Photocatalytic synthesis of highly dispersed Pd nanoparticles on reduced graphene oxide and their application in methanol electro-oxidation†
Haiyan
Li
a,
Guohui
Chang
a,
Yingwei
Zhang
a,
Jingqi
Tian
ab,
Sen
Liu
a,
Yonglan
Luo
a,
Abdullah M.
Asiri
cd,
Abdulrahman O.
Al-Youbi
cd and
Xuping
Sun
*acd
aState Key Lab of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022 Jilin, China. E-mail: sunxp@ciac.jl.cn; Fax: (+86)-431-85262065
bGraduate School of the Chinese Academy of Sciences, Beijing 100039, China
cChemistry Department, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
dCenter of Excellence for Advanced Materials Research, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 22nd March 2012
Abstract
In this communication, we present a green photocatalytic method for the synthesis of highly dispersed Pd nanoparticles (PdNPs) with an average diameter of ca.10 ± 1 nm on the surface of reduced graphene oxide (RGO), using tin(IV) porphyrin (SnP) as a photocatalyst for the reduction of both graphene oxide (GO) and Pd(II). The as-prepared PdNPs–RGO nanocomposites exhibit higher electrocatalytic activities than the commercial Pd/C catalyst for methanol electro-oxidation in alkaline media.
In recent years, the direct methanol fuel cell (DMFC) as a clean-energy conversion device has attracted considerable interest due to its high energy density, low pollutant emission, low operating temperature and ease of handling.1 Among the various electrocatalysts, Pt appears to be the best catalyst and is widely used for the methanol oxidation.2 However, monometallic Pt is susceptible to deactivation or poisoning in the presence of some reaction intermediates during electrocatalytic processes, especially the CO molecules. Moreover, the high cost and limited resource also hindered its practical application.3 To solve these shortcomings, much effort has been directed towards the development of low Pt loading or exploitation of Pt-free materials, and it was found that Pd-based nanomaterial is a better electrocatalyst than Pt due to its high abundance and catalytic activities for methanol oxidation in alkaline media.4
Another way to improve the electrocatalytic activities is to introduce the support materials possessing high specific surface area, excellent electronic conductivity and chemical stability. Graphene as a two-dimensional (2D) nanomaterial made up of a single atomic layer of conjugated sp2 carbon atoms with unique chemical properties is an ideal candidate for support materials.5 Singh and Awasthi utilized graphene nanosheets as catalyst support for Pd nanoparticles (PdNPs) and confirmed that the graphene nanosheet is a much superior support for obtaining highly dispersed and active PdNPs for ethanol/methanol electro-oxidation in alkaline solutions.1a Wen et al. synthesized Pd/SnO2–graphene nanosheet composites by a microwave-assisted reduction process and found that the composites showed superior electrocatalytic activity for ethanol oxidation.6 It is now commonly recognized that the structure–performance relationships of nanomaterial strongly depend on the methods of preparation, size, and dispersion; the smaller size and higher dispersion of PdNPs on thegraphene with large specific surface area are considered as the key factors accounting for the higher catalytic activity.7 Unfortunately, the graphene is generally synthesized through the chemical reduction route, the reduced graphene oxide (RGO) tends to form irreversible agglomerates, leading to a low specific surface area.8 Moreover, the reducing agents used like hydrazine, dimethylhydrazine, and sodium borohydride are either toxic or hazardous,9 may pose environmental problems and health risks, thus hindering their practical applications. Similarly, metal nanoparticles are synthesized through chemical reduction of corresponding metal salts, and some surfactants (usually organic agents) are introduced in order to obtain the small size and well dispersion of nanoparticles.10 Therefore, it is very important to develop a facile, environmentally-friendly method for the synthesis of metal/grephene nanocomposites.
Herein, we present a green photocatalytic method for the synthesis of highly dispersed PdNPs on an RGO surface using SnP as a photocatalyst for the reduction of both graphene oxide (GO) and Pd(II) (Scheme 1). The as-prepared PdNPs–RGO nanocomposites exhibit higher electrocatalytic activities than the commercial Pd/C catalyst for methanol electro-oxidation in alkaline media. Our present method for synthesis of Pd/graphene nanocomposites is significant for the following two reasons: (1) the photocatalytic reduction is simple, environmentally-friendly and time-saving. (2) PdNPs have a small size distribution and well dispersion on an RGO surface without adding any surfactants.
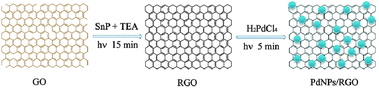 | ||
| Scheme 1 A schematic diagram to illustrate the procedure for preparing PdNPs–RGO nanocomposites by photocatalytic reduction (λ > 400 nm). | ||
Fig. 1a shows the low magnification TEM image of PdNPs–RGO nanocomposites after a 5 min irradiation period, indicating that a large amount of nanoparticles are generated and well dispersed on the surface of RGO. The high magnification TEM image shown in Fig. 1b further reveals that the nanoparticles present near spherical morphology and are well separated from each other, no aggregation is observed on the RGO surface. Their size distribution was evaluated statistically through measuring the diameter of 100 nanoparticles in the selected TEM image. It is noted that the nanoparticle size is distributed mainly between 7.7 nm and 14 nm (with an average diameter of ca.10 ± 1 nm) (Fig. 1d). The HRTEM image in Fig. 1c reveals clear lattice fringes with an interplanar distance of 0.22 nm, corresponding to the (111) planes of the face-centered cubic (fcc) structure of metallic Pd.11 The influence of irradiation time on the reaction course is presented in Fig. S1 (ESI†). It is of importance to note that the size of PdNPs has not changed significantly, but the irradiation time increased, leading to an increased loading amount of PdNPs, indicating that the PdNPs loading amount can be tuned by the irradiation time involved.
 | ||
| Fig. 1 (a) Low, (b) high magnification TEM images and (c) HRTEM image of PdNPs–RGO nanocomposites, (d) the corresponding particle size distribution histograms of the PdNPs. | ||
In our present study, the successful reduction of GO was firstly verified by Raman spectroscopy, as shown in Fig. 2. GO exhibits two characteristic bands: the D band at ∼1355 cm−1, arising from a breathing mode of κ-point photons of A1g symmetry, which is attributed to local defects and disorders; and the G band at 1601 cm−1, arising from the first order scattering of the E2g phonon of sp2 C atoms. After the photocatalytic process, it is obvious that the obtained products show relatively higher intensity of D to G band (0.93) than that of GO (0.83), providing a piece of evidence to support the reduction of GO after the visible-light illumination.12
 | ||
| Fig. 2 Raman spectra of (a) GO and (b) the products obtained after the visible-light illumination. | ||
Fig. 3a and b show the C 1s XPS for GO and PdNPs–RGO nanocomposites, respectively. GO shows four different types of C 1s peaks at 284.5, 285.6, 286.7 and 288.6 eV, corresponding to C–C, C–OH, C–O (epoxy) and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively.13 The peak intensity of C–O in PdNPs–RGO, however, tremendously decreases, and the content of C–C correspondingly increases dramatically at the same time. These results confirm the reduction of GO. In addition, the observed increase in peak intensity of CO is due to the presence of SnP.14 The Pd 3d XPS spectrum in Fig. 3c consists of two asymmetric peaks at around 335.4 and 340.7 eV, corresponding to Pd 3d5/2 and Pd 3d3/2 core levels, respectively, which confirms the formation of metallic Pd.15
O bonds, respectively.13 The peak intensity of C–O in PdNPs–RGO, however, tremendously decreases, and the content of C–C correspondingly increases dramatically at the same time. These results confirm the reduction of GO. In addition, the observed increase in peak intensity of CO is due to the presence of SnP.14 The Pd 3d XPS spectrum in Fig. 3c consists of two asymmetric peaks at around 335.4 and 340.7 eV, corresponding to Pd 3d5/2 and Pd 3d3/2 core levels, respectively, which confirms the formation of metallic Pd.15
 | ||
| Fig. 3 C 1s XPS spectra of (a) GO and (b) PdNPs–RGO, and (c) Pd 3d XPS spectrum of PdNPs–RGO nanocomposites. | ||
Fig. 4a shows the cyclic voltammograms (CVs) of PdNPs–RGO nanocomposites and commercial Pd/C in N2 saturated 0.5 M NaOH solution at a scan rate of 50 mV s−1. The CVs show typical potential regions for hydrogen adsorption/desorption and the formation/reduction of surfaces of Pd oxide, Pd–OHad or Pd(OH)2. The reduction charge of the Pd(OH)2 was employed to estimate electrochemical active area (ECSA) values for the Pd catalysts based on the charge density for the formation of a Pd(OH)2 monolayer (430 μC cm−2).16 The ECSA value of PdNPs–RGO nanocomposites is 13.3 m2 g−1, higher than that of the Pd/C electrode (2.8 m2 g−1), which is most likely due to the much better dispersion of Pd on the RGO surface. This also demonstrates that the PdNPs deposited on the RGO are electrochemically more accessible, which is very important for electrochemical oxidation of methanol. The CVs of methanol oxidation on PdNPs–RGO nanocomposites and commercial Pd/C are displayed in Fig. 4b, which were recorded in 0.5 M NaOH + 1 M CH3OH at a scan rate of 50 mV s−1. Both samples exhibit two peaks of methanol oxidation under anodic conditions: the sharp rising current peak in the forward scan is attributed to the characteristic methanol oxidation on the electrode surface, forming adsorbed carbonaceous intermediates, which can be oxidized at the higher potential and form Pd–OH or Pd–O species; the oxidation peak in the back scan is related to removal of carbonaceous species not completely oxidized in the forward scan.17 Therefore, the peak potential and current density of the forward anodic oxidation peak can be used to evaluate the catalytic activity of the electrocatalyst. Compared with the commercial Pd/C catalyst, the PdNPs–RGO nanocomposites show a significant enhancement of the peak current density and an obvious negative shift of the peak potential for methanol oxidation. It is noted that the forward peak current density for methanol oxidation on the PdNPs–RGO nanocomposites is 1.6 mA cm−2, which is 3.4 times higher than that on the commercial Pd/C catalyst (0.36 mA cm−2). On the other hand, the peak potential for methanol oxidation on the PdNPs–RGO nanocomposites is −0.04 V vs. Ag/AgCl, which is more negative than that on the commercial Pd/C catalyst (0.02 V vs. Ag/AgCl). The above results indicate that PdNPs–RGO catalysts exhibit higher electrocatalytic activities than the commercial Pd/C catalyst.
 | ||
| Fig. 4 Cyclic voltammograms of PdNPs–RGO nanocomposites and commercial Pd/C in N2 saturated (a) 0.5 M NaOH solution and (b) 0.5 M NaOH + 1 M CH3OH solution at a scan rate of 50 mV s−1 at 25 °C, and (c) chronoamperometry of PdNPs–RGO and commercial Pd/C in 0.5 M NaOH + 1 M CH3OH solution at an operation potential of −0.1 V at 25 °C. | ||
The electrochemical stability of the catalysts for methanol electro-oxidation was investigated by chronoamperometric experiments at −0.1 V in 0.5 M NaOH + 1 M CH3OH solution, as shown in Fig. 4c. The rapid current decay can be explained by the poisoning mechanism of intermediate and some poisoning species during the methanol oxidation reaction in alkaline media.18 Obviously, the current decay for the reaction on the PdNPs–RGO nanocomposites is significantly slower than that on the commercial Pd/C catalyst, indicating that the PdNPs–RGO nanocomposites significantly enhance the electrochemical stability for methanol electro-oxidation in alkaline media. The above results suggest that RGO sheet plays a vital role in promoting the methanol oxidation, which could be attributed to the intrinsic electron transfer characteristic of RGO sheet, the small size distribution and well dispersion of PdNPs on the RGO surface.19
In summary, the PdNPs–RGO nanocomposites were successfully synthesized via an environmentally-friendly phtotcatalytic method without adding any surfactants. The PdNPs have a small size distribution and are well dispersed on the RGO surface, which results in much better electrocatalytic activity and stability than commercial Pd/C toward methanol electro-oxidation in alkaline media. Our present study is important because it provides us a general methodology for facile and green preparation of metal nanoparticles–RGO nanocomposites for applications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21175129) and the National Basic Research Program of China (No. 2011CB935800).Notes and references
- (a) R. N. Singh and R. Awasthi, Catal. Sci. Technol., 2011, 1, 778 Search PubMed; (b) S. Wasmus and A. Küver, J. Electroanal. Chem., 1999, 461, 14 CAS; (c) W. C. Choi, M. K. Jeon, Y. J. Kim, S. I. Woo and W. H. Hong, Catal. Today, 2004, 93, 517 Search PubMed.
- (a) A. S. Arico, S. Srinivasan and V. Antonucci, Fuel Cells, 2001, 1, 133 CAS; (b) S. Rousseau, C. Coutanceau, C. Lamy and J. M. Léger, J. Power Sources, 2006, 158, 18 CAS; (c) Z. Wei, L. Li, Y. Luo, C. Yan, C. Sun, G. Yin and P. Shen, J. Phys. Chem. B, 2006, 110, 26055 CAS; (d) Y. Li, W. Gao, L. Ci, C. Wang and P. M. Ajayan, Carbon, 2010, 48, 1124 CAS.
- (a) Z. Zhang, Y. Wang and X. Wang, Nanoscale, 2011, 3, 1663 CAS; (b) H. Ren, M. Humbert, C. Menning, J. Chen, Y. Shu, U. Singh and W. Cheng, Appl. Catal., A, 2010, 375, 303 CAS; (c) F. Kadirgan, S. Beyhan and T. Atilan, Int. J. Hydrogen Energy, 2009, 34, 4312 CAS.
- (a) P. Shen and C. Xu, Electrochem. Commun., 2006, 8, 184 CAS; (b) V. Rao, H. Hariyanto, C. Cremers and U. Stimming, Fuel Cells, 2007, 7, 417 CAS; (c) G. Cui, S. Song, P. Shen, A. Kowal and C. Bianchini, J. Phys. Chem. C, 2009, 113, 15639 CAS.
- (a) E. J. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255 CAS; (b) Y. Li, L. Tang and J. Li, Electrochem. Commun., 2009, 11, 846 CAS; (c) B. Seger and P. V. Kamat, J. Phys. Chem. C, 2009, 113, 7990 CAS.
- Z. Wen, S. Yang, Y. Liang, W. He, H. Tong, L. Hao, X. Zhang and Q. Song, Electrochim. Acta, 2010, 56, 139 CAS.
- B. Wu, D. Hu, Y. Kuang, Y. Yu, X. Zhang and J. Chen, Chem. Commun., 2011, 47, 5253 CAS.
- S. Stankovich, D. Dikin, R. Piner, K. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CAS.
- (a) S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CAS; (b) W. Gao, L. B. Alemany, L. Ci and P. M. Ajayan, Nat. Chem., 2009, 1, 403 CAS.
- Y. Zhu, Y. Kang, Z. Zou, Q. Zhou, J. Zheng, B. Xia and H. Yang, Electrochem. Commun., 2008, 10, 802 CAS.
- S. Yuan, Q. Sheng, J. Zhang, F. Chen, M. Anpo and W. Dai, Catal. Lett., 2006, 107, 19 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558 CAS.
- S. Gilje, S. Han, M. Wang, K. L. Wang and R. B. Kaner, Nano Lett., 2007, 7, 3394 CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CAS.
- Y. Chen, H. Zheng, Z. Guo, C. Zhou, C. Wang, A. Borgna and Y. Yang, J. Catal., 2011, 283, 34 CAS.
- L. Xiao, L. Zhuang, Y. Liu, J. T. Lu and H. D. Abruna, J. Am. Chem. Soc., 2009, 131, 602 CAS.
- C. Xu, R. Wang, M. Chen, Y. Zhang and Y. Ding, Phys. Chem. Chem. Phys., 2010, 12, 239 CAS.
- (a) Y. Qin, H. Yang, X. Zhang, P. Li and C. Ma, Int. J. Hydrogen Energy, 2010, 35, 7667 CAS; (b) R. N. Singh, A. Singh and R. Anindita, Int. J. Hydrogen Energy, 2009, 34, 2052 CAS; (c) R. N. Singh, A. Singh and R. Anindita, Carbon, 2009, 47, 271 CAS; (d) C. Xu, H. Wang, P. Shen and S. Jiang, Adv. Mater., 2007, 19, 4256 CAS.
- (a) J. Shi, G. Yang and J. Zhu, J. Mater. Chem., 2011, 21, 7343 CAS; (b) Y. Zhang, G. Chang, S. Liu, J. Tian, L. Wang, W. Lu, X. Qin and X. Sun, Catal. Sci. Technol., 2011, 1, 1636 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, characterization and supplementary figure. See DOI: 10.1039/c2cy20099f |
| This journal is © The Royal Society of Chemistry 2012 |