diffraction patterns of (a) Ca–Al–Cl hydrocalumite (Ca2Al(OH)6Cl·2H2O) as a reference, (b) as-synthesized slagHC, (c) slagHC(400), (d) slagHC(600) and (e) slagHC(800). The main crystallographic planes are indicated for pure hydrocalumite. Unindexed peaks seen in (d) and (e) are identical to those of mayenite (Ca12Al14O33). | ||
The SEM micrograph of as-synthesized slagHC revealed a plate-like morphology with the particle size of 0.2–1.0 μm, which well resembles those of pure Ca–Al–Cl hydrocalumite (Fig. 2(a) and (c)). These observations demonstrate that a single-phase hydrocalumite is successfully synthesized from BFS and that the interfusion of impurity metals has little influence on the crystal structure and morphology of the resulting material; the minor metal cations such as Mg2+, Fe3+, Ti4+ and Mn4+ are considered to be incorporated in the hydroxyl layers by isomorphorously substituting either Ca2+ or Al3+ sites depending on their valence states (tetravalent metals may have a possibility to be present as oxoanion species in the interlayer or as aggregated species on the crystallite surface; for detailed elemental and structural analyses, readers are referred to our previous report, ref. 19) (the chemical composition of the material is given in the Experimental section).
 | ||
| Fig. 2 FE-SEM images of (a) as-synthesized slagHC, (b) slagHC(800), (c) as-synthesized Ca–Al–Cl hydrocalumite and (d) Ca–Al–Cl(800). | ||
The slagHC(400) exhibited a broad peak in the 2θ range 25–40° attributable to the amorphous CaO phase (Fig. 1(c)), indicating the total collapse of layered structure and the transformation into Ca(Al)–O mixed oxide above 400 °C (Scheme 2). It is speculated that Mg2+ replaces Ca2+ sites and other minor elements such as Fe3+, Ti4+ and Mn4+ isomorphorously substitute Al3+ sites. Calcination above 600 °C led to the generation of new crystalline phases assignable to CaO and mayenite (Ca12Al14O33) (Fig. 1(d) and (e)). Calcination at 800 °C gave similar XRD patterns with increased intensities. Along with calcination up to 800 °C, the plate-like morphology was transformed into vesicle-like morphology with particle size of 0.05–0.5 μm, but the three different crystalline phases, CaO, MgO and mayenite, were visually undistinguishable (Fig. 2(b) and (d)). These crystallographic and morphological changes along with calcination well agree with those for pure Ca–Al–Cl hydrocalumite previously reported elsewhere.18,29,31 In general, thermal decomposition of Mg–Al hydrotalcite yields Mg(Al)–O mixed oxide with higher surface area,14 however, such an increase in surface area upon calcination was not observed for Ca–Al hydrocalumite compounds; BET surface area was determined to be 21.1 m2 g−1 for as-synthesized slagHC, 18.9 m2 g−1 for slagHC(400) and 2.8 m2 g−1 for slagHC(800). Likewise, BET surface area of the Ca–Al–Cl hydrocalumite continuously decreased along calcination (11.9 m2 g−1 for as-synthesized Ca–Al–Cl, 10.7 m2 g−1 for Ca–Al–Cl(400) and 5.7 m2 g−1 for Ca–Al–Cl(800).
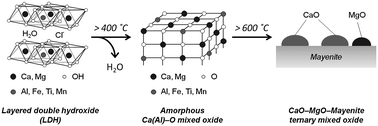 | ||
| Scheme 2 Changes in structure of hydrocalumite synthesized from BFS (slagHC). | ||
The decomposition dynamics typical of LDH compounds were tracked by thermogravimetric analysis. Fig. 3 shows the TG curves and the corresponding first derivatives of slagHC and Ca–Al–Cl hydrocalumite. The TG curve reveals three main weight losses over the temperature ranges of 25 °C ≤ T ≤ 200 °C, 200 °C ≤ T ≤ 400 °C, and 400 °C ≤ T, which are commonly ascribed to the following processes: dehydration of the water molecules intercalated in the interlayer, dehydroxylation of the hydroxyls and decomposition of intercalated anions.29 Both samples showed similar weight losses (17.1–17.7 wt%) at 317–318 °C, which closely correspond to the mass fraction of 3 water molecules; Ca2Al(OH)6Cl → Ca2AlO3Cl + 3H2O, indicative of structural similarity. Additional exothermic peaks seen at above 600 °C can be ascribed to the transition from amorphous Ca(Al)–O mixed oxide into CaO and mayenite binary phases involving the anion decomposition. Elemental analysis revealed different anion decomposition behavior at this stage; an appreciable loss in the quantity of Cl− ions was observed for the Ca–Al–Cl hydrocalumite, whereas slagHC retained most of Cl− ions (see also Table 2 below). This result indicates that most of Cl− ions in slagHC remain inside the crystals even after the calcination over 800 °C probably due to the presence of impurity transition metals, which can stabilize Cl− ions. Contrarily, Cl− ions in pure Ca–Al–Cl hydrocalumite are likely to be readily eliminated along the thermal treatment. This difference in anion decomposition behavior brings about the unique CO2/H2O-tolerance property as will be discussed later.
 | ||
| Fig. 3 (A) Weight loss curves and (B) their first derivatives of slagHC (black) and Ca–Al–Cl hydrocalumite (grey). | ||
Catalytic properties of calcined materials
To evaluate the influences of the structural changes on catalytic performance, we preliminarily examined the catalytic activities by employing base-catalyzed transesterification of ester with alcohol as a model reaction (Scheme 3). Transesterification reaction is one of the industrially important chemical reactions typically found in the synthesis of polyester, plastic recycling and the production of biodiesel fuels from fatty/vegetable oils as well.8,32 | ||
| Scheme 3 Transesterification of n-ethyl butyrate with methanol. | ||
Fig. 4A shows the activities of slagHC calcined at different temperatures in the transesterification of n-ethyl butyrate with methanol. Although as-synthesized slagHC is active for this reaction (Fig. 4A (a), 41% conversion after 12 h), a higher conversion rate was attained after thermal treatment in air. This is likely due to its improved basic properties involving phase transition from LDH structure to mixed oxide form, as previously reported as well.33 For example, in an amorphous Ca(Al)–O mixed oxide form, 77.3% conversion was obtained after 12 h (Fig. 4A (b)), which was considerably higher than that of Mg(Al)–O mixed oxide derived from Mg–Al–CO3 hydrotalcite (prepared with Mg/Al = 3.0; Fig. 4B (c), 9.2% conversion in 12 h). Such a superior activity of slagHC(400) is primarily due to the stronger basic nature of Ca atoms than that of Mg. A further higher catalytic performance (Fig. 4A (c), 78.7% conversion after 6 h) was attained over slagHC(800), which possesses CaO, MgO and mayenite ternary phases.
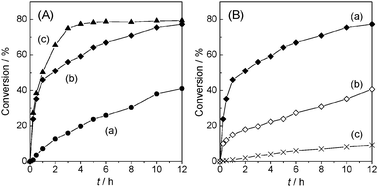 | ||
Fig. 4 Transesterification between n-ethyl butyrate and methanol catalyzed by (A) slagHC calcined at different temperatures: (a) as-synthesized slagHC, (b) slagHC(400) and (c) slagHC(800), and (B) the related mixed oxides: (a) slagHC(400), (b) Ca(Al)–O mixed oxide and (c) Mg(Al)–O mixed oxide. The Ca(Al)–O mixed oxide and Mg(Al)–O mixed oxide were obtained from Ca–Al–Cl hydrocalumite and Mg–Al–CO3 hydrotalcite, respectively, by calcination at 400 °C for 6 h in air. Reaction conditions: catalyst (20 mg), n-ethyl butyrate (12.4 mmol), methanol (2 mL), ester![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH = 1:4, 60 °C. Maximum conversion in this reaction is 80%. :MeOH = 1:4, 60 °C. Maximum conversion in this reaction is 80%. | ||
Particular attention needs to be paid to the superior catalytic activity of slagHC(400) compared with that of pure Ca(Al)–O mixed oxide, which is crystallographically the same analogue (Fig. 4B (b), 40.7% conversion after 12 h), implying a role of the slag-derived impurity elements as catalyst promoters. To clarify the changes in basicity of the catalysts, TPD measurement using CO2 as probe molecule was carried out. Fig. 5 displays the CO2-TPD profiles of slagHC calcined at various temperatures. As-synthesized slagHC showed several desorption peaks at the low temperature region, 97 and 298 °C (Fig. 5(a)), which are attributable to weak basic sites related to surface hydroxyl groups and medium basic sites derived from O2− ions adjacent to the surface hydroxyl groups, respectively.31,33 The slagHC calcined at 400 °C exhibited defined CO2 desorption peaks at around 730 °C (Fig. 5(b)), reflecting the generation of strong basic sites on the amorphous Ca(Al)–O mixed oxide.15,31 Calcination at 800 °C afforded a single CO2 desorption band at 580 °C, which is identical to that of the CaO crystalline phase (Fig. 5(c)).34 These drastic changes in basicity upon the thermal decomposition are fairly consistent with the crystallographic changes confirmed by XRD and TG as well as the catalytic results shown in Fig. 4A. More significantly, the number of basic sites formed on slagHC(400) is more than that of pure Ca–Al hydrocalumite calcined at the same temperature, and its desorption peak appeared at 30 °C higher position (compare Fig. 5(b) and (d)). Based on the results of CO2-TPD measurement and mean surface areas obtained from N2 adsorption, the surface densities of basic sites were calculated to be 85.4 and 73.6 μmol m−2 for slagHC(400) and Ca–Al–Cl(400), respectively, clearly indicating that the interfusion of impurity elements leads to the enhancement of surface basicity.
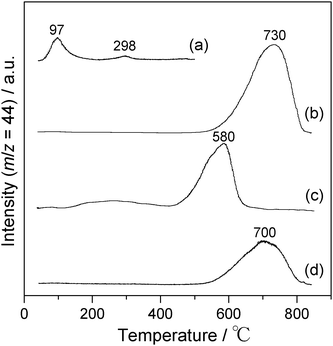 | ||
| Fig. 5 CO2-temperature programmed desorption profiles of (a) as-synthesized slagHC, (b) slagHC(400), (c) slagHC(800) and (d) Ca(Al)–O mixed oxide obtained from the Ca–Al hydrocalumite. | ||
To provide more explicit evidence for this, an additional catalytic experiment using Mg-, Fe- and Mn-doped Ca(Al)–O mixed oxides, whose doping amounts were adjusted to be the same as those of slagHC (10.6, 1.0 and 0.4 mol% as metallic elements, respectively), was carried out. The catalytic activity increased in the order of: Ca(Al)–O mixed oxide = Mg-doped Ca(Al)–O mixed oxide < Mg/Mn-doped Ca(Al)–O mixed oxide < Mg/Fe-doped Ca(Al)–O mixed oxide < Mg/Fe/Mn-doped Ca(Al)–O mixed oxide < slagHC(400) (for reaction kinetics, see Fig. S1 in ESI†), undoubtedly verifying that the addition of Mg has little contribution to the activity but the doping of transition metals (Fe and Mn) improves its catalytic activity corresponding to their doping amounts. These results agree well with the similar study on Mg–Al mixed oxides previously done by Macala et al., who have found that Fe3+ and Ga3+-doped Mg(Al)–O mixed oxides exhibit increased catalytic activity in the transesterification of triacetin.35 These are possibly attributed to the high electronegativity of transition metals incorporated within the Ca(Al)–O mixed oxide matrix which is able to strengthen the Lewis basicity of the O2− anions forming Ca–O+–M− pairs (M = Fe and Mn).
It is also worth noting that the XRD diffraction patterns of the mixed oxides were unchanged upon the reaction. Furthermore, the reaction was immediately quenched after the removal of the catalysts by filtration, and leaching of the slag-derived metal elements during the reactions was negligible, thereby demonstrating that slagHC can be used as an active, reusable and stable solid base catalyst for this type of reaction.
Biodiesel synthesis from vegetable oil
In the previous section, it was clarified that the mixed oxides obtained from slag-made hydrocalumites work as efficient base catalysts with improved basicities due to the creation of Lewis base sites. The catalysts were next tested for the transesterification of soybean oil with methanol under relatively moderate reaction conditions, i.e., with the methanol/oil molar ratio of 12, reaction temperature of 60 °C and use of 1 wt% of the catalyst (Scheme 4). In this study, soybean oil was used as a model vegetable oil, because prices of soybean oil are currently at the lowest among the major vegetable oils produced today. All the pretreatments of the catalysts were performed in air and the pretreated catalysts were added to the reaction medium immediately after cooling to room temperature. | ||
| Scheme 4 Transesterification of triglyceride with methanol. | ||
Table 1 summarizes the results of transesterification of soybean oil with methanol using slagHC and the related solid catalysts. We first examined the influence of calcination temperatures of the catalyst to determine the optimal calcination temperature since basic strength of hydrocalumites strongly depends on the calcination temperatures as demonstrated above. The slagHC catalyst pretreated below 400 °C was almost inactive for this reaction (entries 1 and 2), while it was active for the transesterification of n-ethyl butyrate. The slagHC calcined at 600 °C yielded 65.8% FAME after 6 h (entry 3), and the highest catalytic activity was attained with slagHC(800) having CaO, MgO and mayenite crystalline phases, giving 97.0% FAME after 6 h (entry 4). Additionally, when slagHC(800) was used as a catalyst, markedly increased conversion rates were attained by increasing the catalyst weight; >99% FAME yield was afforded within 2.5 h by using 5 wt% of the catalyst (entry 5). Furthermore, its activity was reproducible in a 30 times large-scale operation, where 210 g of soybean oil was converted into 180 g of FAME (as a purified product) within 9 h, which corresponds to 95% of FAME yield (entry 6). While slagHC exhibits different basicity depending on its crystalline structure as evidenced by CO2-TPD, no clear correlation between the catalytic activities and the basic strength was observed in this catalysis, being indicative of different reaction kinetics. XRD identified that slagHC(800) consists of CaO, MgO and mayenite phases, the former two phases are well-known to have strong basicity and the latter shows acid–base properties. However, MgO, mayenite as well as the raw BFS alone were inactive (entries 9–12), but an activated CaO gave 74.6% FAME yield after 6 h of reaction (entry 8), indicating that crystalline CaO generated by the thermal decomposition is responsible for this reaction. Nevertheless, slagHC(800) exhibited superior catalytic performance to that of conventional bulk CaO under the same reaction conditions, despite its low CaO content (slagHC(800) contains ca. 52 wt% of CaO).
| Entry | Catalyst | Phase | t/h | FAME yield/% |
|---|---|---|---|---|
| a Reaction conditions: catalyst (0.1 g), soybean oil (7.0 g, 8 mmol), methanol (4 mL, 96 mmol), 60 °C. b Preheated at 200 °C to remove physisorbed water molecules. c 5 wt% catalyst was used. d Operated at a 30 times larger scale. e Prepared from CaCO3 by calcination at 900 °C for 6 h in air. f Pretreated at 800 °C in air for 6 h. g Synthesized with the Mg/Al molar ratio of 3 and with Na2CO3 at 65 °C. | ||||
| 1 | slagHCb | Hydrocalumite | 6 | 0.4 |
| 2 | slagHC(400) | Amorphous Ca(Al)–O mixed oxide | 6 | 0.8 |
| 3 | slagHC(600) | CaO + Mayenite + MgO(trace) | 6 | 65.8 |
| 4 | slagHC(800) | CaO + Mayenite + MgO(trace) | 6 | 97.0 |
| 5 | slagHC(800)c | 2.5 | >99 | |
| 6 | slagHC(800)d | 9 | 95.0 | |
| 7 | Ca–Al–Cl(800) | CaO + Mayenite | 6 | 97.5 |
| 8 | CaOe | CaO | 6 | 74.6 |
| 9 | Mayenitef | Mayenite | 6 | No reaction |
| 10 | MgOf | MgO | 6 | <0.1 |
| 11 | Mg–Al–CO3f,g | Mg(Al)–O mixed oxide | 6 | 1.8 |
| 12 | Raw-slagf | Amorphous mixed oxide | 6 | 1.0 |
| 13 | NaOH | Homogeneous | 1.5 | >99 |
A plausible reason for the low catalytic performance of CaO is surface poisoning by deactivating agents in the atmosphere such as CO2 and H2O. Fig. 6 shows the reaction kinetics of transesterification of soybean oil under standard reaction conditions. As depicted in Fig. 6, when slagHC(800) was used, the reaction was initiated immediately after adding the catalyst (Fig. 6(a)) and reached 95% FAME yield within 5 h of reaction, whereas air-activated CaO exhibited a quite slow reaction rate in the initial stage (Fig. 6(c)). More significantly, slagHC(800) still remained active even after air-exposure for 1 day (Fig. 6(d), 92.0% FAME yield after 6 h), contrarily air-activated CaO lost its whole activity after air-exposure for 1 day (Fig. 6(f)), suggesting that the slagHC(800) catalyst is less poisoned during the pretreatment/exposure procedures in air compared with bulk CaO.
 | ||
| Fig. 6 Reaction kinetics in transesterification of soybean oil with methanol using (a,d) slagHC(800), (b,e) Ca–Al–Cl(800) and (c,f) CaO. Filled symbols (a, b and c) represent FAME yields of the catalysts used immediately after the pretreatment, and opened symbols (d, e and f) represent those used after air-exposure for 1 day. Reaction conditions: catalyst (0.1 g), soybean oil (7.0 g, 8 mmol), methanol (4 mL, 96 mmol), soybean oil:methanol = 1:12, 60 °C. | ||
One of the crucial drawbacks of alkaline earth metal-based catalysts is poisoning by CO2 and moisture in air derived from their strong basicity. A number of studies utilizing CaO as a base catalyst have commonly reported that CaO easily suffers from CO2 poisoning in the atmosphere during its preparation, operation and storage, which leads to the formation of undesirable superfacial calcium carbonate species. Therefore, it requires pretreatment at a high temperature under an inert gas atmosphere (e.g. at 800 °C under a He gas flow) to remove the adsorbed species.34,36 Regarding the reaction kinetics of CaO in transesterification of triglyceride, Kouzu et al. have reported that calcium diglyceroxide (Ca-Gly), which is produced via a reaction between CaO and by-produced glycerol, is the main active species for this reaction.37 We also confirmed the transformation of CaO into Ca-Gly phase in the initial stage of the reaction by XRD (see Fig. S2 in ESI†). Considering this, the induction period seen in CaO (Fig. 6(c)) is likely to correspond to the period for CaO to be transformed into Ca-Gly, and it can be deduced that CO2- and H2O-poisoning during the pretreatment retards the formation of active Ca-Gly phase and ultimately causes a complete loss of its activity.
Similar reaction kinetics were also observed for Ca–Al–Cl(800). Although Ca–Al–Cl(800) showed an induction period of ca. 2 h to initialize the reaction, it afforded FAME yield comparable to that of slagHC(800) after 6 h of reaction (Fig. 6(b), 97.5% FAME yield after 6 h). However, a significant decrease in activity was observed after air-exposure for 1 day (Fig. 6(e), 3.1% FAME yield after 6 h), presenting different CO2- and H2O-poisoning behavior from that of the slagHC(800) catalyst.
Mechanism of CO2/H2O-tolerance property
The progress of the adsorption of carbonate anions and water molecules on the solid surface was monitored by infrared spectroscopy. The peaks located at 1410 and 3643 cm−1 in CaO are assignable to O–C–O vibrations for adsorbed carbonate anions (CO32−) and to superficial OH groups associated with Ca(OH)2, respectively (Fig. 7A (c)).38 Such absorption bands were scarcely seen in the IR spectra of slagHC(800) and Ca–Al–Cl(800) taken immediately after the calcination treatment (Fig. 7A (a) and (b)). From independent CO2-TPD measurements, the amounts of CO2 adsorbed during the pretreatment process were estimated to be 0.40, 0.51 and 1.67 mmol g−1 for slagHC(800), Ca–Al–Cl(800) and CaO, respectively (for TPD profiles, see Fig. S3 in ESI†). This order is fairly consistent with the IR results and agrees well with the lengths of induction period seen in the initial stage of soybean oil transesterification. | ||
| Fig. 7 IR spectra of (a) slagHC, (b) Ca–Al–Cl hydrocalumite and (c) CaO: (A) after calcination at 800 °C in air for 6 h and (B) after exposure to air for 1 day. | ||
After exposure to air for 1 day, intensity of the peak assignable to Ca(OH)2 was significantly increased for CaO. Such a peak was, on the other hand, less pronounced for Ca–Al–Cl(800) and was hardly observed for slagHC(800). These two hydrocalumite-derived catalysts, instead, exhibited several absorption bands assignable to the stretching and bending vibration of OH at around 3500 cm−1 and at 1620 cm−1, respectively, reflecting the presence of physisorbed water molecules on the solid surface (Fig. 7B (a) and (b)). The infrared bands seen at 1420 and 1480 cm−1 are characteristic of the C–O stretching of physisorbed carbonate groups,38 whose peaks were less pronounced for slagHC(800) and Ca–Al–Cl(800) compared with that of CaO. These results explicitly indicate that hydrocalumite-derived mixed oxides are more tolerant to atmospheric CO2 and H2O compared with bulk CaO. One of the plausible explanations for this is the synergism between CaO and mayenite phases, however it cannot provide any decisive reasons for the prominent catalytic and CO2/H2O-tolerance properties of the slagHC(800) catalyst. The most predominant difference between slagHC(800) and other catalysts to be conceivable is the presence of slag-derived impurity elements.
FE-SEM observations confirmed that there is no appreciable difference in morphology, particle size and surface area between slagHC(800) and Ca–Al–Cl(800) (see Fig. 2), indicating that the slag-derived impurity elements have little influence on the structures. However, an elemental analysis demonstrated a different anion decomposition behavior during the calcination treatment between slagHC and Ca–Al–Cl catalysts, as mentioned above. As summarized in Table 2, the Cl/Al ratio slightly decreased from 1.03 to 0.642 after the calcination at 800 °C for slagHC, whereas the Ca–Al–Cl hydrocalumite lost a major part of Cl− ions (decreased from 0.917 to 0.276) upon the calcination. Furthermore, the spent slagHC(800) catalyst had a decreased Cl− content (Cl/Al = 0.279), indicating the consumption of Cl− ions during the 1st cycle of soybean oil transesterification. On the other hand, the Cl/Al ratio of Ca–Al–Cl(800) was scarcely changed even after the 1st cycle of reaction (Cl/Al: 0.276 → 0.269), suggesting that the remaining Cl− ions in Ca–Al–Cl(800) are mainly present in mayenite phase. From these results, we assume that the Cl− ions remaining on the CaO phase of slagHC(800) act as surface-protecting species, preventing the CaO surface from being poisoned by atmospheric CO2 and H2O, and thus the CO2/H2O-tolerance property is conferred (Scheme 5A). The retention of Cl− ions in CaO phase is probably attributed to the stabilizing effect of the slag-derived Fe and Mn atoms uniformly distributed in slagHC (TEM image and elemental mapping results are given in Fig. S4 and Table S1 in ESI†). Contrarily, in the case of Ca–Al–Cl(800), the loss of Cl− ions is likely to lead to the formation of superfacial calcium hydroxide and calcium carbonate species on the CaO phase, which retards the subsequent transformation from CaO into active Ca-Gly phase acting as a physical barrier (Scheme 5B).
| Sample | Phasea | Molar ratiob | |
|---|---|---|---|
| Ca/Al | Cl/Al | ||
| a Determined by XRD. b Determined by ICP analysis. c Values after 1 cycle (6 h) of the soybean oil transesterification. | |||
| slagHC | Hydrocalumite | 2.03 | 1.01 |
| slagHC(400) | Amorphous mixed oxide | 2.33 | 1.03 |
| slagHC(600) | CaO + mayenite + MgO(trace) | 2.49 | 1.03 |
| slagHC(800) | CaO + mayenite + MgO(trace) | 2.39 | 0.642 |
| slagHC(800)c | Ca-Gly + mayenite + MgO(trace) | 2.01 | 0.279 |
| Ca–Al–Cl | Hydrocalumite | 2.43 | 0.917 |
| Ca–Al–Cl(800) | CaO + mayenite | 2.51 | 0.276 |
| Ca–Al–Cl(800)c | Ca-Gly + mayenite | 1.84 | 0.269 |
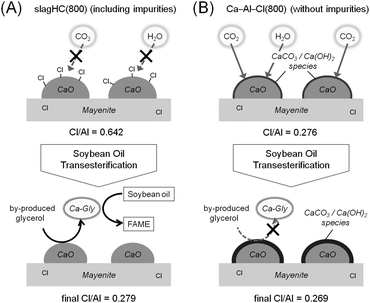 | ||
| Scheme 5 Possible surface environment of (A) slagHC(800) and (B) Ca–Al–Cl(800) catalysts before (above) and after (below) the soybean oil transesterification. | ||
Moreover, an additional catalytic experiment using slagHC(NO3)(800), a slagHC catalyst prepared by using HNO3 instead of HCl (intercalating NO3− ions instead of Cl− ions), resulted in 1.2% FAME yield after 6 h under the same reaction conditions, whilst it has the same crystalline structure and morphology as those of slagHC(Cl)(800). This marked difference can be explained by the different decomposition dynamics of intercalated anions identified by TG and elemental analysis, in which the intercalated NO3− ions were completely eliminated at around 540 °C for slagHC(NO3), whereas slagHC(Cl) retained most of intercalated Cl− ions even after calcination at 800 °C. In the case of slagHC(Cl)(800), the Cl− anions remaining in the catalyst appear to function as superfacial protective species as discussed above, which prohibit the active CaO surface from being deactivated by air. Contrarily, slagHC(NO3)(800) is likely to readily suffer from H2O and CO2 poisoning because of the elimination of protective anion species during the calcination procedure, which thereby results in a marked loss of catalytic activity. These results revealed that the slag-made solid base catalyst, slagHC(800), hardly suffers from air due to the unique CO2/H2O-tolerance property which arises from the combination of both intercalated Cl− ions and impurity transition metals such as Fe and Mn. Besides, this catalyst can be simply prepared by calcination of the slag-made hydrocalumite precursor at 800 °C in air without the activation treatment conventionally required for bulk CaO, which may lead to the reduction of manufacturing costs and high feasibility.
Recyclability test
An important aspect to be taken into account for developing solid catalysts for biodiesel production is the recyclability of the catalyst. As shown in Fig. 8, although the slagHC(800) catalyst still remained active until 3rd use when 1 wt% of the catalyst was used, appreciable loss of catalytic activity was observed over the repeated catalyst use; the conversion rate gradually decreased with repeated use, where 85.7% and 68.6% FAME yields (6 h) were obtained for 2nd and 3rd catalytic runs, respectively. This significant decrease in catalytic activity is the consequence of leaching of the active Ca-Gly species during the reaction. As shown in Table 2, the Ca/Al ratio decreased from 2.39 to 1.72 after 3 times of repeated use (corresponding to 44% loss of the generated Ca-Gly species). XRD analysis also confirmed decreased intensity of the diffraction peaks assignable to the Ca-Gly phase after each cycle (for the changes in XRD patterns over multi-cycle catalytic runs, see Fig. S2 in ESI†). | ||
| Fig. 8 Influence of repetitive cycles on FAME yield and Ca/Al ratio. Reaction conditions: catalyst (slagHC(800), 0.1 g), soybean oil (7.0 g, 8 mmol), methanol (4 mL, 96 mmol), soybean oil:methanol = 1:12, 60 °C. | ||
A homogeneous process by the leached Ca-Gly species contributes to the generation of calcium carboxylates and leads to separation problems. From the calculation based on elemental analysis, the concentration of the eluted Ca in the FAME product after 1 cycle of soybean transesterification can be roughly estimated to be 15–30 ppm, whose value seems rather low compared with those ever reported in the other literature using CaO as a catalyst.12 The remaining fraction of the eluted Ca is believed to be present in the by-produced glycerol phase as Ca-Gly species. Although the CaO phase had been supported by the simultaneously formed mayenite phase, we unfortunately could not circumvent the dissolution of Ca into the reaction medium. For practical biodiesel production, purification of the produced biodiesel using cation-exchange resin which is capable of removing the greatest part of the soluble Ca would be required because elution of alkaline or alkaline earth metal cations significantly decreases the quality of biodiesel fuel. Developing an alternative strategy to fully stabilize the CaO phase against leaching is one of the future tasks to be solved.39
Conclusions
This work demonstrated that the slag-made hydrocalumite and its derivatives can be used as useful solid base catalysts for transesterifications including biodiesel synthesis. The slag-made hydrocalumite was transformed into a CaO, MgO and mayenite ternary mixed oxide after calcination at 800 °C, and the thus prepared mixed oxide catalyst afforded up to 97% FAME under relatively mild reaction conditions, i.e. with the methanol/oil molar ratio of 12, reaction temperature of 60 °C and use of 1 wt% of the catalyst. The most significant characteristic feature of the slag-made catalyst is a prominent CO2/H2O-tolerance property derived from the combination of intercalated Cl− ions as superfacial protecting species and slag-derived impurity elements as stabilizing agents, by which the catalyst remained active even after air-exposure for 1 day.BFS can be used as an inexpensive and abundant precursor for fabricating active materials of particular catalytic interest. Although consideration needs to be given to the potential influence of compositional variation on catalytic performance, the slag-made hydrocalumite can be an alternative solid base catalyst for scalable and cost-effective biodiesel production. We believe that this engineering strategy will surely contribute to the sustainable chemical processes in an economical and ecological way.
Experimental section
Materials
All experiments were performed using the same batch of BFS, which consists of an amorphous mixed oxide phase with SBET = 8.1 m2 g−1 and Vtotal = 0.016 cm3 g−1. The chemical composition of BFS is as follows; CaO: 40.09, SiO2: 34.58, Al2O3: 14.78, MgO: 5.29, Fe2O3: 1.53, TiO2: 0.78, MnO: 0.27, others: 2.68 (wt%). This chemical composition is fairly common in BFS produced in the Japanese iron-making plant. The minor components such as Fe, Ti and Mn are inevitably included in every BFS. All commercially available chemical reagents for synthesis, solvents, and organic compounds for catalytic reactions were purified using standard procedures.The synthesis of slagHC
The slag-made hydrocalumites (slagHC) were synthesized according to the method we previously reported.19 Typically, 10.0 g of the ball-milled slag was dissolved in 200 mL of 3 M HCl aqueous solution and was stirred at 100 °C for 2 h to fully pectize the Si component. The obtained suspension was separated into hydrated silica gel (SiO2 content >92 wt% as a dried product) and leaching-solution containing other elements by filtration and washing with 200 mL of deionized water. To this leaching solution, 2 M NaOH aqueous solution was added dropwise and pH was adjusted to 11.5 ± 0.1. The suspension was vigorously stirred for 1 h and then aged at 100 °C for another 18 h to carry out crystallization, followed by filtration, washing with deionized water and drying at 100 °C overnight to yield 6.2 g of pale brown-colored slagHC. The nominal chemical composition of slagHC determined by elemental and thermogravimetric analyses is presented as Ca2.03Mg0.49Fe0.046Mn0.019Si0.025Ti0.026Al1.00 (OH)7.15Cl1.01(CO3)0.11·2.05H2O. The samples calcined at T °C in air were named as slagHC(T).The synthesis of comparative materials
To provide a comparison, the Ca–Al–Cl hydrocalumite (Ca2Al(OH)6Cl·2H2O) was also prepared by a conventional coprecipitation method. In a typical method, 2 M NaOH aqueous solution was added dropwise to a mixed aqueous solution containing a calcium salt (CaCl2; Wako Pure Chemical Ind. Ltd.) and an aluminium salt (AlCl3·6H2O; Nacalai Tesq Inc.) with the Ca/Al molar ratio of 2.0 at 65 °C under vigorous stirring. The metal concentration in the solution was adjusted to 0.66 mol L−1, and pH of the solution was adjusted to 11.5 ± 0.1. The resulting precipitate was aged in the mother solution for 18 h at the same temperature, and then filtered, washed with decarbonated water several times and dried overnight at 100 °C. A mayenite (Ca12Al14O33) was also obtained by the similar procedure but with the Ca/Al molar ratio of 12:14, where the obtained precipitate was finally calcined at 800 °C in air for 12 h to afford the mayenite phase.
Characterization
Powder X-ray diffraction (XRD) patterns were recorded using a Rigaku Ultima IV diffractometer with CuKα radiation (λ = 1.54056 Å). Scans were performed at step size 0.02° over the 2θ range 10–70°. The morphology and particle size of the crystals were observed by Field Emission-scanning electron microscopy (FE-SEM) in a JEOL, JSM-6500. Nitrogen adsorption–desorption isotherms were measured at −196 °C using a BELSORP-max system (BEL Japan, Inc.). For layered double hydroxide compounds, the samples were outgassed under vacuum at 200 °C to eliminate physisorbed water molecules prior to measurement, whereas oxide compounds were outgassed under vacuum at 350 °C. Specific surface areas were calculated by the BET (Brunauer–Emmett–Teller) method using N2 adsorption data ranging from P/P0 = 0.05 to 0.25. Thermal analysis was carried out with differential thermal analysis-thermogravimetry (DTA-TG) (MAC Science Co. Ltd., TG-DTA2000S) from room temperature to 1000 °C at a heating rate of 10 °C min−1 in N2 flow of 50 mL min−1 using α-Al2O3 as standard. Infrared spectra were recorded with a JASCO FTIR-6100 instrument in the spectral range 4000–400 cm−1 under vacuum with a resolution of 4 cm−1 using samples diluted with KBr.The basicity of catalysts was studied by temperature programmed desorption using CO2 as probe molecule (CO2-TPD) and using a BELCAT-B system (BEL Japan, Inc.). Samples (50 mg) were preheated under a He flow (100 mL min−1) for 6 h at the maximum temperature at which sample calcination was performed (as-synthesized slagHC was exceptionally pretreated at 200 °C), subsequently were allowed to cool down to 40 °C and exposed to flowing CO2 (50 mL min−1) for 30 min. Then, the system was purged at 40 °C for 30 min with He in order to eliminate weakly adsorbed CO2. The CO2-TPD was carried out between 40 and 850 °C under a He flow (30 mL min−1) with a pumping rate of 10 °C min−1, and CO2 was detected by on-line mass spectrometry. To quantify the adsorbed amount of CO2 and H2O molecules during the catalyst pretreatment in air, TPD measurement was also performed for the samples pretreated under the same conditions as those before the soybean oil transesterification (800 °C under an air flow for 6 h).
Transesterification of n-ethylbutyrate
A Pyrex glass reactor containing the catalyst (20 mg), n-ethyl butyrate (12.4 mmol) and methanol (2 mL) with the molar ratio of n-ethyl butyrate:MeOH = 1:4 was magnetically stirred at 60 °C for up to 12 h. When calcined samples are used, catalysts were added into the reaction solution immediately after the calcination procedure. A portion of the reaction mixture was taken at appropriate intervals by filtration and then analyzed using a gas chromatograph (Shimadzu GC-14B) with a flame ionization detector equipped with a Rtx®-5 column. The conversion was calculated based on the ethyl butyrate:methyl butyrate molar ratio.
Biodiesel synthesis from soybean oil
The reaction was performed in a Pyrex glass batch reactor with a water-cooled condenser. Typically, the catalysts were calcined at 800 °C in air for 6 h with a heating rate of 3 °C min−1 before the reaction. After cooling to ambient temperature, the catalyst (0.1 g, corresponding to 1 wt% of the reaction mixture) was immediately added to the reaction mixture containing soybean oil (7.0 g, 8 mmol; edible grade, acid value <0.1 mg of KOH per gram, Nacalai Tesq Inc.) and anhydrous methanol (4 mL, 96 mmol; anhydrous, Aldrich Inc.) with the molar ratio of methanol:oil = 12:1. The mixture was magnetically stirred at 60 °C with stirring rate of 1000 rpm. A portion of the reaction mixture was withdrawn at appropriate intervals to determine the yield of FAME. Prior to the analysis, the catalyst and the by-produced glycerol were removed from the withdrawn mixture through decantation, followed by filtration. The produced amount of FAME, monoglyceride, diglyceride, and the residual amounts of soybean oil were quantified using a gas chromatograph (Shimadzu GC-14B) with a flame ionization detector equipped with a DB-HT-SIMDIS stainless capillary column. To examine the catalyst recyclability, the spent catalyst was three times repeatedly used after recovering by filtration, washing with acetone and drying at room temperature in a vacuum.
Acknowledgements
This work was financially supported by the Global COE Program for Chemistry Innovation (Project: Center of Excellence for Advanced Structural and Functional Materials Design), and the Grant-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology (No. 40200688). Y.K. thanks the JSPS Research Fellowships for Young Scientists.References
- International Energy Agency, World Energy Outlook 2011 - Executive Summary, IEA publications, 2007.
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148 CAS.
- M. Balakrishnan, V. S. Batra, J. S. J. Hargreaves and I. D. Pulford, Green Chem., 2011, 13, 16 RSC.
- (a) R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542 RSC; (b) R. Luque, J. C. Lovett, B. Datta, J. Clancy, J. M. Campelo and A. A. Romero, Energy Environ. Sci., 2010, 3, 1706 RSC.
- For overviews, see: (a) F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1 CrossRef CAS; (b) L. G. Schumacher, J. V. Gerpen and B. Adams, Encyclopedia of Energy, 2004, 1, 151 CrossRef; (c) M. Lapuerta, O. Armas and J. Rodríguez-Fernández, Prog. Energy Combust. Sci., 2008, 34, 198 CrossRef CAS; (d) A. Demirbas, Energy Convers. Manage., 2009, 50, 14 CrossRef CAS.
- (a) Y. Zhang, M. A. Dube, D. D. McLean and M. Kates, Bioresour. Technol., 2003, 89, 1 CrossRef CAS; (b) Y. Zhang, M. A. Dube, D. D. McLean and M. Kates, Bioresour. Technol., 2003, 90, 229 CrossRef CAS.
- N. N. A. N. Yusuf, S. K. Kamarudin and Z. Yaakub, Energy Convers. Manage., 2011, 52, 2741 CrossRef CAS.
- (a) B. Smith, H. C. Greenwell and A. Whiting, Energy Environ. Sci., 2009, 2, 262 RSC; (b) S. Lestari, P. Mäki-Arvela, J. Beltramini, G. Q. M. Lu and D. Y. Murzin, ChemSusChem, 2009, 2, 1109 CrossRef CAS; (c) A. Sivasamy, K. Y. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278 CrossRef CAS; (d) A. K. Endalew, Y. Kiros and R. Zanzi, Biomass Bioenergy, 2011, 35, 3787 CrossRef CAS.
- (a) H. J. Kim, B. S. Kang, M. J. Kim, Y. M. Park, D. K. Kim, J. S. Lee and K. Y. Lee, Catal. Today, 2004, 93, 315 CrossRef; (b) T. Ebiura, T. Echizen, A. Ishikawa, K. Murai and T. Baba, Appl. Catal., A, 2005, 283, 111 CrossRef CAS; (c) W. Xie and H. Li, J. Mol. Catal. A: Chem., 2006, 255, 1 CrossRef CAS; (d) W. Xie, H. Peng and L. Chen, Appl. Catal., A, 2006, 300, 67 CrossRef CAS.
- (a) S. Gryglewicz, Bioresour. Technol., 1999, 70, 249 CrossRef CAS; (b) S. Gryglewicz, Appl. Catal., A, 1999, 192, 23 CrossRef; (c) M. Di Serio, R. Tesser, L. Pengmei and E. Santacesaria, Energy Fuels, 2008, 22, 207 CrossRef CAS.
- (a) C. Ngamcharussrivichai, P. Totarat and K. Bunyakiat, Appl. Catal., A, 2008, 341, 77 CrossRef CAS; (b) A. K. Singh and S. D. Fernando, Energy Fuels, 2008, 22, 2067 CrossRef CAS.
- (a) M. Kouzu, T. Kasuno, M. Tajika, Y. Sugimoto, S. Yamanaka and J. Hidaka, Fuel, 2008, 87, 2798 CrossRef CAS; (b) M. Kouzu, J. Hidaka, Y. Komichi, H. Nakano and M. Yamamoto, Fuel, 2009, 88, 1983 CrossRef CAS; (c) M. Kouzu, S. Yamanaka, J. Hidaka and M. Tsunomori, Appl. Catal., A, 2009, 355, 94 CrossRef CAS.
- C. Reddy, V. Reddy, R. Oshel and J. G. Verkade, Energy Fuels, 2006, 20, 1310 CrossRef CAS.
- For reviews, see: (a) F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS; (b) K. Kaneda, K. Yamaguchi, K. Mori, T. Mizugaki and K. Ebitani, Catal. Surv. Jpn., 2000, 4, 31 CrossRef CAS; (c) K. Takehira and T. Shishido, Catal. Surv. Asia, 2007, 11, 1 CrossRef CAS; (d) D. P. Debecker, E. M. Gaigneaux and G. Busca, Chem.–Eur. J., 2009, 15, 3920 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183 CrossRef CAS.
- W. Xie, H. Peng and L. Chen, J. Mol. Catal. A: Chem., 2006, 246, 24 CrossRef CAS.
- M. Di Serio, M. Ledda, M. Cozzolino, G. Minutillo, R. Tesser and E. Santacesaria, Ind. Eng. Chem. Res., 2006, 45, 3009 CrossRef CAS.
- M. J. Campos-Molina, J. Santamaría-González, J. Mérida-Robles, R. Moreno-Tost, M. C. G. Albuquerque, S. Bruque-Gámez, E. Rodríguez-Castellón, A. Jiménez-López and P. Maireles-Torres, Energy Fuels, 2010, 24, 979 CrossRef CAS.
- Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, J. Mater. Chem., 2010, 20, 5052 RSC.
- H. G. V. Oss, Iron and Steel Slag, in 2008 Minerals Yearbook, U.S. Department of the Interior, U.S. Geological Survey, 2010, pp. 69.1 Search PubMed.
- (a) B. Das, S. Prakash, P. S. R. Reddy and V. N. Misra, Resour., Conserv. Recycl., 2007, 50, 40 CrossRef; (b) Z. Huaiwei and H. Xin, Resour., Conserv. Recycl., 2011, 55, 745 CrossRef.
- S. Wang, Environ. Sci. Technol., 2008, 42, 7055 CrossRef CAS.
- (a) Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, J. Mater. Chem., 2009, 19, 7263 RSC; (b) Y. Kuwahara, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, Chem. Lett., 2009, 38, 626 CrossRef CAS.
- K. Hosni and E. Srasra, Appl. Clay Sci., 2009, 43, 415 CrossRef CAS.
- K. Wilson, C. Hardacre, A. F. Lee, J. M. Montero and L. Shellard, Green Chem., 2008, 10, 654 RSC.
- Y. Kuwahara, K. Tsuji, T. Ohmichi, T. Kamegawa, K. Mori and H. Yamashita, ChemSusChem, 2012 DOI:10.1002/cssc.201100814 , in press.
- S. J. Ahmed and H. F. W. Taylor, Nature, 1967, 215, 622 CrossRef CAS.
- I. Rousselot, C. Taviot-Guého, F. Leroux, P. Léone, P. Palvadeau and J. P. Besse, J. Solid State Chem., 2002, 167, 137 CrossRef CAS.
- L. Vieille, I. Rousselot, F. Leroux, J. P. Besse and C. Taviot-Guého, Chem. Mater., 2003, 15, 4361 CrossRef CAS.
- A. C. Vieira, R. L. Moreira and A. Dias, J. Phys. Chem. C, 2009, 113, 13358 CAS.
- E. López-Salinas, M. E. L. Serrano, M. A. C. Jácome and I. S. Secora, J. Porous Mater., 1996, 2, 291 CrossRef.
- (a) J. Otera, T. Yano, A. Kawabata and H. Nozaki, Tetrahedron Lett., 1986, 27, 2383 CrossRef CAS; (b) D. Seebach, E. Hungerbühler, R. Naef, P. Schnurrenberger, B. Weidmann and M. Züger, Synthesis, 1982, 138 CrossRef CAS.
- M. J. Climent, A. Corma, S. Iborra, K. Epping and A. Velty, J. Catal., 2004, 225, 316 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537 CrossRef CAS.
- G. S. Macala, A. W. Robertson, C. L. Johnson, Z. B. Day, R. S. Lewis, M. G. White, A. V. Iretskii and P. C. Ford, Catal. Lett., 2008, 122, 205 CrossRef CAS.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaría and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317 CrossRef CAS.
- M. Kouzu, T. Kasuno, M. Tajika, S. Yamanaka and J. Hidaka, Appl. Catal., A, 2008, 334, 357 CrossRef CAS.
- (a) H. D. Lutz, H. Möller and M. Schmidt, J. Mol. Struct., 1994, 328, 121 CrossRef CAS; (b) J. C. Lavalley, Catal. Today, 1996, 27, 377 CrossRef CAS.
- For example, see: (a) M. Kim, C. DiMaggio, S. Yan, S. O. Salley and K. Y. Simon Ng, Green Chem., 2011, 13, 334 RSC; (b) W. Thitsartarn and S. Kawi, Green Chem., 2011, 13, 3423 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed reaction kinetics in n-ethyl butyrate transesterification, XRD patterns after repetitive soybean oil transesterification, TPD profiles and TEM images of the synthesized materials. See DOI: 10.1039/c2cy20113e |
| This journal is © The Royal Society of Chemistry 2012 |