Synthesis of 5-substituted 1H-tetrazoles using a nano ZnO/Co3O4 catalyst†
Sandeep M.
Agawane
and
Jayashree M.
Nagarkar
*
Department of Chemistry, Institute of Chemical Technology, Nathalal Parikh Marg, Matunga, Mumbai 400 019, Maharashtra, India. E-mail: smagawane@gmail.com; jm.nagarkar@ictmumbai.edu.in; jayashreenagarkar@yahoo.com; Fax: +91-2233611020; Tel: +91-223361111/2222
First published on 25th April 2012
Abstract
Zinc salts have catalytically active sites suitable for synthesis of substituted 1H-tetrazoles. Herein we report the synthesis of 5-substituted 1H-tetrazoles catalyzed by nano ZnO/Co3O4. This is a novel heterogeneous catalyst which showed excellent efficiency, affording good to excellent yield of products.
Tetrazoles are a class of nitrogen containing heterocyclic compounds which are studied extensively due to their wide range of applications.1 The importance of tetrazole and its derivatives has increased because of their use as an isosteric replacement for carboxylic acids in drug design.2 The metabolically stable tetrazole group has a pKa similar to that of –CO2H. When it is a part of a drug molecule, it has the potential of offering a longer in vivo half-life. The presence of the tetrazole moiety in a drug molecule improves oral bioavailability and cell penetration. The angiotensin II receptor antagonist Losartan (Fig. 1) is one example where tetrazole is being used in replacement of –CO2H.3 Application of tetrazoles involves their use in pharmaceuticals, in speciality explosives4 and as precursors of a variety of nitrogen containing heterocyclic compounds like imidoylazides.5
 | ||
| Fig. 1 Losartan. | ||
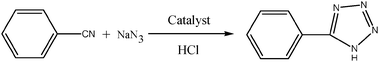
Synthesis of 5-substituted 1H-tetrazoles involves [3+2] cycloaddition of an azide (NaN3/TMSN3) to the corresponding nitrile. Several methods have been reported for the preparation of 5-substituted 1H-tetrazoles. They suffer from various disadvantages such as the use of toxic metals, expensive reagents, explosive hydrazoic acid and severe reaction conditions.6 The use of stoichiometric amounts of zinc(II) salt,7a–d metal complex,7e CdCl2,6 Fe(OAc)2,7f TBAF,7g micellar media7h and ionic liquid7i has sorted out the above said problems.
However these homogeneous catalysts pose difficulties in separation, recovery and reusability. Therefore many researchers have developed various heterogeneous catalyst systems, such as silica supported FeCl3,8a ZnS,8b Zn/Al hydrotalcite,8c Cu2O,8d antimony trioxide,8e ZnHAP8f and metal tungstates.8g It was proved that the function of zinc compounds is to activate the –CN triple bond of nitriles. This activation is only due to the coordination of the nitrogen atom with the Lewis acid metal like zinc.9 We are interested in developing heterogeneous metal oxide catalysts for various organic transformations.10 The use of ZnO alone could not fetch good yield of the 5-substituted 1H-tetrazoles. Hence we tried to increase the acidity of zinc oxide by mixing it with other metal oxides such as TiO2, CeO2 and Co3O4. It was observed that the ZnO/TiO2, ZnO/CeO2 and ZnO/Co3O4 combinations were not explored for the synthesis of 5-substituted 1H-tetrazoles. The mixed oxide of ZnO/Co3O4 shows good optical and magnetic properties. The catalytic activity of Co3O4/ZnO was well studied for carbonylation of glycerol.11 Herein we report the synthesis of nano ZnO/Co3O4 mixed metal oxide and its application as a heterogeneous catalyst for the synthesis of 5-substituted 1H-tetrazoles.
All the mixed metal oxides were prepared by taking a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio of their precursors. Nano ZnO/Co3O4 mixed metal oxide is synthesized by a simple precipitation method under ultrasonication. Zinc acetate and cobalt acetate in a 1:1 mole ratio were dissolved in 0.1 N HCl and a precipitate is obtained by dropwise addition of aqueous ammonia. The whole procedure of precipitation was carried out under ultrasonication. Titanium tetraisopropoxide and cerium nitrate were used as precursors for preparation of TiO2 and CeO2. The bulk metal oxides and mixed metal oxides were prepared by adding an aqueous ammonia solution to the solutions of respective precursors. This addition was carried out without ultrasonication. The pH of the solution was adjusted between 8–9.
:1 mole ratio of their precursors. Nano ZnO/Co3O4 mixed metal oxide is synthesized by a simple precipitation method under ultrasonication. Zinc acetate and cobalt acetate in a 1:1 mole ratio were dissolved in 0.1 N HCl and a precipitate is obtained by dropwise addition of aqueous ammonia. The whole procedure of precipitation was carried out under ultrasonication. Titanium tetraisopropoxide and cerium nitrate were used as precursors for preparation of TiO2 and CeO2. The bulk metal oxides and mixed metal oxides were prepared by adding an aqueous ammonia solution to the solutions of respective precursors. This addition was carried out without ultrasonication. The pH of the solution was adjusted between 8–9.
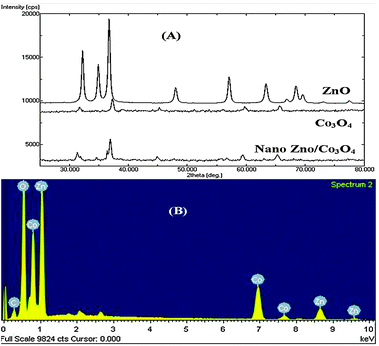
The major phase of the obtained particle was analyzed by X-ray diffraction. Fig. 2A shows the XRD spectra of ZnO, Co3O4 and Nano ZnO/Co3O4. The XRD spectrum of ZnO is indexed to the hexagonal close packed wurtzite structure. The peaks at about 31°, 34°, 35°, 47°, 56°, 62°, 67° and 68° correspond to 100, 002, 101, 102, 110, 103, 200, and 112 crystal planes respectively.12a The XRD spectrum of Co3O4 is indexed to the cubic spinels. The peaks at 31° and 37° in the spectrum of Co3O4 correspond to (220) and (311) planes and confirm the cubic spinel structure of Co3O4.12b The prepared nano ZnO/Co3O4 did not show any extra peak other than the peaks observed for ZnO and Co3O4. EDAX analysis shown in Fig. 2B confirms that the prepared nano ZnO/Co3O4 is composed of zinc (40.45%), cobalt (32.41%) and oxygen (27.14%) elements only. The zinc X-ray energies are 8.63 keV (K-line) and 1.01 (L-line), the cobalt X-ray energies are 6.92 keV (K-line) and 0.77 (L-line) and the oxygen X-ray energy is 0.523 keV (K-line).
The FTIR spectra of the obtained Co3O4, ZnO and nano ZnO/Co3O4 are shown in Fig. 3A. In the spectrum of Co3O4 peaks at 663.48 cm−1 and 569.39 cm−1 are assigned to the ν(Co–O) modes, which confirm the formation of Co3O4. The broad bands at 3456 cm−1 and 1626 cm−1 are assigned to OH stretching and bending modes of water respectively.12c In the spectrum of ZnO, the peak at 3497 cm−1 corresponds to the vibration mode of the OH group of water. It is due to the presence of a small amount of adsorbed water on the surface of the ZnO molecule. The band at 1627 cm−1 is due to the OH bending of water. Zn–O stretching is observed at 526 cm−1.12d The FTIR spectrum for nano ZnO/Co3O4 shows peaks at 3599 cm−1 and 1646 cm−1, which may be because of vibration mode of water adsorbed on the surface of the catalyst. The new peaks are observed at 1250.80 cm−1 and 979.46 cm−1. They may be assigned to Co bonding with ZnO, due to insertion of the Co in the ZnO structure which gives local vibration in ZnO. This is proved on the basis of the Raman spectrum. The Raman spectrum of ZnO/Co3O4 is shown in Fig. 3B. It is reported that if the concentration of Co in ZnO/Co3O4 is more than 0.2 mole fraction, the Raman active modes for ZnO are not observed. As the Co concentration increases, ZnO loses its translational symmetry and Co3O4 gains the translational symmetry toward F2g symmetry. The modes related to Co are due to the secondary phase of Co3O4 and this indicates the local vibrations of Co in ZnO. The modes of Co3O4 dominate over the ZnO modes.13 The mole ratio of ZnO/Co3O4 used in the current experiment is 1:1 (0.5 mole fraction). The Raman active modes observed at 487 cm–1 for Eg, 524 and 625 cm–1 are assigned to F2g modes of Co3O4. The Raman active mode observed near 709 cm–1 appears to be due to Co doping.
 | ||
| Fig. 3 FT-IR of Co3O4, ZnO and ZnO/Co3O4 (A) and Raman spectra (B) of nano ZnO/Co3O4. | ||
The size and shape of the nano ZnO/Co3O4 particles and their degree of agglomeration were obtained by TEM (Fig. 4A) and SEM (Fig. 4B and C). TEM shows that the size of nano ZnO/Co3O4 is in the range of 40–50 nm. The SEM image depicts that the crystals have cubic structure. TEM and SEM images are in good agreement with each other in terms of size and shape.

A model reaction was carried out by taking the mixture of benzonitrile, sodium azide and catalyst in the presence of DMF solvent. Table 1 gives the details about particle size, total acid sites and catalytic activity of ZnO, Co3O4, bulk ZnO/Co3O4 and nano ZnO/Co3O4 (Table 1, entries 1–4). The catalytic activity of ZnO/Co3O4 was found to be more because of better acidity than either ZnO or Co3O4.14 More interestingly Co3O4 has higher acidity than ZnO, but shows less catalytic activity than ZnO. This may be due to activation of the CN triple bond of the nitrile by coordination of the nitrogen atom with the Lewis acid metal like zinc. In addition, nano ZnO/Co3O4 shows better catalytic activity than bulk ZnO/Co3O4 because it has smaller particle size and hence has high surface area. Table 2 shows the comparison of the activity of the different heterogeneous catalysts with that of prepared ZnO/Co3O4. The comparison of the different catalysts (previously reported and used in the present study) was done by considering the quantity of the catalyst used for the reaction. We observed that the nano ZnO/Co3O4 catalyst gives better yield than FeCl3/SiO2, ZnHAP and Zn/Al-HT whereas a similar yield is obtained in the case of BaWO3. Nano ZnO/Co3O4 proved to be a better catalyst than BaWO3 with respect to recyclability. Table 3 gives the details of optimized conditions of the model reaction. It can be seen that 95% yield of the product is obtained at 140 °C whereas 90% yield is obtained at 120 °C. Since there is a very marginal difference in the yield, all the reactions are carried out 120 °C. Prolonged heating beyond 12 h did not show significant increase in the yield of the desired product.
| Sr. No. | Catalyst | Size | Tot. no. of acid sites | Yieldb (%) | |
|---|---|---|---|---|---|
| mL g−1 | mmol g−1 | ||||
| a Reaction conditions: 1 mmol benzonitrile, 1.5 mmol NaN3, 50 mg catalyst and 3 ml DMF for 12 h at 120–130 °C. b Isolated yield. | |||||
| 1 | No catalyst | — | — | — | 0 |
| 2 | ZnO | 19 μm | 1.3377 | 0.0548 | 45 |
| 3 | TiO2 | 53 μm | — | — | 10 |
| 4 | CeO2 | 73 μm | — | — | 41 |
| 5 | Co3O4 | 2 μm | 2.4814 | 0.1017 | 31 |
| 6 | ZnO/TiO2 | 444 μm | — | — | 5 |
| 7 | ZnO/CeO2 | 56 μm | — | — | 58 |
| 8 | ZnO/Co3O4 | 18 μm | — | — | 80 |
| 9 | Nano ZnO/Co3O4 | 40–50 nm | 4.5352 | 0.1859 | 90 |
| Sr. No. | Catalyst (mg) | Time/h | Temp/°C | Solvent | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 1 mmol benzonitrile, 1.5 mmol NaN3, and 3 ml solvent. b Isolated yield. c Catalyst after four cycles. | |||||
| 1 | 50 | 24 | 120 | DMF | 90 |
| 2 | 50 | 18 | 120 | DMF | 90 |
| 3 | 50 | 12 | 120 | DMF | 90 |
| 4 | 50 | 6 | 120 | DMF | 55 |
| 5 | 50 | 12 | 80 | DMF | 14 |
| 6 | 50 | 12 | 100 | DMF | 30 |
| 7 | 50 | 12 | 140 | DMF | 95 |
| 8 | 50 | 12 | 120 | DMSO | 49 |
| 9 | 50 | 12 | 120 | NMP | 75 |
| 10 | 50 | 12 | 120 | H2O | ≤10 |
| 11 | 40 | 12 | 120 | DMF | 70 |
| 12 | 30 | 12 | 120 | DMF | 62 |
| 13 | 20 | 12 | 120 | DMF | 38 |
| 14 | 10 | 12 | 120 | DMF | 33 |
| 15 | 50 | 12 | 120 | DMF | 83c |
Solvent has a remarkable effect on the yield of the desired product. DMF was found to be the most suitable solvent giving a maximum yield of 90% whereas NMP and DMSO gave 75% and 49% yield respectively. Nano ZnO/Co3O4 also showed good recyclability for four cycles giving 83% of product yield.
To study the scope and applicability of the developed protocol, different benzonitriles were used as the starting material and the results are shown in Table 4. The results show that all nitriles react efficiently with sodium azide and give excellent yield of desired products. The presence of an electron donating group at the para position gave an excellent yield of more than 92% (Table 4, entries 3–5). The reactions of benzonitrile having an electron withdrawing group at the para position gave a similar yield (Table 4, entry 2). Thus, the observations suggest that an electron donating group or an electron withdrawing group on benzonitrile does not have a notable effect and gives good to excellent yield.








In conclusion, we have prepared nano ZnO/Co3O4 as a novel catalyst for 5-substituted 1H-tetrazoles from nitriles and sodium azide. This nano sized heterogeneous catalyst showed excellent catalytic activity than ZnO or Co3O4. The given methodology is efficient and environmentally benign. This recyclable catalyst offers advantages like simple work-up and high yields. The developed concept is expected to be more general and easily applicable.
The authors are thankful to UGC-Green Technology Centre, New Delhi, India, for awarding the fellowship.
Notes and references
- R. N. Butler, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, UK, 1996, vol. 4 Search PubMed.
- R. Herr, Bioorg. Med. Chem., 2002, 10, 3379 CrossRef CAS.
- B. C. Bookser, Tetrahedron Lett., 2000, 41, 2805 CrossRef CAS.
- (a) V. A. Ostrovskii, M. S. Pevzner, T. P. Kofmna, M. B. Shcherbinin and I. V. Tselinskii, Targets Heterocycl. Syst., 1999, 3, 467 CAS; (b) M. Hiskey, D. E. Chavez, D. L. Naud, S. F. Son, H. L. Berghout and C. A. Bome, Proc. Int. Pyrotech. Semin., 2000, 27, 3 Search PubMed.
- (a) R. Huisgen, J. Sauer, H. J. Sturn and J. H. Markgraf, Chem. Ber., 1960, 93, 2106 CrossRef CAS; (b) D. J. Moderhack, J. Prakt. Chem., 1988, 340, 687 CrossRef; (c) A. R. Modarresi-Alam, H. Keykha, F. Khamooshi and H. A. Dabbagh, Tetrahedron, 2004, 60, 1525 CrossRef CAS; (d) A. R. Modarresi-Alam, H. Keykha, F. Khamooshi, M. Rostamizadeh, M. Nasrollahzadeh, H. R. Bijanzadeh and E. Kleinpeter, J. Mol. Struct., 2007, 841, 67 CrossRef.
- G. Venkateshwarlu, A. Premalatha, K. C. Rajanna and P. K. Saiprakash, Synth. Commun., 2009, 39, 4479 CrossRef CAS.
- (a) Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS; (b) Z. P. Demko and K. B. Sharpless, Org. Lett., 2002, 4, 2525 CrossRef CAS; (c) O. G. Mancheño and C. Bolm, Org. Lett., 2007, 9, 2551 Search PubMed; (d) L.-Z. Wang, Z.-R. Qu, H. Zhao, X.-S. Wang, R.-G. Xiong and Z.-L. Xue, Inorg. Chem., 2003, 42, 3969 CrossRef CAS; (e) L. Bosch and J. Vilarrasa, Angew. Chem., 2007, 119, 4000 CrossRef; (f) J. Bonnamour and C. Bolm, Chem.–Eur. J., 2009, 15, 4543 CrossRef CAS; (g) D. Amantini, R. Beleggia, F. Fringuelli, F. Pizzo and L. Vaccaro, J. Org. Chem., 2004, 69, 2896 CrossRef CAS; (h) B. S. Jursic and B. W. Leblanc, J. Heterocycl. Chem., 1998, 35, 405 CrossRef CAS; (i) B. Schmidt, D. Meid and D. Kieser, Tetrahedron, 2007, 63, 492 CrossRef CAS.
- (a) D. Habibi and M. Nasrollahzadeh, Synth. Commun., 2010, 40, 3159 CrossRef CAS; (b) L. Lang, B. Li, W. Liu, L. Jiang, Z. Xu and G. Yin, Chem. Commun., 2010, 46, 448 RSC; (c) M. Lakshmi Kantam, K. B. Shiva Kumar and K. Phani Raja, J. Mol. Catal. A: Chem., 2006, 247, 186 CrossRef; (d) T. Jin, F. Kitahara, S. Kamijo and Y. Yamamoto, Tetrahedron Lett., 2008, 49, 2824 CrossRef CAS; (e) G. Venkateshwarlu, K. C. Rajanna and P. K. Saiprakash, Synth. Commun., 2009, 39, 426 CrossRef CAS; (f) M. Laxmi Kantam, V. Balasubramanyam and K. B. Shiva Kumar, Synth. Commun., 2006, 36, 1809–1814 CrossRef; (g) J. He, B. Li, F. Chen, Z. Xu and G. Yin, J. Mol. Catal. A: Chem., 2009, 304, 135 CrossRef CAS.
- (a) F. Himo, Z. P. Demko and L. Noodleman, J. Org. Chem., 2003, 68, 9076 CrossRef CAS; (b) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210 CrossRef CAS; (c) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2003, 125, 9983 CrossRef CAS.
- (a) S. M. Agawane and J. M. Nagarkar, Tetrahedron lett., 2011, 52, 3499–3504 CrossRef CAS; (b) S. M. Agawane and J. M. Nagarkar, Tetrahedron lett., 2011, 52, 5220–5223 CrossRef CAS.
- (a) E. Kandjani, S. E. Hashemi Amiri, M. R. Vaezi and S. K. Sadrnezhaad, J. Optoelectron. Adv. Mater., 2010, 12, 2057–2062 Search PubMed; (b) F. Rubio-Marcos, V. Calvino-Casilda, M. A. Banares and J. F. Fernandez, J. Catal., 2010, 275, 288–293 CrossRef CAS.
- (a) Y. Tak and K. Yong, J. Phys. Chem. C, 2008, 112, 74 CrossRef CAS; (b) Y.-P. Yang, K.-L. Huang, R.-S. Liu, L.-P. Wang, W.-W. Zeng and P.-M. Zhang, Trans. Nonferrous Met. Soc. China, 2007, 17, 1082 CrossRef CAS; (c) D. Zou, C. Xu, H. Luo, L. Wang and T. Ying, Mater. Lett., 2008, 62, 1976 CrossRef CAS; (d) N. Faal Hamedani and F. Farzaneh, J. Sci., Islamic Repub. Iran, 2007, 17, 23 Search PubMed.
- R. Bhargava, P. K. Sharma, S. Kumar, A. C. Pandey and N. Kumar, J. Raman Spectrosc., 2011, 42, 1802–1807 CrossRef CAS.
- See TPD in ESI†.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20094e |
| This journal is © The Royal Society of Chemistry 2012 |