DOI:
10.1039/C2CY20110K
(Communication)
Catal. Sci. Technol., 2012,
2, 1556-1559
Asymmetric, biocatalytic labeled compound synthesis using transaminases†
Received
24th February 2012
, Accepted 19th March 2012
First published on 20th March 2012
Abstract
The use of transaminases for the incorporation of deuterium and tritium in chiral amines starting from prochiral ketone precursors has been demonstrated. Efficient labeling was demonstrated using a variety of ketone substrates. Amplification of the labeled product was observed, with tritium incorporation enriched up to 2.8X the statistically expected levels of labeling.
Introduction
The asymmetric synthesis of deuterium and tritium labeled compounds is an essential tool in understanding drug metabolism and pharmacodynamics.1 Radioactive tritium labels allow for a detailed understanding of drug absorption and distribution during pre-clinical drug development activities. The usual means for installing such labels begin with synthesis from readily available deuterium and tritium sources, notably hydride reagents and enriched hydrogen gas.1a Most often, racemic chemical transformations are utilized followed by preparative chiral chromatography. While reliable, the radiolabel's half life along with tracking, accounting, and disposal of all associated radioactive waste materials often poses a significant challenge. Catalytic, asymmetric transfer hydrogenation2 and hydride reductions3 along with diastereoselective reductions of imines,4 insertion reactions,5 and a variety of base mediated exchange reactions6 have all been shown to access labeled chiral amines. Less exploited is the use of biocatalysis to install deuterium/tritium labels in an asymmetric fashion.7 Deuterium oxide has been used to elucidate the reaction mechanism of glutamate-alanine transaminase, but this approach has never been exploited as a general means to convert ketones to labeled, enantiopure amines.8
2. Experimental
Detailed experimental procedures can be found in the ESI.†
3. Results and discussion
3.1. Deuterium incorporation into sitagliptin free base
During the development of a biocatalytic approach to Januvia® suitable for commercial production,9 we conducted the transamination of ketoamide 1 under representative transamination conditions with isopropyl amine and one of the advanced transaminase variants in a mixture of D2O and DMSO (Scheme 1). After 1H NMR analysis, it appeared that the sitagliptin free base (2) generated in the reaction had nearly complete deuterium incorporation both adjacent to the amine stereocenter and at the newly formed center.
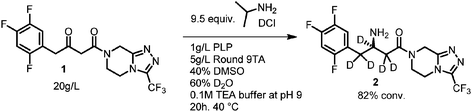 |
| | Scheme 1 Deuterium incorporation into sitagliptin free base. | |
The results of the aforementioned experiment conform with the generally accepted equilibrium based mechanism for transaminase enzymes wherein the pyridoxal-5′-phosphate (PLP) forms an imine with the ketone in the enzyme's active site.8a This example suggested that the imine/enamine intermediates and the catalytically active residues in the active site are in rapid exchange with the aqueous solvent (Scheme 2). Not surprisingly, we also observed background exchange at the alpha-carbon in solutions containing the substrate ketone and D2O. However, incorporation of deuterium at the newly formed amine center was solely catalyzed by the transaminase. Under our operating conditions, no ND2 and NHD species were detected.
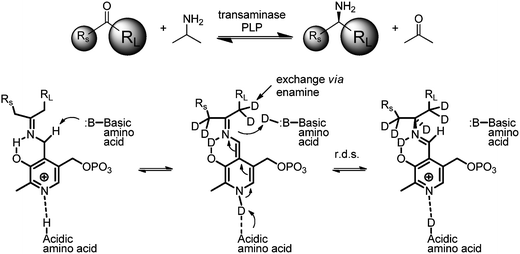 |
| | Scheme 2 Equilibrium mechanism. | |
3.2. General deuterated amine synthesis via ketone transamination
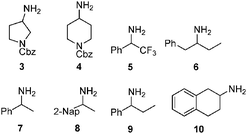
We found these results to be general with regard to the transamination and deuteration of a number of ketones (Table 1). Deuterium incorporation was >98% at the indicated positions as determined by either MS or 1H NMR analysis under similar transamination conditions using either an R-selective transaminase variant developed for sitagliptin9 or an S-selective transaminase.10
Table 1 Full deuteration of amines
| Amine |
Sitagliptin Rnd. 11 |
P1G5 |
| config.a |
eeb % |
%conv.c |
% dd |
config.a |
eeb % |
%conv.c |
% dd |
|
Assigned by analogy.
Determined by chiral phase chromatography.
Conversion by HPLC.
Determined by LC/MS.
|
|
3
|
(R) |
99 |
94 |
90 (d5) |
(S) |
99 |
97 |
90 (d5) |
|
4
|
NA |
NA |
90 |
90 (d5) |
NA |
NA |
90 |
90 (d5) |
|
5
|
(S) |
99 |
90 |
90 (d1) |
(R) |
99 |
90 |
90 (d1) |
|
6
|
(R) |
99 |
90 |
90 (d5) |
ND |
ND |
< 5 |
90 (d5) |
|
7
|
(R) |
99 |
90 |
90 (d4) |
(S) |
99 |
90 |
90 (d4) |
|
8
|
(R) |
99 |
90 |
90 (d4) |
(S) |
99 |
90 |
90 (d4) |
|
9
|
(R) |
99 |
90 |
90 (d3) |
ND |
ND |
< 5 |
90(d3) |
|
10
|
(R) |
99 |
90 |
99 (d5) |
(S) |
99 |
90 |
99 (d5) |
3.2. Deuterium amplification
In preparation of incorporating radioactive tritium into the amine product, we needed to demonstrate that the enzyme would still insert the label with dilute tritium oxide in water at levels that would be deemed useful. In order to study the effects of dilute tritium oxide in water, we first studied the transamination with varying amounts of D2O in water with a variety of available transaminase enzymes (Fig. 1). In all cases, enrichment and amplification above the statistically expected amount was observed in the amine product. For example, 14 vol% D2O with isopropyl amine and S-selective Cambrex S16 transaminase gave 32% labeling primarily at the newly formed stereocenter. The same effect was observed using other R- and S-selective transaminases from Codexis. This dramatic amplification effect indicated that high levels of enrichment could be achieved with diluted amounts of deuterium or tritium oxide in water.
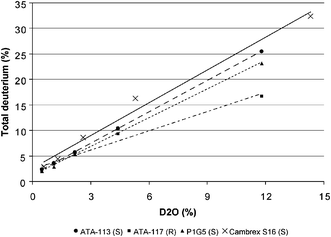 |
| | Fig. 1 Deuterium amplification. | |
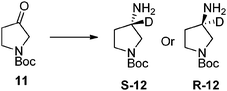
In order to gain a better understanding of the various reaction parameters' influence on both ketone conversion and levels of isotope incorporation under dilute deuterium oxide conditions, a series of experiments were conducted examining enzyme loading, PLP concentration, temperature, pH, and D2O concentration. We found that although enzyme loading, PLP concentration, and temperature did impact the rate of reaction and conversion, the overall deuterium incorporation remained constant across these factors for the transamination of N-Cbz pyrrolidinone (13) catalyzed by CDX-017. The reaction pH had a significant influence on conversion and a modest impact on the extent of deuteration with pH 10 giving complete conversion and 36% deuteration using 6 vol% D2O, whereas pH 8 gave only 67% conversion and 31% deuteration. D2O concentration had little to no effect on reaction rate or conversion, however the amount of labeling increased with increasing D2O content (Fig. 2). The weighted enrichment average indicates that the deuterium enrichment remains relatively constant regardless of D2O concentration. Increasing D2O concentration also leads to a wider distribution of the number of deuteriums incorporated into the product amine, while the lowest D2O concentration afforded mostly mono-deuterated amine product at the amine stereocenter (Fig. 3).
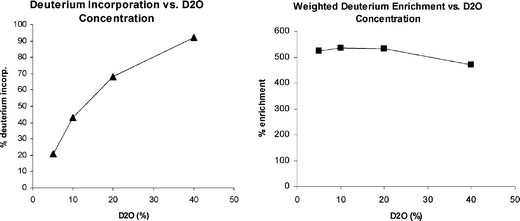 |
| | Fig. 2 Isotope Incorporation vs. D2O Concentration. | |
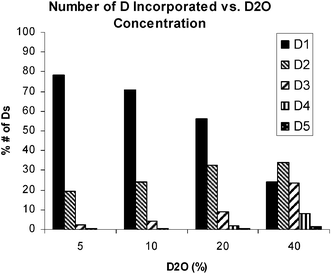 |
| | Fig. 3 Number of D incorporated in amine Product vs. D2O Concentration. | |
3.3. Tritium incorporation and amplification
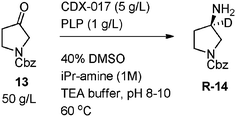
The results from the aforementioned deuterium experiments indicated that conducting the transamination with dilute tritium oxide at pH 9–10 and running reactions to complete conversion would lead to maximum levels of mostly mono-tritiated amine product at the amine stereocenter. This was proved to be accurate when reactions run with tritium dioxide exhibited a similar enrichment phenomenon, with 280% enrichment observed instead of the 100% statistically expected level of labeling (Scheme 3). In addition, a reaction and work-up protocol was developed which minimized operations, contamination, and waste. First, an enzyme stock solution containing 10 mg mL−1 enzyme and 10 mg mL−1 PLP co-factor dissolved in 125 mg mL−1 isopropylamine hydrochloride at pH 9.5 in 0.2 M triethanolamine buffer was prepared along with a 25 mg mL−1 solution of substrate in DMSO. Then, 0.2 mL of this enzyme stock solution was added to 1mL of 0.2% THO in water followed by 0.8 mL of the substrate stock solution. The reaction was then stirred at ambient conditions for two days. A simple work-up was devised in which the reaction was simply diluted with water and extracted with isopropyl acetate. After concentration of the organics, the crude amine was purified by reverse phase chromatography.
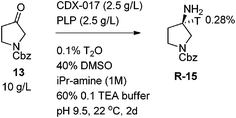 |
| | Scheme 3 Tritium Incorporation Using a Transaminase. | |
The observed enrichment effects during transamination can be explained by the thermodynamic preference for C–D (or C–T) bonds versus a C–H bond. In addition, the isotopic labeling occurs predominantly at the newly formed amine stereocenter with scrambling to the adjacent positions over time under reversible reaction conditions (isopropylamine donor). While a late transition state kinetic isotope effect may be operative, the magnitude of this enrichment suggests a strong thermodynamic preference as the dominant effect.
4. Conclusions
In summary, transaminases have been shown to be an effective way to generate chiral amines from prochiral ketone precursors. We have demonstrated that this same transformation can be used to efficiently generate deuterium and tritium labeled amines. The methodology appears to be general and has been demonstrated on several ketone substrates. Finally, deuterium and tritium enrichment has been observed, with 280% tritium enrichment observed instead of the 100% statistically expected level of labeling.
References
-
(a)
R. Voges, J. R. Heys and T. Moenius, Preparation of Compounds with Tritium and Carbon-14, John Wiley and Sons, Ltd, United Kingdom, 2009 Search PubMed
 ;
(b)
J. Allen and R. Voges, Synthesis and Applications of Isotopically Labelled Compounds 1994, John Wiley and Sons, Inc, New York, 1995 Search PubMed .
;
(b)
J. Allen and R. Voges, Synthesis and Applications of Isotopically Labelled Compounds 1994, John Wiley and Sons, Inc, New York, 1995 Search PubMed .
-
(a) C. J. Cobley and J. P. Henschke, Adv. Synth. Catal., 2003, 345, 195 CrossRef CAS ;
(b) C. R. Graves, K. A. Scheidt and A. T. Nguyen, Org. Lett., 2006, 8, 1229 CrossRef CAS .
- D. Miyazaki, K. Nomura, T. Yamashita, I. Iwakura, T. Ikeno and T. Yamada, Org. Lett., 2003, 5, 3555 CrossRef CAS .
-
(a) M. A. T. Maughan, I. G. Davies, T. D. W. Claridge, S. Courtney, P. Hay and B. G. Davis, Angew. Chem., 2003, 115, 3918 CrossRef ;
(b) V. P. Miller, D. Yang, T. M. Weigel, O. Han and H. Liu, J. Org. Chem., 1989, 54, 4175 CrossRef CAS .
- E. C. Lee and G. C. J. Fu, J. Am. Chem. Soc., 2007, 129, 12066 CrossRef CAS .
-
(a) P. J. M. Taylor and S. D. Bull, Tetrahedron: Asymmetry, 2006, 17, 1170 CrossRef CAS ;
(b) B. Lygo and L. D. Humphreys, Tetrahedron Lett., 2002, 43, 6677 CrossRef CAS .
- For transaminases used to make labeled amino acids, see:
(a) J. Raap, S. Nieuwenhuis, A. Creemers, S. Hexspoor, U. Kragl and J. Lugtenburg, Eur. J. Org. Chem., 1999, 2609 CrossRef CAS ;
(b) S. D. Dimitrijevich, M. D. Scanlon and M. Anbar, J. Labelled Compd. Radiopharm., 1982, 19, 573 CrossRef CAS . For other enzymatic labeled compound synthesis, see
(c) K. Edegger, C. C. Gruber, T. M. Poessl, S. R. Wallner, I. Lavandera, K. Faber, F. Niehaus, J. Eck, R. Oehrlein, A. Hafner and W. Kroutil, Chem. Commun., 2006, 2402 RSC ;
(d) C.-H. Wong and G. M. J. Whitesides, J. Am. Chem. Soc., 1983, 105, 5012 CrossRef CAS ;
(e) R. S. Phillips, B. Sundararju and N. G. Faleev, J. Am. Chem. Soc., 2000, 122, 1008 CrossRef CAS .
-
(a) A. J. L. Cooper, J. Bio. Chem., 1976, 251, 1088 CAS ;
(b) P. H. Yu, D. A. Durden, B. A. Davis and A. A. Boulton, J. Neurochem., 1987, 48, 440 CrossRef CAS ;
(c) M. M. Mahon, R. Graber, P. Christen and J. P. G. Malthouse, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1999, 1434, 191 CrossRef CAS .
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. Hughes, Science, 2010, 329, 305 CrossRef CAS .
- Codexis panel transaminase P1G5.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20110k |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.