A non-hydrolytic sol–gel route to highly active MoO3–SiO2–Al2O3 metathesis catalysts†
Damien P.
Debecker
*a,
Karim
Bouchmella
b,
Mariana
Stoyanova
c,
Uwe
Rodemerck
c,
Eric M.
Gaigneaux
a and
P.
Hubert Mutin
*b
aInstitute of Condensed Matter and Nanoscience–MOlecules, Solids and reactiviTy (IMCN/MOST), Université catholique de Louvain, Croix du Sud 2/17, 1348 Louvain-La-Neuve, Belgium. E-mail: damien.debecker@uclouvain.be; Fax: +3210473649; Tel: +3210473648
bInstitut Charles Gerhardt, UMR 5253, CNRS-UM2-ENSCM-UM1, Université Montpellier 2, cc 1701, Montpellier 34095, France. E-mail: hubert.mutin@univ-monpt2.fr; Fax: +334 6714 3852; Tel: +334 6714 4943
cLeibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 21st December 2011
Abstract
MoO3-based materials are known to be appropriate catalysts for the heterogeneous metathesis of light olefins. Classical preparation methods involve the deposition of a Mo oxide phase on the surface of preformed support via impregnation, grafting or thermal spreading. An alternative sol–gel approach for the elaboration of Mo-based catalysts is presented in this article. Mesoporous ternary Si–Al–Mo mixed oxides are prepared in one step, via non-hydrolytic condensation of chloride precursors in non-aqueous media. After calcination, effective catalysts with very good textures and highly dispersed surface molybdenum species are obtained. The Si/Al ratio influences both the texture and the acidity of the materials, which significantly affects the propene self-metathesis activity. The activity also increases with the MoO3 content. The best catalysts with optimized composition significantly outperform the catalysts prepared by other methods.
1. Introduction
The metathesis of light olefins is a crucial reaction for the petro-chemical industry as it allows the conversion of low-value alkenes (or alkenes produced in excess in the refinery) into more demanded ones.1 Historically, the metathesis reaction was discovered when propene was reacted on supported MoO3 catalysts and the production of ethene, cis-2-butene and trans-2-butene was observed.2 It was then found that supported W and Re oxides can also catalyse the self-metathesis of propene. The reaction is athermal and reversible. Nowadays, the reverse reaction (cross-metathesis of ethene and butene) is of particular interest because propene is highly demanded in the market.3The self-metathesis of propene is a convenient model reaction largely used to evaluate the metathesis activity of heterogeneous catalysts. Recent works on MoO3-based metathesis catalysts have highlighted their high activity, especially if supported on silica–alumina.4–7 MoO3/SiO2–Al2O3 catalysts, as opposed to MoO3/SiO2 or MoO3/Al2O3 catalysts, develop a surface acidity which is beneficial to the reaction.4,5 In addition, the superior activity of highly dispersed Mo species was supported by many experimental data.8–10 Consequently, much effort has been directed toward finding preparation methods which promote the stabilization of highly dispersed MoOx species at the surface of silica–alumina.
Traditional catalyst preparation methods (impregnation, thermal spreading, grafting) usually consist of two-step procedures in which the support—prepared in the first step—is covered by the active phase in the second step. This strategy implies limitations such as the non-uniform deposition of the active phase (e.g. not deposited inside the pores of the support11), the formation of inactive species (e.g. Al2(MoO4)3 and MoO3 crystals11,12) or the lack of interaction between the active phase and the support (e.g. incomplete spreading of MoO3via thermal spreading13,14). Impregnation methods can be improved somewhat by changing the nature of the Mo precursor, with the aim to increase the Mo dispersion, thereby increasing the activity.9,15 Nevertheless, obtaining a molecular-scale dispersion of Mo species at the surface of a pre-existing support remains difficult. Recently, remarkable turn-over frequencies were reached with catalysts prepared via flame spray pyrolysis (FSP) because this method allows the production of highly dispersed species at low MoO3 loading.8 However, as the specific surface area remains relatively low with this method, the MoO3 loading has to be kept very low (1 wt%) to prevent the formation of condensed molybdena phases, thus limiting the actual specific activity. Outstanding cross-metathesis activity was recently obtained with highly porous MoSiAl mixed oxides prepared via an aerosol-assisted sol–gel method,16 showing again that high specific surface area and good Mo dispersion are the decisive factors for the design of efficient metathesis catalysts.
In this context, a new one-step process based on a non-hydrolytic sol–gel route and leading to MoO3–SiO2–Al2O3 mixed oxides was proposed recently.17 Preliminary catalytic assays showed that these materials were active catalysts for the self-metathesis of propene. The present paper presents a systematic approach to optimize the preparation method and reach high metathesis activity. The materials were obtained by the direct reaction of low-cost Mo, Si and Al chloride precursors with diisopropyl ether (see Experimental section). Two parameters of the preparation were screened: the Al2O3 and the MoO3 contents. The physico-chemical properties of the solids produced were studied and correlated with the catalytic performance. Particular attention was devoted to the description of the MoOx species and to the acidity of the solids.
The choice of the non-hydrolytic sol–gel route is justified by the difficulties encountered in the synthesis of mesoporous mixed oxides by conventional sol–gel processes. Such conventional sol–gel routes are based on the hydrolysis and condensation of molecular precursors, usually alkoxides.18–21 However, in the case of SiO2-based mixed oxides, the very different reaction rates of metal and silicon precursors make the simultaneous control of composition, structure and texture of the gels problematic. Thus, complicated experimental procedures are required to prepare homogeneous mesoporous materials, including prehydrolysis of the less reactive precursors or modification with chelating agents of the more reactive ones, and low-temperature extraction with supercritical CO2 (aerogels).22 Conversely, non-hydrolytic routes23 based on the reaction of chloride precursors with alkoxide precursors or diisopropyl ether were shown to provide an excellent control over the stoichiometry and the homogeneity of mixed oxide gels. Furthermore, the generally high degree of condensation of non-hydrolytic gels allows the formation of mesoporous xerogels with high surface area and pore volumes after simple evaporative drying, thus avoiding the supercritical drying step.24 Accordingly, these non-hydrolytic routes are attracting increasing attention for the preparation of mixed oxide catalysts.25–33
2. Results
In this study, two parameters of the catalysts preparation have been varied systematically: the alumina content (in other words, the Si/Al ratio) and the MoO3 content. The samples are labelled NH_Y_X, where NH stands for “non-hydrolytic sol–gel”, Y represents the Al2O3 content (in wt%) and X represents the MoO3 content (in wt%). The effect of these parameters on the texture and acidity of the solids was correlated to their catalytic activity in the metathesis of propene.A first set of ten samples (Table 1, first ten entries) with various Si/Al ratios and a constant MoO3 content was examined with the aim to identify the most appropriate Al2O3 content. The MoO3 content was kept constant at 10 wt%. The range of SiO2/Al2O3 ratio explored varied from infinite (no alumina in NH_0_10) to zero (no silica in NH_90_10). Then, mixed oxides with the most promising Si/Al ratio (Si/Al ≈ 17) and various MoO3 contents ranging from 5 to 20 wt% were examined in a search for higher catalytic performances (Table 1, last four entries).
| Sample | Composition SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al2O3:MoO3 (wt%)a :Al2O3:MoO3 (wt%)a |
SSA/m2 g−1 | V p/cm3 g−1 | D p/nm | SSAμ/m2 g−1 |
|---|---|---|---|---|---|
| a Determined by ICP-OES and considering that all Si, Al and Mo atoms are in the form of SiO2, Al2O3 and MoO3 respectively. | |||||
| NH_0_10 | 90.3:0.0:9.7 |
740 | 1.1 | 5.8 | 70 |
| NH_5_10a | 83.9:5.4:10.7 |
520 | 1.5 | 12.0 | 40 |
| NH_10_10 | 79.9:10.0:10.1 |
330 | 0.6 | 7.8 | 30 |
| NH_15_10 | 74.0:15.4:10.5 |
200 | 0.3 | 6.5 | 30 |
| NH_20_10 | 68.8:19.4:11.8 |
190 | 0.3 | 6.7 | 30 |
| NH_25_10 | 64.5:25.4:10.1 |
210 | 0.3 | 6.2 | 26 |
| NH_45_10 | 44.2:45.5:10.3 |
320 | 0.4 | 5.3 | 59 |
| NH_65_10 | 26.3:62.9:10.8 |
290 | 0.2 | 3.3 | 100 |
| NH_80_10 | 10.6:78.9:10.5 |
260 | 0.1 | 2.2 | 130 |
| NH_90_10 | 0.0:89.5:10.5 |
480 | 0.6 | 5.1 | — |
| NH_5_5 | 90.3:5.2:4.6 |
500 | 0.9 | 7.2 | 40 |
| NH_5_10b | 86.4:4.1:9.5 |
480 | 1.6 | 13.1 | 20 |
| NH_5_15 | 79.9:4.7:15.4 |
470 | 1.6 | 13.3 | 0 |
| NH_5_20 | 77.0:4.2:18.8 |
490 | 1.6 | 13.3 | 10 |
2.1. Composition and texture of MoO3–SiO2–Al2O3 catalysts prepared by non-hydrolytic sol–gel
ICP-OES results showed that the final composition of the Mo–Si–Al mixed oxides prepared by non-hydrolytic sol–gel was always very close to the nominal one (Table 1). This means that no metallic species were lost in the liquid phase or in the gas phase during the drying and calcination steps. The reliability of the synthesis in terms of sample composition was also demonstrated for other catalytic systems.32,33Nitrogen physisorption indicated that the SiO2–MoO3 (NH_0_10) and Al2O3–MoO3 (NH_90_10) samples were mesoporous with type IV isotherms (according to the BDDT classification) typical of mesoporous solids (see Fig. S1, ESI†) and specific surface areas (SSA) of 740 m2 g−1 (NH_0_10) and 480 m2 g−1 (NH_90_10) (Table 1). Conversely, the SiO2–Al2O3–MoO3 catalysts exhibited Type II isotherms, except NH_80_10 which was mainly microporous with a type I isotherm (Fig. 1). The NH_5_X samples with a 5 wt% alumina content and various MoO3 loadings all showed similar specific surface areas around 500 m2 g−1 and high pore volumes (Vp), from 0.9 to 1.6 cm3 g−1. Thus, the MoO3 content had no significant impact on the texture (see Fig. S2, ESI†). The samples with higher alumina contents had significantly lower surface areas (from 190 to 330 m2 g−1) and pore volumes (from 0.1 to 0.6 cm3 g−1). t-Plot analysis indicated that the surface area developed by micropore walls (denoted SSAμ in Table 1) was low, except for the catalysts with a high alumina content (NH_65_10 and NH_80_10). Average pore diameter (Dp) ranged between 5 and 14 nm. TEM micrographs showed highly porous particles, with polydispersed and apparently interconnected pores (Fig. 2).
 | ||
Fig. 1 N2 adsorption–desorption isotherms for SiO2–Al2O3–MoO3 samples. ( ) NH_5_10 adsorption and desorption respectively, ( ) NH_5_10 adsorption and desorption respectively, ( ) NH_10_10, ( ) NH_10_10, ( ) NH_15_10, ( ) NH_15_10, ( ) NH_65_10 and ( ) NH_65_10 and ( ) NH_80_10. The isotherms of NH_20_10, NH_25_10 and NH_45_10 catalysts are very similar to those of NH_15_10 and are not shown for the sake of clarity of the figure (see Fig. S3, ESI†). ) NH_80_10. The isotherms of NH_20_10, NH_25_10 and NH_45_10 catalysts are very similar to those of NH_15_10 and are not shown for the sake of clarity of the figure (see Fig. S3, ESI†). | ||
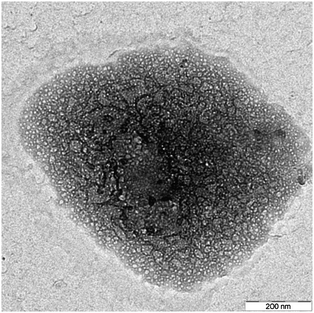 | ||
| Fig. 2 Transmission electron micrograph of the NH_5_10 sample (scale bar = 200 nm). | ||
2.2. Structural characterization and surface acidity
Raman spectroscopy was tentatively used to characterize the MoOx species present in the solids. However, the background of the spectra, originating from complex vibration modes of the silica–alumina matrix and from a broad and intense signal presumably due to the fluorescence of impurities made the observation of MoOx species complicated. Fig. 3 shows that for the silica-rich samples the intensity of the background increased with the alumina content, coherent with the increasing amount of defects in the structure of the silica–alumina matrix as the Al content increased. Thus, in Al-containing samples, dispersed and amorphous molybdenum species with weak Raman response35 were not observable. Such fluorescence problems could possibly be minimized using another excitation source or analysing the sample under dehydrated conditions.8,36 In the conditions used here, however, it can already be stressed that no bands related to crystalline MoO3 species were detected in any spectra (signals expected at 996, 819 and 666 cm−137), even though such crystalline species are known to be strong Raman scatterers compared to amorphous and dispersed species. In similar samples with strong fluorescence background and analysed under similar conditions, small amounts of crystalline Mo oxide (even small crystallites) were easily detected (e.g. in MoO3/SiO2–Al2O3 catalysts prepared via wet impregnation11 or thermal spreading13). So from Raman spectroscopy the presence of MoO3 crystallites can be discarded for catalysts prepared by non-hydrolytic sol–gel. The NH_0_10 catalyst is the only sample for which the background was not too intense and for which two broad bands could be distinguished. Their maxima appear around 945–950 cm−1 and 871–878 cm−1. The most intense peak is classically attributed to the stretch vibration of terminal Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups in amorphous polymeric MoOx species,37,38 while the other band is attributed to the stretch vibration of Mo–O–Mo bonds in polymers of octahedral MoO6 units39 or to isolated tetrahedrally coordinated Mo atoms.38
O groups in amorphous polymeric MoOx species,37,38 while the other band is attributed to the stretch vibration of Mo–O–Mo bonds in polymers of octahedral MoO6 units39 or to isolated tetrahedrally coordinated Mo atoms.38
 | ||
| Fig. 3 Raman spectra of (a) NH_0_10, (b) NH_5_10, (c) NH_10_10, (d) NH_15_10 and (e) NH_20_10. The scale of the left axis changes from one spectrum to another and shows that the intensity of the background increases when the Al content increased. | ||
| Sample | NH3 desorbed/mmol g−1 | Density of acid site/nm−2 | Density of weak sitesa/nm−2 | Density of medium + strong sitesa/nm−2 |
|---|---|---|---|---|
| a In brackets is given the percentage of each type of sites. | ||||
| NH_0_10 | 0.5 | 0.4 | 0.4 (100%) | 0 (0%) |
| NH_5_10a | 0.5 | 0.6 | 0.5 (82%) | 0.1 (18%) |
| NH_5_20 | 0.8 | 1.0 | 0.8 (79%) | 0.1 (28%) |
| NH_10_10 | 0.9 | 1.6 | 1.0 (58%) | 0.7 (42%) |
| NH_15_10 | 0.9 | 2.7 | 1.4 (51%) | 1.3 (49%) |
| NH_20_10 | 0.8 | 2.5 | 1.5 (61%) | 1.0 (39%) |
| NH_25_10 | 0.8 | 2.3 | 1.1 (49%) | 1.2 (51%) |
| NH_45_10 | 1 | 1.9 | 1.5 (79%) | 0.4 (21%) |
| NH_65_10 | 1.2 | 2.5 | 1.4 (55%) | 1.1 (45%) |
| NH_80_10 | 1 | 2.3 | 1.3 (56%) | 1.0 (44%) |
| NH_90_10 | 0.6 | 0.8 | 0.7 (90%) | 0.1 (10%) |
2.3. Metathesis activity
The samples were tested in the self-metathesis of propene at 40 °C (Fig. 4). All samples catalyzed the conversion of propene to ethene and butene with ∼99% selectivity. The catalyst activity appeared to be highly dependent on the alumina and on the MoO3 contents. For some samples, the activity increased during the first 20 minutes of the reaction and then slow deactivation took place. For some other samples, no activation period was observed and deactivation started from the beginning of the reaction. | ||
Fig. 4 Specific metathesis activity versus time on stream. The effect of the alumina content can be visualized within the series: ( ) NH_0_10, ( ) NH_0_10, ( ) NH_5_10, (×) NH_10_10, ( ) NH_5_10, (×) NH_10_10, ( ) NH_25_10, ( ) NH_25_10, ( ) NH_90_10. For the sake of clarity, the activity of NH_65_10 and NH_80_10 (lower than that of NH_0_10) and the activity of NH_15_10 and NH_20_10 (curves similar to NH_10_10) are not shown. The effect of the MoO3 content can be visualized within the series: ( ) NH_90_10. For the sake of clarity, the activity of NH_65_10 and NH_80_10 (lower than that of NH_0_10) and the activity of NH_15_10 and NH_20_10 (curves similar to NH_10_10) are not shown. The effect of the MoO3 content can be visualized within the series: ( ) NH_5_5, ( ) NH_5_5, ( ) NH_5_10, ( ) NH_5_10, ( ) NH_5_15 and ( ) NH_5_15 and ( ) NH_5_20. Reaction carried out with 200 mg of catalyst (100 mg only for NH_5_15 and NH_5_20 to remain far enough from thermodynamic equilibrium) at 40 °C, in a propene and N2 flow (90 : 10 vol.%) of 8 ml min−1, after activation at 550 °C under N2. ) NH_5_20. Reaction carried out with 200 mg of catalyst (100 mg only for NH_5_15 and NH_5_20 to remain far enough from thermodynamic equilibrium) at 40 °C, in a propene and N2 flow (90 : 10 vol.%) of 8 ml min−1, after activation at 550 °C under N2. | ||
The activity of the samples with a constant 10 wt% MoO3 content strongly depended on the alumina content. The ternary catalysts with high alumina content (NH_65_10 and NH_80_10) as well as the binary samples (NH_90_10 and NH_0_10) showed low activities and relatively fast deactivation, in agreement with previous observations.4 The ternary catalysts with intermediate alumina content (from 5 to 45 wt%) were more active, the best catalyst being the one with only 5 wt% of Al2O3. The activity of the NH_5_X samples increased with the MoO3 charge.
The specific metathesis activity (taken at 14 min on stream) of the different NH catalysts is compared in Fig. 5. In terms of activity per gram of catalyst, the best catalysts in the NH_X_10 series were those with alumina content ranging from 5 to 45 wt%. The specific activity of the samples with no alumina or high alumina contents was much lower. The specific activity of the NH_5_X samples increased roughly linearly with the MoO3 content, reaching the very high specific activity of ∼45 mmol g−1 h−1 for NH_5_20.
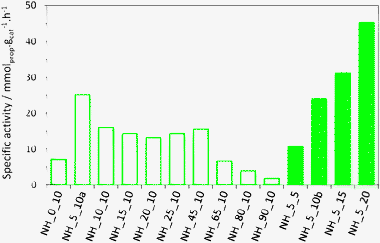 | ||
| Fig. 5 Specific metathesis activity after 14 min on stream. Empty bar shows the influence of the alumina content. Full bar shows that the metathesis activity increases linearly with the MoO3 content. NH_5_10a and NH_5_10b exhibited very similar activity (24 and 25 mmol g−1 h−1 respectively), showing that the preparation method is reproducible. | ||
3. Discussion
3.1. Composition, texture, structure and acidity of the catalysts
Non-hydrolytic sol–gel provides a simple one-step route to mesoporous MoO3–SiO2–Al2O3 materials with relatively high surface areas and pore volumes. The specific surface area is maximum for the silica–molybdena catalyst, and then it decreases markedly upon introduction of 5 and 10 wt% alumina, and remains roughly constant for higher alumina contents. This effect seems to be intrinsic to silica–alumina based systems, regardless of the preparation method.42–44 Thus, the samples with a low alumina content (ca. 5 wt%) presented the best textures, with specific surface area around 500 m2 g−1 and very high pore volumes (0.9–1.6 cm3 g−1). Conversely, the alumina-rich samples offered less interesting textures, with lower pore volume and a significant proportion of micropores.According to Raman spectroscopy and XRD, all samples appeared amorphous. This is encouraging, since MoO3 crystals were identified as inactive in the olefin metathesis.4,11,45 The absence of MoO3 crystals in silica-rich samples and particularly NH_0_10 (no alumina) is noteworthy, as it is well recognized that bulky molybdenum oxide crystals usually tend to form during calcination in MoO3/SiO2 catalysts prepared via classical methods.38 The preferential formation of Mo–O–Mo bridges instead of Mo–O–Si bonds has been ascribed to the fact that both silica and molybdena are acidic, resulting in weak interactions between both materials,46 so that calcination usually leads to sintering and formation of crystalline species. This process seems avoided here.
The catalytic activity of supported molybdenum oxide metathesis catalysts is known to be strongly influenced by the nature of the support4–6,47 and the high activity of catalysts supported on silica–alumina has been ascribed to the acidic character of the support. In the present study, the composition of the samples has a marked influence on the acidity of the samples. The Al-free catalyst is the least acidic in terms of both the absolute amount and the density of acidic sites. Furthermore, it only exhibits weak acidity. As Al is introduced in silica-rich formulations, the density of both weak and medium/strong acidic sites increases and reaches a maximum for alumina contents of 15–25 wt%.
3.2. Correlation with metathesis activity
To take into account the differences in specific surface area or MoO3 content of the catalysts, we plotted in Fig. 6 the specific activity normalized to the specific surface area (“areal activity” in mmol of propene converted per square metre of catalyst and per hour) and the apparent turnover frequency (“TOF” in mmol of propene converted per mole Mo and per second). On the basis of areal activity, the most active NH catalysts are the catalysts with 10 to 20 wt% Al2O3. The catalysts containing no alumina exhibit poor areal activity. Such a trend was previously reported for catalysts prepared by wet impregnation.4 The samples with 65 to 90 wt% Al2O3 also showed poor areal activity, even though no significant difference was observed in terms of acidity with respect to other Al-containing samples. The very high specific activity (Fig. 4) of the NH_5_10 catalyst compared to the other NH_Y_10 catalysts with intermediate Al2O3 contents (10 to 45 wt%) can clearly be explained by a textural effect. The very low activity of NH_0_10 can be ascribed to the absence of medium/strong acidic sites in the silica matrix. On the other hand, the correlation between acidity, texture and activity in alumina containing catalysts is poor. In particular, the relatively low activity of the samples with high alumina content cannot be directly correlated to differences in texture or surface acidity. Possibly, the nature of the Mo surface sites is affected by the high alumina content, as it is well known that Mo can interact strongly with alumina (to form aluminium molybdates, for instance) while the interaction with silica is poor. Moreover, the nature of the acid sites is probably affected. To better understand the influence of the Al content and the role of the acidity of the catalysts in the metathesis reaction it would be necessary to differentiate Brönsted and Lewis acidic sites, for instance using adsorption of pyridine and FTIR. | ||
| Fig. 6 Metathesis activity after 14 min on stream expressed as turnover frequency, “TOF” (left axis, full bars: mmol of propene converted per mol of Mo per second) or “areal activity” (right axis, empty bars: mmol of propene converted per m2 of catalyst per hour). | ||
The different NH_5_X catalysts (which have similar specific surface areas) display similar TOF values, around 10 mmol g−1 s−1. This confirms the good dispersion of Mo species even at high MoO3 content, in agreement with XRD and Raman spectroscopy. It suggests that increasing the loading leads to a proportional increase in the active sites density. This was not the case for catalysts prepared by classical impregnation with ammonium heptamolybdate, where the activity did not improve for loadings above ca. 8 wt%, due to the formation of crystalline molybdenum oxide and Al2(MoO4)3.11
One recurrent problem when comparing data from the literature is the disparity in the experimental conditions (activation and reaction) used by different authors to evaluate the activity of different catalysts. Also, activity data can be expressed in many different ways. Here the catalysts can easily be compared with other systems already described by us4,8,11,13–15 and tested under the exact same experimental conditions (Table 3). The highest performance reported by Handzlik et al. (who used very similar conditions) is shown as well. The specific activity of NH_5_20 is more than 2 times higher than that of MoO3/SiO2–Al2O3 catalysts prepared by optimized impregnation methods15 or flame spray pyrolysis (FSP).8
| Catalyst | Mo source | MoO3 content/wt% | Specific activity/mmol gcat−1 h−1 | TOF/mmolpropene molMo−1 s−1 | Ref. |
|---|---|---|---|---|---|
| a With oxalic acid as an additive. b Mo2-EH = Mo 2-ethylhexanoate. c All catalysts were tested in the same set-up, with exactly the same experimental conditions except “TS-acac”, tested by Handzlik et al. under similar conditions but not in the same set-up. d Activity data published in ref. 17 were underestimated due to diffusional limitations. | |||||
| NH_5_10 | MoCl5 | 9.5 | 25 | 10.5 | This workd |
| NH_5_20 | MoCl5 | 18.8 | 45 | 9.6 | This workd |
| WI-AHM | AHM | 8.0 | 12 | 6.0 | 11 |
| WI-OXA | AHMa | 19.2 | 19 | 3.9 | 15 |
| WI-MoH | Mo hydrate | 12.9 | 21 | 6.5 | 15 |
| FSP 1 | Mo2-EHb | 1.0 | 18 | 71.3 | 8 |
| FSP 3 | Mo2-EHb | 3.0 | 9 | 11.4 | 8 |
| FSP 5 | Mo2-EHb | 5.0 | 2 | 1.9 | 8 |
| TS-acacc | MoO2(acac)2 | 2.0 | 10 | 20.0 | 5 |
| TS-MoO3 | MoO3 | 11.5 | 12 | 4.2 | 13 |
The TOF values found for the best NH catalysts are around 10 mmolpropene mol−1Mo s−1, significantly higher than the values reported for MoO3/SiO2–Al2O3 catalysts prepared by impregnation methods. Previously reported XPS results17 showed that migration of Mo species toward the surface took place during calcination of NH catalysts; however, the proportion of Mo atoms at the surface of NH catalysts cannot be higher than in the catalysts prepared by impregnation. Thus, the higher TOF values observed indicate that the MoOx surface species formed in the non-hydrolytic samples are more active. Indeed, time-of-flight secondary ion mass spectroscopy suggested that highly dispersed Mo (isolated) surface sites were present at the surface of NH catalysts,17 whereas the catalysts prepared by impregnation contained a significant amount of oligomeric Mo species.
An extremely high TOF value (71 mmolpropene mol−1Mo s−1) was recently reported for a MoO3/SiO2–Al2O3 catalyst prepared by flame spray pyrolysis with a very low MoO3 loading (1 wt%).8 In this case, a clear correlation between the proportion of isolated molybdates and the metathesis activity was established and the activity decreased drastically with the MoO3 loading: for a 2 wt% MoO3 loading the TOF value (11.4 mmolpropene mol−1Mo s−1) becomes comparable to the TOF values found for NH catalysts (Table 3). These results support the idea that highly dispersed, possibly isolated, MoOx species are the most active metathesis centres, as earlier suggested by Ono et al.48 However, one must keep in mind that the active sites in supported metathesis catalysts represent a very small proportion of the present transition metal atoms.49
The outstanding specific activity that can be reached for the optimized NH catalysts would thus result from a combination between their excellent texture and the good dispersion of Mo species even at high Mo contents. Note that high metathesis activity levels were also very recently obtained in the reverse reaction (production of propene from ethene and butene) with aerosol-made catalysts.16 These systems might compete with NHSG-made catalysts, but proper comparison (in the same reaction and same conditions) is still lacking.
4. Conclusions
An innovative non-hydrolytic sol–gel route has been investigated for the preparation of MoO3–SiO2–Al2O3 mixed oxides dedicated to the catalysis of light olefin metathesis. This method, based on the etherolysis-condensation of chloride precursors, allows preparing in one step mesoporous mixed oxide samples with well-controlled compositions. Such a sol–gel route differs markedly from classical impregnation methods: instead of depositing a catalytically active phase on a preformed support, the active phase is directly incorporated with the other components of the catalyst. After calcination, well-dispersed MoOx species are stabilized at the surface of the mixed oxides. The influence of the composition was systematically investigated. The Al2O3 content (or Si/Al ratio) strongly influences the texture, the surface acidity and the catalytic performance. Increasing the MoO3 content and keeping the Si/Al ratio to ∼17 allow maintaining the most favourable textural properties and increase the activity of the catalysts. On the basis of specific activity (per mass of catalyst), the best catalysts prepared via the non-hydrolytic route are more than two times more active in the self-metathesis of propene than the best MoO3–SiO2–Al2O3 catalysts prepared via other methods (impregnation, flame spray pyrolysis, thermal spreading) reported in the literature.Experimental section
Catalyst preparation
The non-hydrolytic sol–gel syntheses23 were performed under an argon atmosphere using a glove box. SiCl4 (Alfa Aesar 99.9%), AlCl3 (Alfa Aesar 99.9%), and MoCl5 (Alfa Aesar 99.6%) were used as received. Diisopropyl ether (iPr2O) was purchased (Aldrich, 99%) and was further dried by distillation over sodium wire. Table S1 (ESI†) gives the precise amount of reactants involved in each preparation. The chloride precursors were introduced first in an 80 ml Pyrex glass tube or an autoclave, and then the stoichiometric amount of iPr2O was added. More precisely, the number of moles of iPr2O is calculated so that number of moles of iPr groups is equal to the total number of Cl groups in the precursors. In the end, the solvent (CH2Cl2) was introduced. The amounts of reactants involved in the preparation of each catalyst are reported in Table S1 (ESI†). The solution obtained was heated at 110 °C for 4 days under autogeneous pressure (ca. 0.7 MPa). After cooling to room temperature, the gel was washed with CH2Cl2 and dried at 20 °C under vacuum (0.00001 MPa) for 1 h and then for 4 h at 120 °C. The xerogel was then crushed in a mortar and calcined for 5 hours at 500 °C in flowing air (10 ml min−1) leading to white to brown powders. These samples are labelled NH_Y_X, where NH stands for “non-hydrolytic sol–gel”, where Y represents the Al2O3 weight content (in %) and where X represents the MoO3 weight content (in %). Actually, in the NH_5_X samples (when the MoO3 content is varied), the nominal Al2O3 content is approximately 5 wt%, because it was chosen to keep the SiO2/Al2O3 ratio constant (∼17, as in NH_5_10). The precise nominal Al2O3 content is 5.3, 5.0, 4.7 and 4.4 wt% for NH_5_5, NH_5_10, NH_5_15 and NH_5_20 respectively. The preparation of NH_5_10 was done twice and independently to check the reproducibility of the preparation route (samples noted NH_5_10a and NH_5_10b).Catalyst characterization
The weight percentages of Mo, Si and Al were measured by Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) on an Iris Advantage apparatus from Jarrell Ash Corporation. The materials were dried at 105 °C prior to measurements. N2 physisorption experiments were performed at −196 °C on a Micromeritics Tristar. The samples were outgassed overnight at 150 °C under vacuum (2 Pa). The specific surface area was determined from the BET method in the 0.05–0.30 P/P0 range. The pore size distribution was derived from the desorption branch using the BJH method. The average pore diameter is calculated as (4 × pore volume/BET specific surface area). The micropore volume was estimated using t-plot analysis in the 0.35–0.05 nm range. X-Ray diffraction (XRD) measurements were performed on the fresh catalysts with a Siemens D5000 diffractometer using the Kα radiation of Cu (λ = 1.5418 Å). The 2θ range was recorded between 5° and 75° at a rate of 0.02° s−1. The ICDD-JCPDS database was used to identify the crystalline phases. Raman characterization was done on the InVia Raman microscope (Renishaw) equipped with a diode light (785 nm). Resolution was set to 4 cm−1. Acquisition time was 10 s and 10 scans were recorded and averaged for each sample. The acidity was evaluated by NH3-temperature programmed desorption (NH3-TPD) on a Micromeritics AutoChem 2910 apparatus with a thermal conductivity detector (TCD). A 100 mg sample was preheated in helium at 500 °C for 60 min (ramp 10 °C min−1). Adsorption of NH3 (5 vol% in helium; flow rate 30 ml min−1) was done at 100 °C for 45 min. Physisorbed NH3 was removed by purging with helium at 100 °C for 1 hour (flow rate 30 ml min−1). The TPD measurement was conducted by heating the sample from 100 to 500 °C at a 10 °C min−1 rate.Metathesis reaction
The activity of the catalysts in the self-metathesis of propene was measured in a multi-channel apparatus which allows fully automated control of gas flows and of three temperature zones (gas pre-heating, reactor, and post reactor lines with 16-port valve) along with reactor switching and product sampling.50 All catalysts were sieved and selected in the 200–315 μm granulometric size range. The catalysts (200 mg or 100 mg for the most active samples, NH_5_10, NH_5_15 and NH_5_20) were introduced in quartz straight reactors (5 mm i.d.). Prior to reaction, the catalysts were activated by heating up to 550 °C (temperature ramp of 5 °C min−1) in N2 (14 ml min−1 flow in each reactor) and keeping this temperature for 2 h. Afterwards the system was cooled down to the reaction temperature (40 °C) under the same N2 flow. The reaction was carried out at 40 °C in a propene and N2 flow (90:10 vol.%) of 8 ml min−1. Propene (99.95% purity) and N2 (99.995% purity) were further purified over Molsieve 3A (Roth) filters. N2 was also purified by an oxygen filter (Oxysorb-glass, Linde). The composition of the reaction gas was analysed by an Agilent 6890 GC. The separation of hydrocarbons was performed on a HP-AL/M column (30 m length, 0.53 mm i.d., 0.15 μm film thickness) by applying a temperature ramp between 90 and 140 °C and FID detection. Product analysis took about 6.5 min for each injection. N2, used as an internal standard, was analysed on a HP Plot-Q column with TCD detection. The experiments were carried out at atmospheric pressure. Under these experimental conditions, the selectivity to metathesis products always proved to be close to 100% (typically 99%). Only traces of secondary metathesis products (1-butene, pentenes, and hexenes) and isomerization products (isobutene) were detected. The activity was calculated on the basis of metathesis products (ethene and trans- and cis-butene) formation. The specific activity was defined as the number of moles of propene converted to metathesis products per gram of catalyst and per hour. The apparent turnover frequency (TOF) is defined as the number of moles of propene converted per mole of Mo (considering the total bulk Mo content) and per second. The areal activity was defined as the number of moles of propene converted to metathesis products per square metre of catalyst and per hour. The standard deviation for activity measurements was less than 3%.
Acknowledgements
The authors from ICG acknowledge the Ministère de l'Enseignement Supérieur et de la Recherche and the Centre National de la Recherche Scientifique (CNRS) in France for financial support. D.P.D. thanks the FNRS for his Research Fellow and Postdoctoral Researcher positions. The authors from UCL are involved in the “Inanomat” IUAP network (Service public fédéral de programmation politique scientifique, Belgium), in the Cost Action D41 (European Science Foundation) and in the European Multifunctional Material Institute (EMMI) built on the basis of the former “FAME” Network of Excellence, EU 6th FP.Notes and references
- J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- R. L. Banks and G. C. Bailey, Ind. Eng. Chem. Prod. Res. Dev., 1964, 3, 170–173 CrossRef CAS.
- H. Liu, L. Zhang, X. Li, S. Huang, S. Liu, W. Xin, S. Xie and L. Xu, J. Nat. Gas Chem., 2009, 18, 331–336 CrossRef CAS.
- D. P. Debecker, D. Hauwaert, M. Stoyanova, A. Barkschat, U. Rodemerck and E. M. Gaigneaux, Appl. Catal., A, 2011, 391, 78–85 CrossRef CAS.
- J. Handzlik, J. Ogonowski, J. Stoch, M. Mikolajczyk and P. Michorczyk, Appl. Catal., A, 2006, 312, 213–219 CrossRef CAS.
- J. Handzlik, J. Ogonowski, J. Stoch and M. Mikolajczyk, Catal. Lett., 2005, 101, 65–69 CrossRef CAS.
- H. Aritani, O. Fukuda, T. Yamamoto, T. Tanaka and S. Imamura, Chem. Lett., 2000, 66–67 CrossRef CAS.
- D. P. Debecker, B. Schimmoeller, M. Stoyanova, C. Poleunis, P. Bertrand, U. Rodemerck and E. M. Gaigneaux, J. Catal., 2011, 277, 154–163 CrossRef CAS.
- J. Handzlik, J. Ogonowski, J. Stoch and M. Mikolajczyk, Appl. Catal., A, 2004, 273, 99–104 CrossRef CAS.
- X. Li, W. Zhang, S. Liu, L. Xu, X. Han and X. Bao, J. Phys. Chem. C, 2008, 112, 5955–5960 CAS.
- D. P. Debecker, M. Stoyanova, U. Rodemerck, A. Leonard, B.-L. Su and E. M. Gaigneaux, Catal. Today, 2011, 169, 60–68 CrossRef CAS.
- X. Li, W. Zhang, S. Liu, X. Han, L. Xu and X. Bao, J. Mol. Catal. A: Chem., 2006, 250, 94–99 CrossRef CAS.
- D. P. Debecker, M. Stoyanova, U. Rodemerck, P. Eloy, A. Léonard, B.-L. Su and E. M. Gaigneaux, J. Phys. Chem. C, 2010, 114, 18664–18673 CAS.
- D. P. Debecker, M. Stoyanova, U. Rodemerck and E. M. Gaigneaux, Stud. Surf. Sci. Catal., 2010, 175, 581–585 CrossRef CAS.
- D. P. Debecker, M. Stoyanova, U. Rodemerck and E. M. Gaigneaux, J. Mol. Catal. A: Chem., 2011, 340, 65–76 CrossRef CAS.
- D. P. Debecker, M. Stoyanova, F. Colbeau-Justin, U. Rodemerck, C. Boissière, E. M. Gaigneaux and C. Sanchez, Angew. Chem., Int. Ed., 2012 DOI:10.1002/anie.201106277.
- D. P. Debecker, K. Bouchmella, C. Poleunis, P. Eloy, P. Bertrand, E. M. Gaigneaux and P. H. Mutin, Chem. Mater., 2009, 21, 2817–2824 CrossRef CAS.
- A. Baiker, J.-D. Grunwaldt, C. A. Mueller and L. Schmid, Chimia, 1998, 52, 517–524 CAS.
- R. D. Gonzalez, T. Lopez and R. Gomez, Catal. Today, 1997, 35, 293–317 CrossRef CAS.
- J. Livage, Catal. Today, 1998, 41, 3–19 CrossRef CAS.
- C. Sanchez, L. Rozes, F. Ribot, C. Laberty-Robert, D. Grosso, C. Sassoye, C. Boissiere and L. Nicole, C. R. Chim., 2010, 13, 3–39 CrossRef CAS.
- D. C. M. Dutoit, M. Schneider and A. Baiker, J. Catal., 1995, 153, 165–176 CrossRef CAS.
- A. Vioux, Chem. Mater., 1997, 9, 2292–2299 CrossRef.
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582–596 CrossRef CAS.
- A. M. Cojocariu, P. H. Mutin, E. Dumitriu, F. o. Fajula, A. Vioux and V. Hulea, Chem. Commun., 2008, 5357–5359 RSC.
- O. Lorret, V. Lafond, P. H. Mutin and A. Vioux, Chem. Mater., 2006, 18, 4707–4709 CrossRef CAS.
- P. Moggi, M. Devillers, P. Ruiz, G. Predieri, D. Cauzzi, S. Morselli and O. Ligabue, Catal. Today, 2003, 81, 77–85 CrossRef CAS.
- P. H. Mutin, A. F. Popa, A. Vioux, G. r. Delahay and B. Coq, Appl. Catal., B, 2006, 69, 49–57 CrossRef CAS.
- A. F. Popa, P. H. Mutin, A. Vioux, G. Delahay and B. Coq, Chem. Commun., 2004, 2214–2215 RSC.
- A. G. Fisch, N. S. M. Cardozo, A. R. Secchi, F. C. Stedile, P. R. Livotto, D. S. de Sa, Z. N. da Rocha and J. H. Z. dos Santos, Appl. Catal., A, 2009, 354, 88–101 CrossRef CAS.
- J. Zhu, J. Yang, Z. F. Bian, H. Ren, Y. M. Liu, Y. Cao, H. X. Li, H. Y. He and K. N. Fan, Appl. Catal., B, 2007, 76, 82–91 CrossRef CAS.
- D. P. Debecker, R. Delaigle, K. Bouchmella, P. Eloy, E. M. Gaigneaux and P. H. Mutin, Catal. Today, 2010, 157, 125–130 CrossRef CAS.
- D. P. Debecker, K. Bouchmella, R. Delaigle, P. Eloy, C. Poleunis, P. Bertrand, E. M. Gaigneaux and P. H. Mutin, Appl. Catal., B, 2010, 94, 38–45 CrossRef CAS.
- A. N. Desikan, L. Huang and S. T. Oyama, J. Phys. Chem., 1991, 95, 10050–10056 CrossRef CAS.
- X. Carrier, E. Marceau and M. Che, Pure Appl. Chem., 2006, 78, 1039–1055 CrossRef CAS.
- H. C. Hu, I. E. Wachs and S. R. Bare, J. Phys. Chem., 1995, 99, 10897–10910 CrossRef CAS.
- G. Mestl and T. K. K. Srinivasan, Catal. Rev. Sci. Eng., 1998, 40, 451–570 CAS.
- O. Collart, P. Van der Voort, E. F. Vansant, E. Gustin, A. Bouwen, D. Schoemaker, R. R. Rao, B. M. Weckhuysen and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 1999, 1, 4099–4104 RSC.
- H. Jeziorowski, H. Knozinger, P. Grange and P. Gajardo, J. Phys. Chem., 1980, 84, 1825–1829 CrossRef CAS.
- N. Katada, H. Igi, J. H. Kim and M. Niwa, J. Phys. Chem. B, 1997, 101, 5969–5977 CrossRef CAS.
- L. Rodriguez-Gonzalez, F. Hermes, M. Bertmer, E. Rodriguez-Castellon, A. Jimenez-Lopez and U. Simon, Appl. Catal., A, 2007, 328, 174–182 CrossRef CAS.
- R. E. Sempels and P. G. Rouxhet, J. Colloid Interface Sci., 1976, 55, 263–273 CrossRef CAS.
- P. O. Scokart, F. D. Declerck, R. E. Sempels and P. G. Rouxhet, J. Chem. Soc., Faraday Trans. 1, 1977, 73, 359–371 RSC.
- S. Rajagopal, J. A. Marzari and R. Miranda, J. Catal., 1995, 151, 192–203 CrossRef CAS.
- B. Zhang, N. Liu, Q. Lin and D. Jin, J. Mol. Catal., 1991, 65, 15–28 CrossRef CAS.
- A. N. Desikan, W. M. Zhang and S. T. Oyama, J. Catal., 1995, 157, 740–748 CrossRef CAS.
- H. Aritani, O. Fukuda, A. Miyaji and S. Hasegawa, Appl. Surf. Sci., 2001, 180, 261–269 CrossRef CAS.
- T. Ono, M. Anpo and Y. Kubokawa, J. Phys. Chem., 1986, 90, 4780–4784 CrossRef CAS.
- J. Handzlik and J. Ogonowski, Catal. Lett., 2003, 88, 119–122 CrossRef CAS.
- U. Rodemerck, P. Ignaszewski, M. Lucas and P. Claus, Chem. Eng. Technol., 2000, 23, 413–416 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00475e |
| This journal is © The Royal Society of Chemistry 2012 |