Effect of molecular structure on the hydrogenation and isomerisation of propenylbenzene isomers
Lorna C.
Begley
,
Kirsty J.
Kakanskas
,
Andrew
Monaghan
and
S. David
Jackson
*
Centre for Catalysis Research, WestCHEM, Department of Chemistry, University of Glasgow, Glasgow, G12 8QQ, Scotland, UK. E-mail: david.jackson@glasgow.ac.uk
First published on 28th March 2012
Abstract
The hydrogenation and isomerisation of allylbenzene (AB), trans-β-methyl styrene (TBMS) and cis-β-methyl styrene (CBMS), in the liquid phase, was investigated over a 2.5% Rh/silica catalyst. When reacted individually, the cis-isomer gave the fastest rate of hydrogenation followed by allylbenzene, with the trans-isomer having the slowest rate giving a ratio of rates of CBMS:AB:TBMS of 4.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8:1. The isomerisation reaction followed thermodynamic control. When co-hydrogenated, allylbenzene inhibited the hydrogenation of both CBMS and TBMS but allowed isomerisation: the rate of allylbenzene hydrogenation was unaffected. This behaviour is in keeping with terminal alkenes hydrogenating at edge/corner sites while internal alkenes hydrogenate on terrace faces. The terminal alkene inhibits fast diffusion of hydrogen into the sub-surface and hence prevents hydrogenation of the CBMS and TBMS. The model of sub-surface hydrogen, observed in palladium catalysis, being key for hydrogenation but not isomerisation, was found to explain the results in rhodium hydrogenation catalysis.
:2.8:1. The isomerisation reaction followed thermodynamic control. When co-hydrogenated, allylbenzene inhibited the hydrogenation of both CBMS and TBMS but allowed isomerisation: the rate of allylbenzene hydrogenation was unaffected. This behaviour is in keeping with terminal alkenes hydrogenating at edge/corner sites while internal alkenes hydrogenate on terrace faces. The terminal alkene inhibits fast diffusion of hydrogen into the sub-surface and hence prevents hydrogenation of the CBMS and TBMS. The model of sub-surface hydrogen, observed in palladium catalysis, being key for hydrogenation but not isomerisation, was found to explain the results in rhodium hydrogenation catalysis.
Introduction
The hydrogenation of alkenes to alkanes is an area of catalysis that has been active for over 100 years and yet our understanding is not complete. As Bond1 remarks in his book “the reaction (alkene hydrogenation) is much more than the simple addition of a molecule of hydrogen or of two hydrogen atoms to the carbon-carbon double bond”. In recent years studies have shown that although the hydrogenation of ethene is structure insensitive, higher homologues such as propene2 and pentene3 do show structure sensitivity. Along with hydrogenation comes isomerisation and in some very elegant work Zaera and co-workers [4 and references therein] investigated alkene isomerisation and showed over Pt that the shape of the crystallite, and hence the crystal face, had a significant effect on trans-cis and cis-trans isomerisation such that the rate of each reaction was different depending on the starting isomer. It is clear that these apparently simple reactions are much more complex and sensitive to surface structure than was once thought.In this study we have examined the hydrogenation of alkenyl aromatics, namely allylbenzene, cis-β-methyl styrene and trans-β-methyl styrene. These species are isomers and, in principle, give us information on how steric and electronic effects may influence hydrogenation and isomerisation. As well as studying their individual hydrogenation we have also examined competitive hydrogenation where, as is often the case, two or three isomers were present in the reaction mix.
Experimental
The catalyst used throughout the study was a 2.5% w/w Rh/SiO2 (Rh dispersion 60%, metal crystallite size 1.8 nm, support surface area 488 m2 g−1). Davison Catalysts supplied the silica support, while the active catalyst was prepared by Johnson Matthey by incipient-wetness using aqueous rhodium chloride salts. The catalyst was dried and reduced in flowing H2/N2. Johnson Matthey performed the catalyst characterisation.All reactants were used without further purification. The reaction was carried out in a 0.5l Buchi stirred autoclave with a hydrogen-on-demand delivery system. Around 0.05 g of catalyst was added to 330 ml of degassed solvent, 2-propanol. Reduction of the catalyst was performed in situ by sparging the system with H2 (300 cm3 min−1) for 30 min at 313 K while stirring the contents of the autoclave at 800 rpm. After reduction, the autoclave was adjusted to the appropriate reaction temperature of between 313 K under a nitrogen atmosphere. For allylbenzene (AB), cis-β-methylstyrene (CBMS) and trans-β-methylstyrene (TBMS), 1.5 ml (AB 11.3 mmoles, TBMS and CBMS 11.6 mmoles) was injected into an unstirred solution, followed by 20 ml of degassed 2-propanol (IPA) to ensure that all the reactant was washed into the reactor. For the competitive reactions, 1.5 ml of each reactant was added simultaneously. The autoclave was then mixed briefly at a stirrer speed of 800 rpm and pressurised to 1 barg with N2 and a sample was taken via a sample valve. The vessel was depressurised and then pressurised with H2 to 1 barg. Following this the stirrer was set to a speed of 1000 rpm and samples taken at regular intervals. Liquid samples were analysed by GC using an FID detector and a 50 m CP-Al2O3/Na2SO4 column. Standard checks were undertaken to confirm that the system was not under mass transport control.
Results
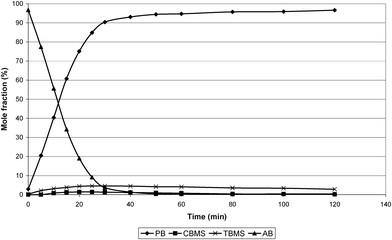
The three isomers were hydrogenated to phenyl propane (PP) at 313 K and 1 barg. The results are shown in Fig. 1–3. | ||
| Fig. 1 Hydrogenation of TBMS at 313 K and 1 barg. | ||
 | ||
| Fig. 2 Hydrogenation of AB at 313 K and 1 barg. | ||
 | ||
| Fig. 3 Hydrogenation of CBMS at 313 K and 1 barg. | ||
It can be seen that all three isomers rapidly hydrogenate to phenyl propane (PP) but AB and CBMS also isomerise to TBMS. The individual reactants were also used over a range of concentrations (3.8–15.2 mmole for AB, 3.9–15.6 mmole for CBMS and TBMS) to determine the order of reaction. The orders of reaction obtained were −1.3 for TBMS, −1.0 for AB and −0.7 for CBMS.
The pair reactions are shown in Fig. 4–6 and the reaction with all three isomers is shown in Fig. 7. In reactions where TBMS is present there is an increase in mole fraction showing that isomerisation is taking place.
 | ||
| Fig. 4 Co-hydrogenation of CBMS and AB at 313 K and 1 barg. | ||
 | ||
| Fig. 5 Co-hydrogenation of TBMS and AB at 313 K and 1 barg. | ||
 | ||
| Fig. 6 Co-hydrogenation of CBMS and TBMS at 313 K and 1 barg. | ||
 | ||
| Fig. 7 Co-hydrogenation of AB, CBMS and TBMS at 313 K and 1 barg. | ||
The initial rate of formation was calculated for PP in each reaction system and is reported in Table 1. An initial rate of isomerisation was also calculated and reported. The loss of the reactant(s) was analysed using first order kinetics and a first order rate constant determined and detailed in Table 1.
| PP | AB | CBMS | TBMS | |
|---|---|---|---|---|
| AB | 11.0 | 0.0912 | 0 | 1.1 |
| CBMS | 23.4 | 0.3 | 0.1339 | 5.9 |
| TBMS | 6.4 | 0 | 0.2 | 0.0321 |
| AB/CBMS | 18.4 | 0.1134 | 0.0085 | 2.6 |
| AB/TBMS | 15.8 | 0.1075 | 0.5 | 0.0138 |
| CBMS/TBMS | 17.3 | 0.3 | 0.0871 | 0.0168 |
| AB/CBMS/TBMS | 18.1 | 0.0817 | 0.0130 | 0 |
When the three reactants were reacted simultaneously, no hydrogenation of TBMS was detected. Rather the concentration of TBMS in the system increased. To further investigate this behaviour the three-reactant reaction was run with a reduced concentration of each isomer, 0.5 ml (3.8 mmole for AB, 3.9 mmole for CBMS and TBMS) for each rather than 1.5 ml. The reaction profile is shown in Fig. 8.
 | ||
| Fig. 8 Conversion (%) of AB, CBMS and TBMS using 0.5 ml of each isomer. | ||
Discussion
Text books will state that the more sterically hindered the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond the harder it is to hydrogenate and hence a slower rate will be seen. A typical ordering will be:
C double bond the harder it is to hydrogenate and hence a slower rate will be seen. A typical ordering will be: 
So we should expect that allylbenzene will hydrogenate faster than CBMS which will hydrogenate faster that TBMS. However when we examine the first order rate constants for the hydrogenation of the individual isomers, we find that CBMS has the fastest rate and we obtain a ratio of rates of CBMS:AB:TBMS of 4.2:2.8:1. This behaviour has been observed with alkenyl aromatics over Pd,5 where a similar ratio of CBMS:AB:TBMS of 3.8:2.2:1 was obtained. Comparable results were also found with pentene hydrogenation over Pd,6 where the cis-isomer was also found to be the most reactive followed by 1-pentene while the trans-isomer was the least reactive and with butene hydrogenation7 where the trans-isomer hydrogenated significantly slower than the cis- and 1-isomers. The slow rate of hydrogenation associated with the trans-isomer has been attributed to its thermodynamic stability, which is reflected in the equilibrium isomer composition at 313 K of approximately 97% trans, 3% cis and <0.1% allylbenzene. The isomerisation rates also are in accord with thermodynamic control. Both allylbenzene and CBMS isomerise to TBMS at a high rate whereas TBMS does not isomerise to AB. Low rates are observed for TBMS isomerisation to CBMS and CBMS to AB. However a thermodynamic argument does not explain why CBMS is more reactive than AB. In a recent study of pentene hydrogenation over a nickel catalyst, McGregor and Gladden8 showed that carbon deposition during pentene hydrogenation followed the sequence cis-2-pentene < 1-pentene < trans-2-pentene and that this was in keeping with the strength of adsorption. If we assume that similar behaviour is occurring with the alkenyl benzenes then the strength of adsorption would be TBMS > AB > CBMS. This ordering would have some support from the reaction orders, which suggests that TBMS is the most strongly adsorbed. Also when TBMS is a reactant in the competitive reactions, there is some evidence for adsorption in preference over the other reactant, however the reaction data from the competitive reactions would be in keeping with a strength of adsorption order of AB > CBMS > TBMS. In either case the differences in hydrogenation activity may be easily explained. Typically the rate of a catalytic reaction may be related to the strength of adsorption through a “volcano” curve. If the strength of adsorption is too weak or too strong then the rate is decreased, only when the strength of adsorption is at an optimum does the maximum rate occur. For an order of AB > CBMS > TBMS we can envisage that CBMS sits at the apex of the volcano curve, whereas for an order of TBMS > AB > CBMS we may envisage a scenario as depicted in Fig. 9.
 | ||
| Fig. 9 Example volcano plot relating strength of adsorption to rate. | ||
However the mode of adsorption is also an important parameter. It is the π-adsorbed state that participates in hydrogenation and isomerisation1,9 and studies of butene adsorption10 have shown that there are differences between the isomers and the mode of adsorption. Over Pt(111) the di-σ adsorbed trans isomer was more strongly bound than the cis whereas with the π-adsorbed species the cis was more stable than the trans. This behaviour was reinforced when hydrogen was co-adsorbed.10 Therefore the trans isomer may be more stable and strongly bound as an inactive di-σ species while the cis isomer is more stable in the active π-adsorbed form.
When CBMS and TBMS are co-hydrogenated, the first order rate constant for each isomer is approximately halved from that observed when each was hydrogenated singly. This is typical for a competitive reaction where the increased concentration of reactants reduces the number of sites available to each. On the contrary, when the pairing is AB/CBMS or AB/TBMS the AB reactivity is unaffected while the reactivity of the other isomer is decreased. This is, at first sight, a surprising result and we see it taken to the extreme when, with all three isomers present, TBMS does not hydrogenate to any significant extent. Given that we have indicated above that TBMS could have the strongest bond to the surface we may have expected that it would strongly inhibit the hydrogenation of AB and CBMS. The fact that the opposite behaviour is observed gives us an insight into the reaction processes occurring on the surface and may suggest that the AB > CBMS > TBMS order for strength of adsorption is correct; however it should be remembered that in a co-adsorption situation the strength of bonding for individual isomers can change.4,10 In all the competitive reactions the rate of AB hydrogenation is barely affected, whereas the activities of both CBMS and TBMS are drastically reduced. Analysing the AB/CBMS reaction in detail reveals that AB hydrogenates more rapidly to phenyl propane, while CBMS isomerises to TBMS until all the AB has been hydrogenated, only then does CBMS hydrogenate to phenyl propane. This can be shown by taking the average of the sum of the number of moles of phenyl propane and AB in solution over the first 25 min of reaction. The value obtained is 1.38 × 10−2 ± 8 × 10−4 moles, this compares to a starting value of AB of 1.33 × 10−2 moles. If a similar process is carried out on the TBMS and CBMS we find that the average sum is 1.19 × 10−2 ± 5 × 10−4 moles, this compares to a starting value of CBMS of 1.24 × 10−2 moles. Hence two separate reactions are taking place on the surface, AB → PP and CBMS → TBMS. After 25 min there is <2% of AB left in solution and CBMS begins to hydrogenate rather than isomerise and the values diverge. Therefore the rate constant for CBMS in the AB/CBMS reaction in Table 1 is for isomerisation rather than hydrogenation. A similar situation applies to the AB/TBMS reaction. Thus AB inhibits hydrogenation but not isomerisation of CBMS and TBMS. This difference between hydrogenation and isomerisation has been observed previously11 where it was found that over single crystal Pd(111) no hydrogenation took place for a pentene/hydrogen system yet hydrogenation occurred over small Pd particles under identical conditions. The reason behind this difference lies in the availability of “sub-surface” hydrogen. In a recent publication12 Schauermann et al. showed that for hydrogenation over palladium, fast diffusion of hydrogen into these sub-surface sites was required, however if this was inhibited, then isomerisation was observed but not hydrogenation. The presence of sub-surface hydrogen in rhodium crystallites has been the subject of some interest.13–17 The sub-surface state was first tentatively assigned by Nieuwenhuys et al.13 who studied hydrogen adsorption on rhodium and concluded “that Rh is able, like Pd, to absorb hydrogen under conditions where the other group VIII metals do not absorb subsurface hydrogen”. A later study in 1991 by Nichtl-Pecher et al.14 suggested that the reconstruction of the surface under hydrogen adsorption opened diffusion channels to the sub-surface, while DFT calculations15,16 suggested that sub-surface hydrogen in rhodium would have a higher energy than surface hydrogen but that stable states were possible. Using hydrogen/deuterium mixtures coupled to TDS and HREELS, Winkler et al.17 revealed the presence of sub-surface hydrogen on Rh(100) and calculated its energy using DFT. Therefore rhodium can accommodate sub-surface hydrogen in a manner similar to palladium. Over palladium it was also shown that rapid diffusion occurred via modified edge and corner sites,12 which may be similar to the diffusion channels proposed by Nichtl-Pecher et al.14 The isomerisation/hydrogenation of internal alkenes has been shown to occur on terrace and plane faces of metal crystallites (Pd) while isomerisation/hydrogenation of terminal alkenes has been shown to occur on edges and corners.3,18,19 Therefore if the adsorbed terminal alkene inhibited the fast diffusion of hydrogen to the sub-surface by reacting with it then we would expect isomerisation of the internal alkene but not hydrogenation – as is indeed observed. This suggests that alkene hydrogenation and isomerisation over rhodium is behaving in an analogous manner to palladium.
The co-hydrogenation of the three isomers behaves in a similar manner: in the initial stages AB is hydrogenated to phenyl propane, while CBMS is isomerised to TBMS; in this case the level of TBMS in the system increases. When the system is run with low concentrations a very clear demarcation of the hydrogenation activity is observed. AB is rapidly hydrogenated while CBMS is preferentially hydrogenated before TBMS, which does not hydrogenate until virtually all the AB and CBMS are hydrogenated. Each reactant was used at this lower concentration individually and the order of activity was found to be the same as at the higher concentration. However as discussed above the competitive regime is significantly different from the individual. AB is hydrogenated at the approximately the same rate in both individual and competitive. There is no competition for the AB sites and hydrogen flux is similar. The rate of CBMS hydrogenation is, as expected, reduced from that of the individual (a first order rate constant of 0.0484 min−1cf. 0.4071 min−1 when non-competitive). The slightly surprising result is the absence of TBMS hydrogenation until the majority of the other reactants have been consumed (AB at 100% conversion, CBMS at ∼70% conversion) and even then the rate is considerably reduced (a first order rate constant of 0.0167 min−1cf. 0.1269 min−1 when non-competitive). However given that TBMS is strongly adsorbed, it would be expected that with reduced concentration and competitive adsorption, it will be the highest energy sites that will have been occupied first, i.e. sites which bind TBMS most strongly. Therefore we may expect a significantly reduced rate.
References
- G. C. Bond, Metal-Catalysed Reactions of Hydrocarbons (Fundamental and Applied Catalysis), Springer, New York, 2005 Search PubMed.
- R. L. Burwell Jr, Langmuir, 1986, 2, 2–11 CrossRef CAS; P. O. Otero-Schipper, W. A. Wachter, J. B. Butt, J. B. Cohen and R. L. Burwell Jr, J. Catal., 1977, 50, 494–507 CrossRef.
- A. M. Doyle, S. K. Shaikhutdinov and H.-J. Freund, Angew. Chem., Int. Ed., 2005, 44, 629–631 CrossRef CAS.
- F. Zaera, Acc. Chem. Res., 2009, 42, 1152–1160 CrossRef CAS.
- L.C. Begley and S. David Jackson, unpublished results.
- A. S. Canning, S. D. Jackson, A. Monaghan and T. Wright, Catal. Today, 2006, 116, 22–29 CrossRef CAS.
- G. C. Bond and J. M. Winterbottom, Trans. Faraday Soc., 1969, 65, 2779–2793 RSC.
- J. McGregor and L. F. Gladden, Appl. Catal., A, 2008, 345, 51–57 CrossRef CAS.
- F. Zaera, Langmuir, 1996, 12, 88–94 CrossRef CAS.
- F. Zaera, J. Am. Chem. Soc., 2008, 130, 14924–14925 CrossRef.
- A. M. Doyle, Sh. K. Shaikhutdinov and H.-J. Freund, J. Catal., 2004, 223, 444–453 CrossRef CAS.
- W. Ludwig, A. Savara, K.-H. Dostert and S. Schauermann, J. Catal., 2011, 284, 148–156 CrossRef CAS.
- V. V. Gorodetskii, B. E. Nieuwenhuys, W. M. H. Sachtler and G. K. Boreskov, Surf. Sci., 1981, 108, 225–234 CrossRef CAS.
- W. Nichtl-Pecher, W. Stammler, K. Heinz and K. Muller, Phys. Rev. B: Condens. Matter, 1991, 43, 6946–6951 CrossRef CAS.
- K. Christmann, Surf. Sci., 2009, 603, 1405–1414 CrossRef CAS.
- P. Ferrin, S. Kandoi, A. U. Nilekar and M. Mavrikakis, Surf. Sci., 2012, 606, 679–689 CrossRef CAS.
- G. Pauer, A. Eichler, M. Sock, M. G. Ramsey, F. Netzer and A. Winkler, J. Chem. Phys., 2003, 119, 5253–5266 CrossRef CAS.
- J. A. Anderson, J. Mellor and R. P. K. Wells, J. Catal., 2009, 261, 208–216 CrossRef CAS.
- P. E. Garcia, A. S. Lynch, A. Monaghan and S. D. Jackson, Catal. Today, 2011, 164, 548–551 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |