Reductive dehalogenation of β-haloacrylic ester derivatives mediated by ene-reductases†
Gábor
Tasnádi
a,
Christoph K.
Winkler
b,
Dorina
Clay
b,
Mélanie
Hall
b and
Kurt
Faber
*b
aACIB GmbH c/o Department of Chemistry, Organic & Bioorganic Chemistry, University of Graz, Heinrichstraße 28, 8010-Graz, Austria
bDepartment of Chemistry, Organic & Bioorganic Chemistry, University of Graz, Heinrichstraße 28, 8010-Graz, Austria. E-mail: kurt.faber@uni-graz.at
First published on 29th March 2012
Abstract
The enzymatic bioreduction of β-halo-α,β-unsaturated carboxylic esters proceeded via sequential enzymatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C reduction—β-elimination to afford saturated carboxylic esters. This novel biodegradation pathway combines the reductive activity of ene-reductases with the spontaneous β-elimination of hydrohalous acid from the unstable (saturated) intermediates. Both enantiomers of methyl 2-chloro-, 2-bromo- and 2-iodopropionate were obtained in good to excellent enantiopurity via enzyme-based stereocontrol using various members of the ‘Old Yellow Enzyme’ family of flavoproteins. Overall, this pathway resembles a reductive dehalogenation of β-halogenated acrylic esters.
C reduction—β-elimination to afford saturated carboxylic esters. This novel biodegradation pathway combines the reductive activity of ene-reductases with the spontaneous β-elimination of hydrohalous acid from the unstable (saturated) intermediates. Both enantiomers of methyl 2-chloro-, 2-bromo- and 2-iodopropionate were obtained in good to excellent enantiopurity via enzyme-based stereocontrol using various members of the ‘Old Yellow Enzyme’ family of flavoproteins. Overall, this pathway resembles a reductive dehalogenation of β-halogenated acrylic esters.
Introduction
The degradation of toxic haloorganic compounds plays an important role in environmental remediation processes. Besides numerous metal-catalyzed processes developed so far,1–3 microbial and enzymatic pathways have been identified and investigated for the degradation of halo-aromatic and -aliphatic compounds.4–6 Carbon–halogen bonds can be cleaved enzymatically by dehalogenases, members of a broad family of structurally and mechanistically diverse enzymes.7 So far, enzymatic dehalogenation is known to proceed via the following mechanisms: oxidation, reduction (via hydrogenolysis), nucleophilic substitution (hydrolytic, thiolytic and intramolecular dehalogenation), dehydrohalogenation, hydration and methyl transfer reaction.4,8 In addition to these entirely enzyme-catalyzed processes, spontaneous dehalogenation of reactive halo-intermediates thus obtained may occur.4 For instance, this has been proposed in the hydrolytic dehalogenation of 3-chloroacrylate catalyzed by trans- and cis-3-chloroacrylic acid dehalogenases (CaaD and cis-CaaD), where the CC-hydration was shown to be enzyme-catalyzed while the elimination of HCl proceeded non-enzymatically via the unstable halohydrin intermediate.9–11 Similarly, 2-haloacrylate hydratase catalyzes the hydration of 2-chloro- and 2-bromoacrylate to the corresponding unstable 2-halo-2-hydroxypropionate, which spontaneously decomposes to yield pyruvate.12 Maleylacetate reductase can degrade 2-chloromaleyl acetate to β-ketoadipate via a sequence of reduction–spontaneous HCl elimination steps.13
Previously we successfully employed ene-reductases, members of the ‘Old Yellow Enzyme’ (OYE) family,14 in the asymmetric bioreduction of a range of alkenes bearing an electron-withdrawing group.15–17 As a continuation of our work, we were interested in the transformation of halogenated acrylic esters. While methyl α-halo-2-alkenoates and 3-arylacrylates are known to be substrates for OYEs,18–21 less is known about α,β-di- and trihalogenated acrylate derivatives. The hydrogenation of β-halo-2-enoates has been investigated with Clostridia sp. under anaerobic conditions and was found to yield the halogen-free saturated carboxylic acids. The O2-sensitive iron–sulfur containing flavoprotein 2-enoate reductase was shown to be responsible for the combined hydrogenation–elimination step, and the whole sequence was proposed to take place in the active site of the enzyme.22,23 Inspired by these investigations, we aimed to study the ability of various isolated oxygen-stable OYEs to reduce di- and trihalogenated acrylic esters. Unexpectedly, various ene-reductases from fungi, bacteria and plants mediated the formal reductive dehalogenation of α,β-haloacrylate esters. Optically active α-halogenated propionic acid esters obtained as end-products of this biodegradation pathway have numerous potential synthetic applications, e.g. in the synthesis of peptidomimetics,24 β2-amino acids,25 chiral ligands26 and oligonucleotide mimics.27 While OYEs have already shown some unusual catalytic promiscuity in biodegradation of nitro-compounds (e.g. via bio-Nef reaction,28 oxazete formation29) and enones (Weitz–Scheffer epoxidation30), their involvement in the biodegradation of halo-organic compounds has not yet been reported.
Results
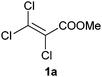
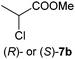
The reduction of methyl 2,3,3-trichloroacrylate (1a) with several ene-reductases (OYE1–3, YqjM, NCR, OPR1 and OPR3) was investigated during the course of a detailed study of the electronic activation of CC-bonds. However, the expected reduction product methyl 2,3,3-trichloropropionate (1b) was not detected, and methyl 2-chloropropionate (7b) was surprisingly identified as the main product with OYE3 and NCR using an excess of NADH (>99% and 15% conversion respectively). Depending on the enzyme, the reaction proceeded in a highly stereocomplementary fashion (OYE3: (S)-7b in 89% ee, NCR: (R)-7b in >99% ee).
Based on additional experiments (see below), the pathway is proposed to proceed as follows: first, 1a was enzymatically reduced to methyl 2,3,3-trichloropropionate (1b), which spontaneously underwent elimination of HCl to furnish methyl 2,3-dichloroacrylate (3a, E/Z-configuration not determined). The latter re-entered the cycle and was further reduced to methyl 2,3-dichloropropionate (3b), which subsequently eliminated HCl to yield methyl 2-chloroacrylate (7a). Finally, 7a was stereoselectively reduced to furnish methyl 2-chloropropionate (7b) as the endproduct of the biodegradation cascade, which required three reducing equivalents in total (Scheme 1). In contrast to the mechanism proposed for the 2-enoate reductase-catalyzed reductive dehydrohalogenation of 3-halogeno-2-enoates,22 the elimination of HCl was shown to be of spontaneous nature: when methyl rac-2,3-dichloropropionate (3b) was incubated in buffer in the absence of enzyme—in (i) Tris buffer (50 mM, pH 7.5), in (ii) buffer supplemented with NADH and in (iii) buffer containing a cofactor recycling system—only methyl 2-chloroacrylate (7a) could be detected as a product with no trace of 3b being left (Table S4, ESI†). As expected, HCl elimination catalyzed by Tris buffer was not observed with the unsaturated compound 1a (Table S3, ESI†). Additionally, all enzymes tested transformed rac-3b into methyl 2-chloropropionate (7b) in the presence of 1.5 equiv. of NADH, confirming that methyl 2-chloroacrylate (7a)—spontaneously produced by HCl elimination from 3b—was nicely accepted by ene-reductases (Table 1, entry 4). Overall, ene-reductases were found to be less active on trichloroacrylate esters, especially YqjM and OPRs (maximum 8% conversion, Table 1, entries 1–3). Low reactivity of intermediates can be excluded since only methyl 2,3-dichloroacrylate (3a) was detected in up to 6% (Table S3, ESI†). The reaction rate of the NCR-catalyzed reduction of 1a, however, could be significantly improved (from 15% up to 50%) using a NAD+–GDH or NADP+–GDH cofactor recycling system (Table 1, entries 2 and 3), without compromising the stereoselectivity [ee (R) > 99%]. Similar significant effects of the cofactor recycling system on the reaction rate of ene-reductases have been observed before.31,32 Overall, the NADP+–GDH recycling system was superior to the NAD+–GDH system, providing generally higher conversions (83–96% vs. 42–71% with OYE1–3, entry 3).
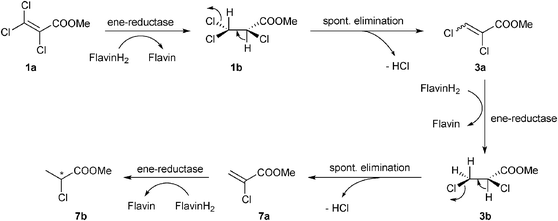 | ||
| Scheme 1 | ||
| Entry | Substrate | Final product | Cofactor | OYE1 | OYE2 | OYE3 | YqjM | NCR | OPR1 | OPR3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Conv. ee (%) | Conv. ee (%) | Conv. ee (%) | Conv. ee (%) | Conv. ee (%) | Conv. ee (%) | Conv. ee (%) | ||||
| Reaction conditions: 10 mM substrate, 15, 25 or 35 mM NADH or cofactor recycling system (see Experimental section) in Tris-HCl-buffer (50 mM, pH 7.5), 30 °C, 120 rpm, 24 h. n/a = not applicable; n.c. = no conversion to product 7–10b; n.d. = not determined; NAD(P)+–GDH = glucose/glucose dehydrogenase was used for cofactor recycling.a Conversion levels are given based on extracted non-polymerized material and reflect the amount of products 7–10b. For by-products distribution, see ESI.b Significant polymerization.c 3% of 3a was detected, E/Z-configuration not determined.d 2% of TBME used as a cosolvent.e 1% of 9a was detected.f 10% of TBME used as a cosolvent.g Intermediates 4a, 4b or 8a were detected up to 19%; E/Z-configurations and enantiomeric excess not determined.h 1% of DMF was used as a cosolvent. | ||||||||||
| 1 |
|
|
NADH | n/ab | n/ab | >99 | n.c. | 15 | n.c. | n.c. |
| 89 (S) | >99 (R) | |||||||||
| 2 | NAD+–GDH | 42 | 64 | 71 | <5 | 42 | <5 | <5 | ||
| 52 (S) | 39 (S) | 89 (S) | n.d. | >99 (R) | n.d. | n.d. | ||||
| 3 | NADP+–GDH | 86 | 83 | 96 | 8 | 50 | n.c.c | <5 | ||
| 45 (S) | 38 (S) | 89 (S) | 91 (R) | >99 (R) | n.d. | |||||
| 4 |

|
NADH | >99 | 78 | >99 | >99 | >99 | >99 | 42 | |
| 43 (S) | 37 (S) | 86 (S) | 89 (R) | >99 (R) | 93 (R) | >99 (R) | ||||
| 5 | NAD+–GDH | 76 | 82 | 74 | 91 | 91 | 77 | 46 | ||
| 41 (S) | 29 (S) | 85 (S) | 89 (R) | >99 (R) | 92 (R) | >99 (R) | ||||
| 6 | NADP+–GDH | 72 | 76 | 83 | 75 | 78 | 79 | 67 | ||
| 45 (S) | 39 (S) | 90 (S) | 89 (R) | >99 (R) | 90 (R) | >99 (R) | ||||
| 7 |
|
|
NADHd | 97 | >99 | 89 | 10 | 83 | <5 | n.c.e |
| 89 (S) | 90 (S) | 92 (S) | 56 (R) | 92 (R) | n.d. | |||||
| 8 | NAD+–GDH | 75 | >99 | 98 | 35 | 97 | 14 | <5 | ||
| 93 (S) | 92 (S) | 93 (S) | 53 (R) | 96 (R) | 75 (R) | n.d. | ||||
| 9 | NADP+–GDH | >99 | >99 | 98 | 69 | 98 | 14 | <5 | ||
| 93 (S) | 93 (S) | 95 (S) | 57 (R) | 96 (R) | 75 (R) | n.d. | ||||
| 10 |
|
NADH | >99 | >99 | >99 | >99 | >99 | 94 | 44 | |
| 92 (S) | 93 (S) | 95 (S) | 61 (R) | 96 (R) | 91 (R) | 90 (R) | ||||
| 11 | NAD+–GDH | >99 | 99 | >99 | >99 | >99 | 94 | 31 | ||
| 93 (S) | 92 (S) | 94 (S) | 59 (R) | 96 (R) | 87 (R) | 90 (R) | ||||
| 12 | NADP+–GDH | 96 | 97 | 98 | 97 | >99 | 74 | 27 | ||
| 94 (S) | 94 (S) | 95 (S) | 59 (R) | 96 (R) | 87 (R) | 91 (R) | ||||
| 13 |
|

|
NADHf | 64 | 20 | 88 | n.c.g | <5 | n.c.g | n.c.g |
| 14 |

|

|
NADHf | 54 | 17 | 98 | <5 | 30 | <5 | <5 |
| 90 (S) | 90 (S) | 86 (S) | n.d. | rac | n.d. | n.d. | ||||
| 15 | NADP+–GDHh | 18 | 13 | 97 | 6 | <5 | 5 | <5 | ||
| 86 (S) | 88 (S) | 70 (S) | 60 (S) | n.d. | 40 (R) | n.d. | ||||
Polymerization of substrates (in the presence or absence of enzymes) occurred to various extents, which is not surprising given the tendency of (α-halo)acrylates to polymerize.33 Interestingly, polymerization was predominant with 1a in the presence of 3.5 equiv. of NADH and OYE1 or OYE2 (Table 1, entry 1). The cofactor recycling system was found to significantly reduce polymerization with 1a and OYE1 and OYE2, thus allowing reduction to 7b in good yield and modest stereoselectivity (up to 86% conversion and 52% ee, entries 2 and 3). This effect is presumably due to altered enzyme kinetics and/or the recycling buffer composition. No traces of by-products arising from hydration could be detected.
The use of a cofactor recycling system resulted in lower conversion levels starting from rac-3b (46–91%) and different product compositions were obtained with NAD+ and NADP+ (Table 1, entries 5 and 6, and Table S4 (ESI†)). Interestingly, spontaneous HCl elimination from 3b was incomplete in the presence of a cofactor recycling system and was more pronounced in the case of NADP+–GDH (3b detected in up to 25% with NADP+, only 8% with NAD+, Table S4 (ESI†)). Overall, methyl 2-chloroacrylate (7a) accumulated in the reaction mixture with both recycling systems, to a larger extent with NAD+ (up to 49% with OPR3, only 27% with NADP+, Table S4 (ESI†)). All ene-reductases displayed comparable stereoselectivities on 1a and 3b, confirming the proposed reaction pathway via the central intermediate 7a (entries 1–6). Good to excellent (R)-stereoselectivity was obtained in the transformation of rac-3b with YqjM/OPR1 (≥89% ee), and NCR/OPR3 (>99% ee), respectively. While OYE1 and OYE2 showed reduced stereoselectivities (max. 45% ee), OYE3 provided the opposite enantiomer (S)-7b in high ee (85–90%).
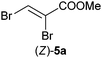
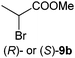
In order to investigate the scope of the biodegradation of halogenated acrylates, brominated analogues were selected. Methyl 2,3-dibromoacrylate [(Z)-5a] was tested with 2.5 equiv. of NADH (Table 1, entry 7 and Table S5 (ESI†)). Overall, 5a and the resulting reduced and monodehydrohalogenated product 9a were well accepted by OYE1–3 and NCR, yielding methyl 2-bromopropionate 9b with 83% to >99% conversion. YqjM and OPRs displayed low activity with (Z)-5a, but conversions could be enhanced in the presence of a cofactor recycling system (entries 8 and 9). Overall, the stereopreference was conserved between methyl 2-bromoacrylate (9a, resulting from spontaneous HBr elimination of rac-5b, Table S6 (ESI†)) and its chlorinated analogue 7a (OYE1–3: (S)-selective, YqjM, NCR and OPRs: (R)-selective; Table 1, entries 4–6 and 10–12). However, OYE1–3 displayed much higher stereoselectivity on the brominated substrate [(S)-9b 89–95% ee] and YqjM showed reduced selectivity toward 9b [(R)-9b 57% eemax].
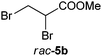
Excellent conversions were obtained in the transformation of methyl 2,3-dibromopropionate (rac-5b) with OYE1–3, YqjM, NCR and OPR1 using molar equivalents of NADH or a cofactor recycling system (entries 10–12). As with monochlorinated analogue 7a, monobrominated compound 9a obtained from spontaneous β-elimination of 5b was more widely accepted by ene-reductases than multibrominated derivatives, especially YqjM and OPRs showed drastic increase in conversion levels compared to those obtained with (Z)-5a (entries 7–9 and Table S6 (ESI†)). The stereoselectivities obtained in the biodegradation of 5a and 5b confirm the reaction pathway described in Scheme 2.
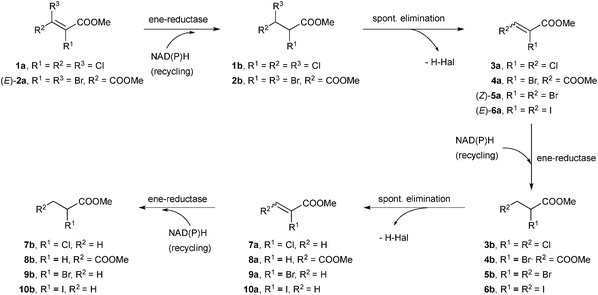 | ||
| Scheme 2 | ||
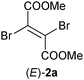
Dimethyl (E)-dibromofumarate [(E)-2a] (Table 1, entry 13 and Table S7 (ESI†)) turned out to be a poor substrate overall. OYE1 and OYE3 showed highest activity and afforded the expected product dimethyl succinate (8b, 64% and 88% conversion, respectively) after a triple bioreduction–elimination sequence, in the presence of 3.5 equiv. of NADH. The presence of a second conjugated ester moiety allows complete dehalogenation via spontaneous β-elimination due to the acidity of both α-H atoms, but renders substrate (E)-2a too bulky to be accepted by the enzymes (except OYE1 and OYE3). Dimethyl esters have been previously shown to be good substrates to ene-reductases.34 The inactivity of 2a is most likely due to the presence of two additional bulky halogen atoms on the CC bond.
Finally, methyl (E)-diiodopropenoate [(E)-6a] was tested (Table 1, entries 14 and 15, and Table S8 (ESI†)). This substrate was well-accepted by OYE3 in the presence of 2.5 equiv. of NADH as well as the NADP+–GDH cofactor recycling system (98% and 97% conversion, respectively), and underwent two bioreduction–elimination cycles yielding methyl (S)-2-iodopropionate [(S)-10b] in 86% and 70% ee, respectively. The ability of OYE3 to accept bulky substrates has been explained by the enlarged binding pocket caused by an exchange of phenylalanine F296 (in OYE1 and OYE2) by serine S297 in OYE3.17,35 OYE1 also converted (E)-6a to (S)-10b with high selectivity (90% ee, 54% conversion), while OYE2 and NCR showed reduced activity (maximum 30% conversion). The iodinated analogue (E)-6a, being the most sterically demanding substrate within this series, was a poor substrate for YqjM and OPRs. Interestingly, YqjM showed opposite stereopreference in the reduction of α-iodoacrylate 10a compared to the chloro- and bromo-analogues and produced (S)-10b in 60% ee (Table 1, entry 15). A similar drastic change in stereopreference was observed with NCR providing end-product 10b in racemic form. The type of halogen had no influence on OYE1–3 stereopreference. Similar effects of the size of the substituents on the stereo-outcome of the reaction have been observed with protected enol ethers for instance,36 and are due to opposite binding modes in the active site.
Conclusion
The biodegradation of chlorinated, brominated and iodinated di- and trihalo-acrylates and -propionates mediated by ene-reductases was achieved via a sequential enzyme-catalyzed bioreduction–spontaneous HX elimination pathway. Di- and trihalogenated acrylates were found to be poor substrates for YqjM and OPRs, while OYE1–3 and NCR were highly active and could catalyze up to three reduction–elimination cycles. Smaller α-monohalogenated analogues were usually well accepted. Sterically hindered methyl (E)-dibromofumarate and methyl (E)-diiodoacrylate could be dehalogenated almost quantitatively by OYE3 to furnish methyl succinate and methyl (S)-2-iodopropionate, respectively. This study shows that ene-reductases are able to play a key role in the reductive dehydrohalogenation, akin to maleyl reductase activity on 2-chloromaleylacetate. Overall, this route provides an additional biodegradation pathway for α,β-unsaturated haloesters.Experimental section
General procedure for the transformation of 1a, rac-3b, (Z)-5a, rac-5b, (E)-2a and (E)-6a
An aliquot of enzyme (OYE1–3, YqjM, NCR, OPR1, OPR3, protein concentration 75–125 μg mL−1, 6.6–31 μL of the stock solution) was added to a Tris-HCl buffer solution (0.8 mL, 50 mM, pH 7.5) containing the substrate (10 mM) and the cofactor NADH (15, 25 or 35 mM). The mixture was shaken at 30 °C and 120 rpm. After 24 h the products were extracted with EtOAc (2 × 0.6 mL). The combined organic phases were dried over Na2SO4 and analyzed by achiral GC to determine the conversion and by chiral GC to determine the enantiomeric excess (see ESI†). For cofactor recycling, the oxidized form of the cofactor (NAD+ or NADP+, 100 μM), the cosubstrate (glucose 35 mM) and the recycling enzyme (glucose dehydrogenase, 10 U) were used.Blank reactions were run in the absence of enzyme by dissolving the substrate in (i) buffer, (ii) buffer containing 1.5, 2.5 or 3.5 equiv. of NADH, and (iii) buffer containing a NAD(P)+–GDH cofactor recycling system.
Products were identified by GC-MS or by comparison with authentic reference materials via co-injection on achiral GC. Conversions were calculated by comparison of the surface area ratio of substrate, intermediates and product peaks by GC.
Acknowledgements
This study was performed in cooperation within the Austrian Centre of Industrial Biotechnology (ACIB, funded through the FFG-COMET-Program) and project P22722 of the FWF and support from the FWF, BMWFJ, BMVIT, SFG, Standortagentur Tirol, ZIT and BASF SE (Ludwigshafen) is gratefully acknowledged. Klaus Ditrich (Ludwigshafen) is cordially thanked for stimulating discussions.References
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS.
- M. A. Keane, ChemCatChem, 2011, 3, 800–821 CrossRef CAS.
- S. Kliegman and K. McNeill, Dalton Trans., 2008, 4191–4201 RSC.
- S. Fetzner and F. Lingens, Microbiol. Rev., 1994, 58, 641–685 CAS.
- S. Fetzner, Appl. Microbiol. Biotechnol., 1998, 50, 633–657 CrossRef CAS.
- A. O. Olaniran and E. O. Igbinosa, Chemosphere, 2011, 83, 1297–1306 CrossRef CAS.
- D. B. Janssen, I. J. T. Dinkla, G. J. Poelarends and P. Terpstra, Environ. Microbiol., 2005, 7, 1868–1882 CrossRef CAS.
- M. M. Häggblom and I. D. Bossert, Dehalogenation Microbial Processes and Environmental Applications, Springer, US, 2004 Search PubMed.
- H. F. Azurmendi, S. C. Wang, M. A. Massiah, G. J. Poelarends, C. P. Whitman and A. S. Mildvan, Biochemistry, 2004, 43, 4082–4091 CrossRef CAS.
- R. M. de Jong, W. Brugman, G. J. Poelarends, C. P. Whitman and B. W. Dijkstra, J. Biol. Chem., 2004, 279, 11546–11552 CrossRef CAS.
- R. M. de Jong, P. Bazzacco, G. J. Poelarends, W. H. Johnson, Y. J. Kim, E. A. Burks, H. Serrano, A.-M. W. H. Thunnissen, C. P. Whitman and B. W. Dijkstra, J. Biol. Chem., 2007, 282, 2440–2449 CrossRef CAS.
- A. M. Mowafy, T. Kurihara, A. Kurata, T. Uemura and N. Esaki, Appl. Environ. Microbiol., 2010, 76, 6032–6037 CrossRef CAS.
- S. R. Kaschabek and W. Reineke, J. Bacteriol., 1993, 175, 6075–6081 CAS.
- R. Stuermer, B. Hauer, M. Hall and K. Faber, Curr. Opin. Chem. Biol., 2007, 11, 203–213 CrossRef CAS.
- M. Hall, C. Stueckler, W. Kroutil, P. Macheroux and K. Faber, Angew. Chem., Int. Ed., 2007, 46, 3934–3937 CrossRef CAS.
- C. Stueckler, N. J. Mueller, C. K. Winkler, S. M. Glueck, K. Gruber, G. Steinkellner and K. Faber, Dalton Trans., 2010, 39, 8472–8476 RSC.
- C. Stueckler, C. K. Winkler, M. Hall, B. Hauer, M. Bonnekessel, K. Zangger and K. Faber, Adv. Synth. Catal., 2011, 353, 1169–1173 CrossRef CAS.
- M. Utaka, S. Konishi, A. Mizuoka, T. Ohkubo, T. Sakai, S. Tsuboi and A. Takeda, J. Org. Chem., 1989, 54, 4989–4992 CrossRef CAS.
- P. Ferraboschi, P. Grisenti, R. Casati, A. Fiecchi and E. Santaniello, J. Chem. Soc., Perkin Trans. 1, 1987, 1743–1748 RSC.
- E. Brenna, F. G. Gatti, A. Manfredi, D. Monti and F. Parmeggiani, Eur. J. Org. Chem., 2011, 4015–4022 CrossRef CAS.
- E. Brenna, G. Fronza, C. Fuganti, D. Monti and F. Parmeggiani, J. Mol. Catal. B: Enzym., 2011, 73, 17–21 CAS.
- H. Sedlmaier, W. Tischer, P. Rauschenbach and H. Simon, FEBS Lett., 1979, 100, 129–132 CrossRef CAS.
- H. Hashimoto and H. Simon, Angew. Chem., Int. Ed. Engl., 1975, 14, 106–107 CrossRef CAS.
- N. Narendra, H. S. Lalithamba and V. V. Sureshbabu, Tetrahedron Lett., 2010, 51, 6169–6173 CrossRef CAS.
- R. Moumné, B. Denise, K. Guitot, H. Rudler, S. Lavielle and P. Karoyan, Eur. J. Org. Chem., 2007, 1912–1920 CrossRef.
- Y. Mei, P. Dissanayake and M. J. Allen, J. Am. Chem. Soc., 2010, 132, 12871–12873 CrossRef CAS.
- A. I. Khan, T. H. S. Tan and J. Micklefield, Chem. Commun., 2006, 1436–1438 RSC.
- K. Durchschein, S. B. Ferreira da Silva, S. Wallner, P. Macheroux, W. Kroutil, S. M. Glueck and K. Faber, Green Chem., 2010, 12, 616–619 RSC.
- K. Durchschein, W. M. F. Fabian, P. Macheroux, K. Zangger, G. Trimmel and K. Faber, Org. Biomol. Chem., 2011, 9, 3364–3369 CAS.
- N. J. Mueller, C. Stueckler, M. Hall, P. Macheroux and K. Faber, Org. Biomol. Chem., 2009, 7, 1115–1119 CAS.
- M. Hall, C. Stueckler, H. Ehammer, E. Pointner, G. Oberdorfer, K. Gruber, B. Hauer, R. Stuermer, W. Kroutil, P. Macheroux and K. Faber, Adv. Synth. Catal., 2008, 350, 411–418 CrossRef CAS.
- M. Hall, C. Stueckler, B. Hauer, R. Stuermer, T. Friedrich, M. Breuer, W. Kroutil and K. Faber, Eur. J. Org. Chem., 2008, 1511–1516 CrossRef CAS.
- C. S. Marvel, E. D. Weil, L. B. Wakefield and C. W. Fairbanks, J. Am. Chem. Soc., 1953, 75, 2326–2330 CrossRef CAS.
- C. Stueckler, M. Hall, H. Ehammer, E. Pointner, W. Kroutil, P. Macheroux and K. Faber, Org. Lett., 2007, 9, 5409–5411 CrossRef CAS.
- K. M. Fox and P. A. Karplus, Structure (London), 1994, 2, 1089–1105 CrossRef CAS.
- C. K. Winkler, C. Stueckler, N. J. Mueller, D. Pressnitz and K. Faber, Eur. J. Org. Chem., 2010, 6354–6358 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General remarks; source of enzymes; synthesis of chiral reference compound (R)-10b; analytical data for the GC-determination of conversion and enantiomeric excess; determination of absolute configuration; tables consisting all results on substrates, intermediates and products. See DOI: 10.1039/c2cy20079a |
| This journal is © The Royal Society of Chemistry 2012 |