Synthesis, characterization and catalytic performance of Mo-TUD-1 catalysts in epoxidation of cyclohexene
Mohamed S.
Hamdy
*ab and
Guido
Mul
a
aPCS Group, Faculty of Science and Technology, MESA+ Institute for Nanotechnology, University of Twente, PO Box 217, 7500 AE Enschede, The Netherlands. E-mail: G.mul@utwente.nl; Fax: +31 53 4892882; Tel: +31 53 4893890
bChemistry Department, Faculty of Science, Helwan University, Cairo, Egypt. E-mail: m.s.hamdy@gmail.com
First published on 25th April 2012
Abstract
A new, 3-D mesoporous amorphous molybdenum-containing silica, denoted Mo-TUD-1, was prepared with variable Mo-loading via direct hydrothermal treatment using triethanolamine as a template. Elemental analysis, N2 sorption measurements, NMR spectroscopy, XRD, UV-Vis spectroscopy, SEM, HR-TEM, and Raman spectroscopy indicated that as a function of increasing loading, the morphology of Mo changes from isolated sites to nano particulates, and finally to crystalline MoO3 at a loading higher than 5 wt%. The catalytic performance of the Mo-TUD-1 samples was investigated in the liquid phase epoxidation of cyclohexene with TBHP as an oxidant. Mo-TUD-1 with a low molybdenum loading showed a higher TON as compared to samples with a high loading, suggesting that isolated Mo-sites are more active than particulate MoO3. Stability and reusability of the catalyst containing isolated sites were also investigated. While leaching was observed initially, stable performance was obtained in as many as three subsequent catalytic runs.
1. Introduction
Oxidation of olefins to the corresponding epoxides is an attractive route for the industrial production of chemicals. The epoxides are well known to be one of the most valuable building blocks to produce fine chemicals.1 However, epoxide selectivity is a challenge, since several side reactions can take place, such as oxidation in the allylic positions, ring-opening of the epoxides or complete oxidation to CO2.2 It is known that compounds containing certain transition metals, such as Mo, W, Ti and V, are able to catalyze the liquid phase epoxidation of olefins using alkyl hydroperoxides as oxidants in homogeneous solution.3 Among these, molybdenum(VI) catalysts are of particular interest because they show the most favourable catalytic performance as reported by Sheldon.4While homogeneous Mo(VI) compounds are very versatile for epoxidation of olefins,5,6 issues of catalyst separation and their reuse are problematic. Hence, the development of heterogeneous catalytic systems for the epoxidation of olefins is highly attractive. Various procedures to prepare active heterogeneous molybdenum-containing catalysts have been reported, such as grafting of molybdenum compounds onto functionalized polymeric supports,7 preparation of amorphous molybdenum silicate via a sol–gel method,8 or incorporation of molybdenum in the framework of zeolites.9
An alternative is the use of mesoporous supports. In particular, mesoporous molecular sieves have attracted great interest in many fields, such as catalysis and materials science, since their disclosure in 1992.10 The most important features for their use are the extremely high surface area and well defined pore size which can be adjusted over a large range (2–20 nm) optimizing accessibility. Materials such as Mo-MCM-4111 and Mo-SBA-1512 were prepared, and used to catalyze the epoxidation of olefins. TUD-113 is also a mesoporous solid material, equipped with a three-dimensional structure, tuneable pore size distribution, and surface area reaching values as high as 900 m2 g−1. The main advantage of TUD-1 is that it is excellent for framework incorporation of different metals.14–16 A high degree of framework incorporation can be achieved without losing dispersion, which is due to the complexing agent used in the synthesis of TUD-1, triethanolamine. It forms atrane complexes with different metal atoms, thus ensuring incorporation as isolated metal centres, rather than as metal oxide clusters, up to relatively high loading.
Here, for the first time we describe the successful synthesis of TUD-1 mesoporous materials containing molybdenum, via a one-step procedure. We report extensive characterization of the prepared materials, and finally demonstrate the catalytic performance of the prepared samples in the liquid phase epoxidation of cyclohexene under mild conditions. The catalysis is discussed with reference to other elements in TUD-1, as well as to other Mo-containing mesoporous silicas.
2. Experimental
2.1. Mo-TUD-1 synthesis
Mo-TUD-1 mesoporous catalysts with different Si/M ratios were synthesized by aging, drying and then calcination of the homogeneous synthesis mixture as follows, with a molar ratio composition of SiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :x MoOy:0.5 TEAOH:1 TEA:11 H2O. In a typical synthesis deionized water was added to triethanolamine (TEA, 97%, ACROS) and a molybdenum ammonium solution ((NH4)6Mo7O24·4H2O, 81–83% Fluka). The mixture was subsequently stirred for 30 minutes, and added dropwise to tetraethylorthosilicate (TEOS, +98%, ACROS) under vigorous stirring, to prevent precipitate formation. Finally, tetraethyl ammonium hydroxide (TEAOH, 35%, Aldrich) was added dropwise and the overall mixture stirred for at least 2 hours at room temperature. The resulting homogeneous mixture was aged at room temperature for 24 hours, and then dried at 371 K for another 24 hours. The obtained solid was ground and hydrothermally treated in a 50 ml Teflon-lined stainless steel autoclave at 451 K under autogenous pressure for 4 hours. The obtained solid was ground again, and then calcined in static air at 873 K for 10 h applying a heating ramp rate of 1 K min−1.
:x MoOy:0.5 TEAOH:1 TEA:11 H2O. In a typical synthesis deionized water was added to triethanolamine (TEA, 97%, ACROS) and a molybdenum ammonium solution ((NH4)6Mo7O24·4H2O, 81–83% Fluka). The mixture was subsequently stirred for 30 minutes, and added dropwise to tetraethylorthosilicate (TEOS, +98%, ACROS) under vigorous stirring, to prevent precipitate formation. Finally, tetraethyl ammonium hydroxide (TEAOH, 35%, Aldrich) was added dropwise and the overall mixture stirred for at least 2 hours at room temperature. The resulting homogeneous mixture was aged at room temperature for 24 hours, and then dried at 371 K for another 24 hours. The obtained solid was ground and hydrothermally treated in a 50 ml Teflon-lined stainless steel autoclave at 451 K under autogenous pressure for 4 hours. The obtained solid was ground again, and then calcined in static air at 873 K for 10 h applying a heating ramp rate of 1 K min−1.
2.2. Characterization
Instrumental neutron activation analysis (INAA) was used for chemical composition determination (elemental analysis), at the THER nuclear reactor at Delft University of Technology with a thermal power of 2 MW and a maximum neutron reflux of 2 × 1017 m−2 s−1. Nitrogen adsorption/desorption isotherms were recorded on a QuantaChrome Autosorb-6B at 77 K. The pore size distribution was calculated from the adsorption branch using the Barret–Joyner–Halenda (BJH) model. The BET method was used to calculate the surface area (SBET) of the samples, while the mesopore volume (Vmeso) was determined using the t-plot method. 29Si NMR experiments were performed at a magnetic field of 9.4 T on a Varian VXR-400S spectrometer operating at 104.2 MHz with a pulse width of 3.2 ms. Powder X-ray diffraction patterns were measured on a Philips PW 1840 diffractometer equipped with a graphite monochromator using CuKα radiation (λ = 0.1541 nm). The samples were scanned over a range of 0.1–80° 2θ with steps of 0.02°. UV-Vis spectra were collected at ambient temperature on a CaryWin 300 spectrometer using BaSO4 as reference. Samples were ground, heated overnight at 180 °C and then the diffuse reflectance spectra were recorded at ambient conditions in the wavelength range of 190–800 nm. Scanning Electron Microscopy images were recorded at 10 kV on a Philips XL 20 microscope. Samples were coated with gold to avoid charging effects and to enhance contrast. High-Resolution Transmission Electron Microscopy was carried out on a Philips CM30UT electron microscope with a field emission gun as the source of electrons, operated at 300 kV. Ambient laser Raman spectra were obtained using a Renishaw Raman Imaging Microscope, system 2000. The green (λ = 514 nm) polarized radiation of an argon ion laser beam of 20 mW was used for excitation. A Leica DMLM optical microscope with a Leica PL L500/5 objective lens was used to focus the beam on the sample. Moreover, in situ Raman spectra were collected under dehydrated conditions by a high-resolution, dispersive Raman spectrometer system (Horiba-Jobin Yvon LabRam HR) equipped with three laser wavelengths (λ = 532, 442, and 325 nm). 5–10 mg of the loose sample was heated in a controlled high-temperature cell reactor up to 773 K with a ramp of 10 K min−1, and maintained for 10 minutes at that temperature. The Raman spectra were collected in the range of 180–1600 cm−1 at 473 K after dehydration using 442 nm exposure.2.3. Catalysis
The liquid phase oxidation of cyclohexene with tert-butyl hydroperoxide (TBHP) was carried out in a magnetically stirred round-bottom flask fitted with a reflux condenser and heated in a temperature controlled oil bath. The reaction was carried out at 313 K under N2 gas to avoid moisture effects. In a typical test, 100 mg of the catalyst (pre-heated at 453 K overnight) was introduced into the flask. The reaction mixture consisted of cyclohexene (10 mmol, 99%, ACROS), TBHP (11 mmol, 70 wt% in water, ACROS, dried over anhydrous MgSO4 before use) in dichloroethane (10 mL, 99%, Merck) as a solvent. Liquid samples (in total never more than 10% of the reaction volume) were withdrawn at regular interval times from the reaction mixture, filtered by micro-filters and then analyzed by a gas chromatograph (Varian Star 3500) equipped with a 0.25 mm diameter, 50 m OV-1701 capillary column and a flame ionization detector. All the catalysis tests were performed at least two times. The turnover number is defined as the number of moles of cyclohexene converted per mole of molybdenum, on the basis of the amount of product formed after 6 h of reaction. Stability and reusability studies were addressed through hot-filtration of the Mo-1 sample after completion of the reaction. The deposited catalyst was washed several times with ethanol to remove all organic substances, and then dried at 110 °C for 10 h before use in consecutive runs.3. Results and discussion
3.1. Catalysts characterization
The elemental analysis of the different Mo-TUD-1 samples is summarized in Table 1.Comparison of the composition of the synthesis gel and that of the obtained powder after calcination indicates that almost all the metal added during synthesis ends up in the final solid product. At low metal loading, i.e. the Mo-1 sample, the obtained Si/Mo ratio was a bit higher than that present in the synthesis gel. This finding was also observed for other metal-incorporated TUD-1 catalysts.15,16 This might be explained by the very small amount of metal added during the synthesis, which increases the experimental error.
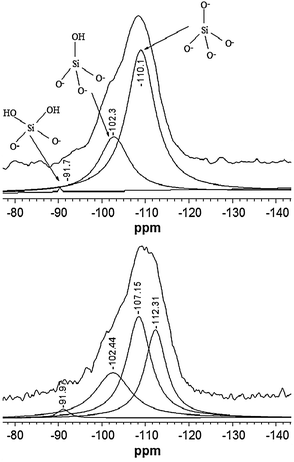
29Si NMR is a powerful tool for the characterization of crystalline zeolites, allowing the detection of hetero-atoms incorporated in the framework.17 Hence, 29Si NMR was performed to study the interaction between Si and Mo. The 29Si MAS NMR spectrum of the calcined siliceous TUD-1 sample is compared with the Mo-TUD-1 spectrum in Fig. 1. Three main peaks were detected after the deconvolution of the TUD-1 spectrum, the first around −110 ppm that is usually assigned to (–O–)4Si with no OH group attached to the silicon atom (Q4). The second peak appearing around −102 ppm is assigned to (–O–)3Si(OH) with one OH group (Q3). The last peak around −90 ppm is assigned to (–O–)2Si(OH)2 with two OH groups (Q2).18 These peaks were also detected in spectra of the Mo-TUD-1 samples, albeit with a shift of the −110 ppm peak to −112 ppm. Moreover, an additional peak is observed at −107 ppm, which might be attributed to the mentioned interaction of Si with Mo incorporated in the TUD-1 framework [(–O–)3Si–O–Mo].
The N2 adsorption/desorption isotherms of the calcined Mo-TUD-1 samples are presented in Fig. 2, left panel. In general, according to IUPAC classification, the hysteresis loops at relative pressure of 0.4–0.9 show characteristic features of type IV isotherms, representative for mesoporous materials,19 and also typical for TUD-1 materials.13 One can distinguish two characteristic types of hysteresis loops. In the first case at low metal loading (Mo-1), the loop is relatively narrow, the adsorption and desorption branches being almost vertical and nearly parallel (H1), which means that the isotherm is governed by delayed condensation. Pore filling and emptying appear to occur in a narrow range indicating uniform, near-cylindrical pores. In addition, no interconnectivity could be observed.
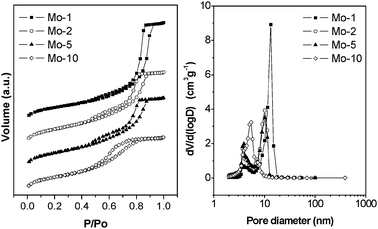 | ||
| Fig. 2 Left panel: N2 sorption isotherms of different Mo-TUD-1 samples. Right panel: the corresponding pore size distribution. | ||
At higher molybdenum loading the hysteresis loop becomes broad, the desorption branch being much steeper than the adsorption one, and the pore filling and emptying profile indicates the presence of a wide range of non-uniform pores (H3). This behaviour is explained by the formation of nano-particles of MoO3 inside the pores at higher loading. The porosity parameters are summarized in Table 1. The surface area of Mo-1 is smaller than that of the parent all silica TUD-1, but increases as a function of increasing molybdenum loading. At the same time, the mesopore diameter and the mesopore volume decrease (Fig. 2, right panel), which is consistent with the formation of molybdenum oxide crystals in the samples of high Mo-loading. Similar trends were also reported for Ti-TUD-1 and Cu-TUD-1.15,16
The high total surface area of samples with a relatively high Mo-loading is presumably the consequence of the introduction of micropores, as confirmed by the pore-size distribution depicted in the right panel of Fig. 2.
Fig. 3 illustrates the XRD patterns of various samples of Mo-TUD-1, as compared to the XRD pattern of MoO3. All Mo-TUD-1 patterns show a single intense diffraction line at 1–2° 2θ, which is common for mesoporous silica. XRD patterns of the samples with a low molybdenum loading (i.e. Si/Mo ratio <20) demonstrate that the prepared samples contain highly dispersed MoOx. Crystalline metal oxide phases, or other bulky dense phases, are absent in most of the samples. However at high molybdenum loading (Mo-10), the diffraction lines assigned to MoO3 can be observed, with a decrease in intensity of the diffraction line assigned to the mesoporous silica structure. This is the result of a decreasing integrity of the mesoporous structure because of the presence of the molybdenum oxide particles.
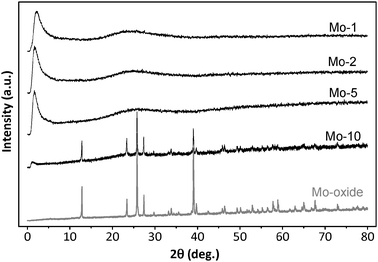 | ||
| Fig. 3 XRD patterns of the different Mo-TUD-1 samples compared with that of MoO3. | ||
The UV-Vis spectra of the calcined Mo-TUD-1 samples are compared to the MoO3 spectrum in Fig. 4. The spectra were collected under ambient (hydrated) conditions. The spectra are dominated by a peak around 245 nm, which is assigned to the ligand to metal charge transfer from O2− to Mo6+,20 and an indication of the presence of isolated Mo6+ moieties, either di-oxo [(O)2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Mo–(O–Si)2] or mono-oxo [OMo–(O–Si)4].21
Mo–(O–Si)2] or mono-oxo [OMo–(O–Si)4].21
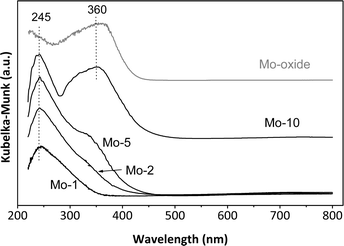 | ||
| Fig. 4 The UV-Vis spectra of the different Mo-TUD-1 samples compared with that of MoO3, spectra collected at ambient conditions. | ||
Moreover, a peak around 360 nm can be observed in the Mo-2, Mo-5 and Mo-10 samples, the intensity of which is increasing with increasing molybdenum loading. This peak is assigned to the presence of MoO3,22 either in the form of nano-crystals inside the pores of TUD-1 or in the form of crystalline MoO3 outside the framework.
The SEM images of a low Mo loading in TUD-1 are very similar to that of TUD-1 (not presented here). The micrographs show that the particles, when present, do not have a well-defined morphology. In general, the samples consist of about 5–50 μm irregularly-shaped particles. However, in samples containing a high metal loading, the Mo-TUD-1 structure is changed to less uniform (Fig. 5) than the appearance of the samples with low Mo loading. This suggests an influence of a high Mo concentration on the TUD-1 structure. In addition, the crystals of molybdenum oxide are clearly visible.
 | ||
| Fig. 5 SEM micrograph of the Mo-10 sample. White arrows indicate crystalline MoO3. | ||
The mesoporous and amorphous structure of the samples was further confirmed by HR-TEM (Fig. 6). The micrograph of the Mo-1 sample shows the irregular sponge-like characteristics of TUD-1.13 The presumably present nano-particles of molybdenum oxide did not appear in the HR-TEM images of Mo-2 or Mo-5 (although their presence was confirmed by UV-Vis and Raman).
 | ||
| Fig. 6 HR-TEM micrographs of Mo-1 sample (left) and Mo-10 sample (right). | ||
This is most likely because these particles reside deep inside the meso-structure. In Mo-10 the HR-TEM micrographs show extra-framework crystalline MoO3, with particle sizes of 200–500 nm.
Fig. 7 presents the Raman spectra of the Mo-TUD-1 samples at ambient (left panel) and under dehydrated conditions (right panel). Under hydrated conditions, two bands around 950 and 875 cm−1 are visible in the spectra of Mo-2 and Mo-5. These bands are characteristic of hydrated molybdate species on various supports, and have been identified on SiO2 supports by de Boer et al.,23 as well as by Wachs et al.24,25 Also Piquemal et al.26 identified Mo species in MCM-41 by Raman spectroscopy. The precise assignment of these bands is not straightforward. It has been proposed that the 950 cm−1 band is related to hydrated isolated MoO4 units dispersed in the pores of various supports (here, TUD-1). The assignment of the 875 cm−1 band is more ambiguous, and could be related to microcrystalline molybdates.27 On the other hand this band is assigned to Mo–O–Mo bridges, as well as to the presence of peroxo species associated with dispersed Mo-species. The spectrum of Mo-10 was dominated by sharp bands characteristic of crystalline MoO3 appearing at 997, 820 and 668 cm−1,28 whereas these bands were not detected in the other spectra (consistent with XRD and UV-Vis spectra).
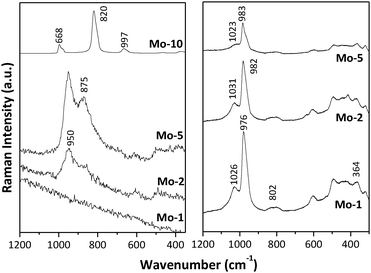 | ||
| Fig. 7 Raman spectra of the different Mo-TUD-1 samples, spectra collected at ambient conditions (left panel) and under dehydration conditions (right panel). | ||
In situ Raman spectra under dehydrated conditions are shown in Fig. 7 (right panel). The spectra of Mo-1, Mo-2 and Mo-5 are dominated by bands appearing at 976, 982, and 983 cm−1, respectively. These bands have been assigned to a dioxo highly dispersed Mo-species (Si–O)2Mo(O)2.24 The small shift in the band position is most likely related to the nature of the silica support.24 The O–Mo–O bending mode exhibits a band at 364 cm−1. The band around 1020–1030 cm−1 is assigned to the mono-oxo (Si–O)4Mo(O) species, again slightly shifted as compared to literature values.24
Combining all the characterization data, one can conclude that Mo-TUD-1 samples are amorphous, mesoporous, and contain a variety of molybdenum sites as a function of loading. Molybdenum is present in the Mo-1 sample predominantly as isolated Mo6+ species. These species are present as dioxo (Si–O)2Mo(O)2, as well as in the form of mono-oxo (Si–O)4Mo(O) species. In Mo-2 and Mo-5, molybdenum is present in isolated form, together with nano-particles of MoO3 presumably located in the mesopores of TUD-1. Finally, in Mo-10, extra-framework crystalline MoO3 particles were detected.
3.2. Catalytic performance
The catalytic activity of the different Mo-TUD-1 samples was evaluated in the epoxidation of cyclohexene with TBHP under similar experimental conditions reported earlier for Ti-TUD-1 at 313 K.14 The major products of the liquid phase epoxidation of cyclohexene with TBHP are cyclohexene oxide (desired), 2-cyclohexen-1-ol, 2-cyclohexen-1-one, 1,2-cyclohexanediol, and 1-(tert-butylperoxy)-2-cyclohexene (undesired).The results of cyclohexene epoxidation over Mo-TUD-1 at 313 K after 6 h are summarized in Fig. 8. When the reaction was performed in the presence of TUD-1, negligible values for the conversion of the cyclohexene, and yield of the epoxide were obtained. This indicates that the solid support employed for the synthesis of Mo-TUD-1 does not promote the epoxidation of cyclohexene. When MoO3 was applied to catalyze the reaction, 22% of cyclohexene and 71% of TBHP were converted after 6 h of reaction with low selectivity (i.e. 14%) towards the epoxide. This is in good agreement with the work of Gould and Rado29 and that of Carreiro and Burke30 where low selectivity of epoxide was obtained in cyclohexene oxidation by TBHP. In the work of Zhou et al.31 0% selectivity towards epoxide was obtained.
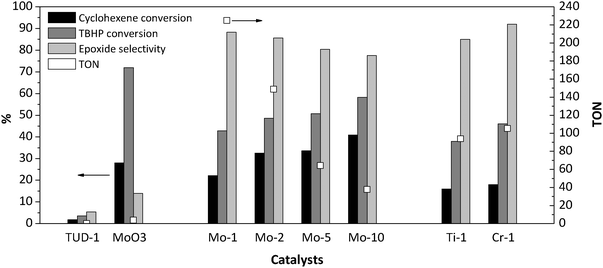 | ||
| Fig. 8 The catalytic performance of Mo-TUD-1 samples in the epoxidation of cyclohexane by TBHP at 313 K after 6 h. | ||
The conversion of both cyclohexene and TBHP is clearly significant when Mo-TUD-1 samples are used. Moreover, Fig. 8 shows that both, the conversion of cyclohexene and TBHP, increase with increasing molybdenum loading. Because the mass of catalyst used in all experiments was kept constant, the TON number decreases with increasing molybdenum loading. In other words, the catalytic activity per molybdenum atom is decreased. This is most likely due to the formation of the MoO3 particles inside and/or outside the TUD-1 framework, hence reducing the number of accessible surface molybdenum sites. The selectivity of cyclohexene oxide is also decreased as a function of Mo-loading. This is likely to be caused by an increasing extent of leaching of molybdenum, which promotes the formation of undesired products.29,30
The Mo-1 sample was chosen to conduct a stability and reusability study, and further evaluate the leaching behavior. Elemental analysis of the recovered solid was carried out to investigate the Si/Mo ratio. After the first run, elemental analysis showed that almost 18% of molybdenum had leached from the framework. However, thereafter the Mo content (wt%) was almost constant in consecutive runs (Fig. 9). In these runs, the conversion was slightly less than that of the fresh sample, whereas the selectivity towards cyclohexene oxide was slightly improved, and the total TON increased. Following the first run, the Mo-TUD-1 catalyst thus showed reproducible performance in consecutive runs, up to the 4th. This is consistent with the leaching of molybdenum from SBA-15 reported by Melero et al.32 The leaching of labile Mo species does not affect the catalytic activity of Mo-TUD-1, and a true heterogeneous catalyst is obtained after the first run.
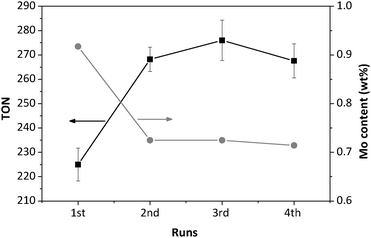 | ||
| Fig. 9 Recycling of the Mo-1 sample; the Mo content and TON as a function of the number of runs. | ||
Comparing the performance of the Mo-1 catalyst to that of other TUD-1 samples containing 1 wt% of active phase (i.e. Ti-1 and Cr-1, see for synthesis details ref. 15 and 33) Mo-1 demonstrates the highest TON (Fig. 8). Mo-1 was almost two times more active than Ti-1 or Cr-1. These results are in agreement with the fact that Mo(VI) is more active than Ti(IV) and Cr(VI) in olefin epoxidation.4 Furthermore, the stability of Cr-supported silica is a major challenge. The activity of the Mo-TUD-1 catalyst cannot be easily compared to other molybdenum containing mesoporous materials, because of differences in reaction conditions. In general, the work of Ziolek and Volta et al. showed that Mo-MCM-41 and Mo-SBA-15 exhibit a very poor epoxide selectivity i.e. approximately 10%, when H2O2 was used as an oxidant and acetonitrile as a solvent.11,12 However, when TBHP was used as an oxidant, a higher selectivity towards epoxide was obtained, albeit at a low rate (8% conversion after 2 h).34 Moreover, a conversion of 36% with 96% selectivity towards epoxide was obtained after 15 hours when cyclohexene was reacted with TBHP over Mo-silicalite,35 while a conversion of 20% and a selectivity of 60% towards epoxide were obtained over NaY.
From the previous data it is obvious that the catalytic performance of Mo-TUD-1 is superior to other metal-containing TUD-1 (e.g. Ti and Cr) catalysts or other Mo-containing mesoporous materials (e.g. Mo-MCM-41 and Mo-SBA-15). A similar conclusion was recently reached by Yang et al.36 where V-TUD-1 showed a higher catalytic performance in cyclo-octene epoxidation as compared to V-MCM-41 or V-SBA-15.
Mo-TUD-1, with its three dimensional structure and great accessibility to Mo(VI) active sites, seems to be a promising heterogeneous catalyst for liquid phase cyclohexene epoxidation with TBHP under mild reaction conditions, although leaching in the first run requires attention.
4. Conclusions
Molybdenum was successfully incorporated into the TUD-1 matrix via a surfactant-free pathway using triethanolamine as a template. Up to a molybdenum loading of 5 wt%, the molybdenum species are homogeneously distributed, without forming a segregated crystalline MoO3 phase, as evidenced by XRD, Raman, and HR-TEM data. In the case of a high molybdenum content >5 wt%, a separate phase of MoO3 was detected. A low loading of Mo-TUD-1 results in the highest TON in cyclohexene epoxidation with TBHP and yields the best selectivity, i.e. 88% towards cyclohexene oxide. Molybdenum is partially leached from the framework of TUD-1 in the first run. Despite this phenomenon, the catalyst performed efficiently up to a fourth run.Acknowledgements
The authors thank Dr S. Choi, and Dr I. E. Wachs, Lehigh University, USA, for assistance in recording and interpretation of the in situ Raman measurements under dehydrated conditions. Dr P. J. Kooyman, TU Delft, The Netherlands, is greatly acknowledged for the HR-TEM micrographs and Dr J. Groen for the N2 sorption measurements.References
- M. Beller and C. Bolm, Transition Metals for Fine Chemicals and Organic Synthesis, Wiley-VCH, Weinheim, 1998, vol. 2, pp. 261–266 Search PubMed.
- T. Sreethawong, Y. Yamada, T. Kobayashi and S. Yo-Shikaw, J. Mol. Catal. A: Chem., 2005, 241, 23–32 CrossRef CAS.
- R. A. Sheldon and J. A. Kochi, Metal-catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- R. A. Sheldon, J. Mol. Catal., 1980, 7, 107–126 CrossRef CAS.
- M. Breuer, K. Dietrich, T. Habicher, B. Hauer, M. Kesseler, R. Sturmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788–824 CrossRef CAS.
- Y. Sui, X. R. Zeng, X. N. Fang, Y. A. Ziao, L. Chen, M. H. Li and S. Cheng, J. Mol. Catal. A: Chem., 2007, 270, 61–67 CrossRef CAS.
- St. V. Kotov, St. Boneva and Ts. Kolev, J. Mol. Catal. A: Chem., 2000, 154, 121–129 CrossRef CAS.
- U. Arnold, R. Serpa da Cruz, D. Mandelli and U. Schuchardt, J. Mol. Catal., 2001, 165, 149–158 CrossRef CAS.
- A. Antinollo, P. Canizares and F. Carrillo-Hermosilla, Appl. Catal., A, 2000, 193, 139–146 CrossRef.
- C. T. Kresage, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef.
- B. Kilos, M. Aouine, I. Nowak, M. Ziolek and J. C. Volta, J. Catal., 2004, 224, 314–325 CrossRef CAS.
- B. Kilos, I. Nowak, M. Ziolek, A. Tuel and J. C. Volta, Stud. Surf. Sci. Catal., 2005, 158, 1461–1468 CrossRef.
- J. C. Jansen, Z. Shan, L. Marchese, W. Zhou, N. Puil and T. Maschmeyer, Chem. Commun., 2001, 713–714 RSC.
- Z. Shan, E. Gianaotti, J. C. Jansen, J. A. Peters, L. Marchese and T. Maschmeyer, Chem.–Eur. J., 2001, 7, 1437–1443 CrossRef CAS.
- M. S. Hamdy, O. Berg, J. C. Jansen, T. Maschmeyer, J. A. Moulijn and G. Mul, Chem.–Eur. J., 2006, 12, 620–628 CrossRef.
- M. S. Hamdy, G. Mul, W. Wei, R. Anand, U. Hanefeld, J. C. Jansen and J. A. Moulijn, Catal. Today, 2005, 110, 264–271 CrossRef CAS.
- M. Stocker, Stud. Surf. Sci. Catal., 1994, 85, 429–507 CrossRef CAS.
- K. J. Mackenzie and M. E. Smith, Multinuclear solid-state NMR of inorganic materials, Pergamon, 2002 Search PubMed.
- K. Sing, D. Everett, R. Haul, L. Moscou, R. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- S. Higashimoto, Y. Hu, R. Tsumura, K. Iino, M. Matsuoka, H. Yamashita, Y. Shul, M. Che and M. Anpo, J. Catal., 2005, 235, 272–278 CrossRef CAS.
- Z. Li, L. Gao and S. Zheng, Appl. Catal., A, 2002, 236, 163–171 CrossRef CAS.
- M. S. Morey, J. D. Bryan, S. Schwarz and G. D. Stucky, Chem. Mater., 2000, 12, 3435–3444 CrossRef CAS.
- M. de Boer, A. J. van Dillen, D. C. Koningsberger, J. W. Geus, M. A. Vuurman and I. E. Wachs, Catal. Lett., 1991, 11, 227–240 CrossRef CAS.
- E. L. Lee and I. E. Wachs, J. Phys. Chem. C, 2007, 111, 14410–14425 CAS.
- H. Tian, C. A. Roberts and I. E. Wachs, J. Phys. Chem. C, 2010, 114, 14110–14120 CAS.
- J. Piquemal, J. Manoli, P. Beaunier, A. Ensuque, P. Tougne, P. Legrand and J. Bregeault, Microporous Mesoporous Mater., 1999, 29, 291–304 CrossRef CAS.
- F. D. Hardcastle and I. E. Wachs, J. Raman Spectrosc., 1990, 21, 683–691 CrossRef CAS.
- A. N. Desikan, L. Huang and S. T. Oyama, J. Phys. Chem., 1991, 95, 10050–10056 CrossRef CAS.
- E. S. Gould and M. Rado, J. Catal., 1969, 13, 238–244 CrossRef CAS.
- E. Carreiro and A. J. Burke, J. Mol. Catal. A: Chem., 2006, 249, 123–128 CrossRef CAS.
- Q. N. Zhang, J. Zhang, T. H. Wu and X. P. Zhou, Catal. Commun., 2009, 10, 1279–1283 CrossRef CAS.
- J. A. Melero, J. Iglesias, J. M. Arsuaga, J. Sainz-Pardo, P. de Frutos and S. Blazquez, Appl. Catal., A, 2007, 331, 84–94 CrossRef CAS.
- M. S. Hamdy, O. Berg, J. C. Jansen, Th. Maschmeyer, A. Arafat, J. A. Moulijn and G. Mul, Catal. Today, 2006, 117, 337–342 CrossRef CAS.
- J. Jarupatrakorn, M. P. Coles and T. D. Tilley, Chem. Mater., 2005, 17, 1818–1828 CrossRef CAS.
- M. Masteri-Farahani, F. Farzaneh and M. Ghandi, J. Mol. Catal. A: Chem., 2003, 192, 103–111 CrossRef CAS.
- Z. Guo, C. Zhou, S. Hu, Y. Chen, X. Jia, R. Lau and Y. Yang, Appl. Catal., A, 2012, 419–420, 194–202 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |