Syngas production by CO2 reforming of methane using LnFeNi(Ru)O3 perovskites as precursors of robust catalysts
Svetlana
Pavlova
*a,
Leyla
Kapokova
a,
Rimma
Bunina
a,
Galina
Alikina
a,
Nataliya
Sazonova
a,
Tamara
Krieger
a,
Arkadii
Ishchenko
a,
Vladimir
Rogov
ab,
Roman
Gulyaev
a,
Vladislav
Sadykov
ab and
Claude
Mirodatos
c
aBoreskov Institute of Catalysis SB RAS, Novosibirsk, Russia. E-mail: pavlova@catalysis.ru
bNovosibirsk State University, Novosibirsk, Russia
cInstitute de Recherches sur la catalyse et l'environnement de Lyon, France
First published on 13th July 2012
Abstract
LnFeNi(Ru)O3 perovskites synthesized via a modified Pechini method have been studied as catalyst precursors in CO2 reforming of methane. The structural and redox properties along with catalytic performance were found to depend upon Ln nature and Ru content in the perovskite. Active and stable catalysts have been formed from perovskite precursors in reaction media at high temperatures due to Fe–Ni–(Ru) alloy particles segregation from the perovskite lattice and their stabilization on the oxide surface. Coking stability of these catalysts in dry reforming of real natural gas containing admixtures of higher hydrocarbons is explained by combined effects of Ni dilution by Ru in alloy nanoparticles and high mobility and reactivity of surface/lattice oxygen in the disordered Ln ferrite support.
1. Introduction
The carbon dioxide reforming of methane has recently attracted considerable attention as a process generating synthesis gas due to both environmental and commercial reasons.1,2 Dry reforming transforms cheap undesirable greenhouse gases and is particularly important in the case of biogas or gas fields containing a significant amount of methane and carbon dioxide. The synthesis gas produced in this reaction having the H2/CO ratio close to unity is a favorable feedstock for further chemical processes such as the Fischer–Tropsch synthesis and production of oxygenates followed by the liquid fuel synthesis.3–7The most active catalysts for methane dry reforming (MDR) are based on the noble metals (Rh, Ru, Ir, Pt) or nickel.3–12 The major problem of MDR hindering its industrial application is the coking of catalysts and, as a result, their deactivation in the case of Ni-containing catalysts.3–9 Nevertheless, due to much lower cost of nickel compared with noble metals, the development of Ni-based catalysts resistant to coking is profitable from the commercial point of view. Different approaches have been considered up to now to minimize the coke deposition on catalysts.3–9,13–24 Among them an attractive option is the use of perovskite-like oxides ABO3 as the catalyst precursors when formation of highly dispersed active metal fixed in the oxide matrix occurs under MDR reaction conditions as a result of perovskite reduction.13–24 The structural and thermal stability as well as catalytic activity of such systems can be adjusted by the substitution with various cations on A and/or B sites. For Ni-containing perovskite-type oxides of general formula  (A, A′ – La, Ca, Sr, Ce, Pr; B′ – Fe, Co, Ru; x, y – the substitution degree) as precursors of MDR catalysts, a high dispersion of Ni depending on the nature of A, A′ and/or B′ cations as well as participation of mobile oxygen of the oxide matrix in the reaction thus preventing the catalyst coking and increasing its activity and stability have been demonstrated.13–24 For example, in the case of La1−xAxNiO3 (A = Ce, Pr) perovskites, La0.9Pr0.1NiO3 demonstrated the highest catalytic activity and resistance to coking in MDR explained by a higher dispersion of Ni in the reduced catalyst as well as redox properties of praseodymium oxide.16 For Ln1−xCaxRu0.8Ni0.2O3 (Ln = La, Sm, Nd) catalysts, change in the nature and composition of cations in the A-site was shown to strongly affect stability and selectivity of catalysts.22 A partial substitution of Ni with Co,17,18 Fe19–21 and/or Ru22–26 leading to formation of Ni–Me alloys under reducing MDR reaction conditions improves catalyst activity and stability. Thus, for the LaNi1−xRuxO3 (x = 0, 0.1, 0.2, 1) catalysts, partial substitution of Ni with Ru hampers coking.24
(A, A′ – La, Ca, Sr, Ce, Pr; B′ – Fe, Co, Ru; x, y – the substitution degree) as precursors of MDR catalysts, a high dispersion of Ni depending on the nature of A, A′ and/or B′ cations as well as participation of mobile oxygen of the oxide matrix in the reaction thus preventing the catalyst coking and increasing its activity and stability have been demonstrated.13–24 For example, in the case of La1−xAxNiO3 (A = Ce, Pr) perovskites, La0.9Pr0.1NiO3 demonstrated the highest catalytic activity and resistance to coking in MDR explained by a higher dispersion of Ni in the reduced catalyst as well as redox properties of praseodymium oxide.16 For Ln1−xCaxRu0.8Ni0.2O3 (Ln = La, Sm, Nd) catalysts, change in the nature and composition of cations in the A-site was shown to strongly affect stability and selectivity of catalysts.22 A partial substitution of Ni with Co,17,18 Fe19–21 and/or Ru22–26 leading to formation of Ni–Me alloys under reducing MDR reaction conditions improves catalyst activity and stability. Thus, for the LaNi1−xRuxO3 (x = 0, 0.1, 0.2, 1) catalysts, partial substitution of Ni with Ru hampers coking.24
In our previous study of LnFe0.7Ni0.3O3−δ (Ln = La, Pr, Sm) perovskites27 under realistic conditions of MDR, their transformation into composites comprised of Ni–Fe alloy particles and LnOx epitaxially bound with remaining Ln–Fe–O perovskite particles was demonstrated. This microstructure plays an important role in MDR through activation of CO2 on the oxide sites and transfer of active oxygen-containing species to alloy particles where they interact with CHx fragments producing syngas. The nature of Ln cation affects both the composition of the Ni–Fe alloy and oxygen mobility/reactivity in perovskites, PrFe0.7Ni0.3O3−δ being the most active and stable catalyst.
This work presents results on synthesis and characterization of structural and catalytic properties in dry reforming of LnFe1−xRuxNi0.3O3−δ (Ln = La, Pr, x = 0–0.1) perovskites as catalyst precursors. The impact of the Ln cation nature and partial substitution of Fe with Ru on the real structure of perovskites, their reduction features and catalytic activity and stability in MDR has been elucidated.
2. Experimental
Perovskites were synthesized by a Pechini route using metal (Me) nitrates, Ru(OH)Cl3, citric acid (CA), ethylene glycol (EG) and ethylenediamine (ED) as reagents.28,29 The molar ratio of CA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EG:ED:Me was 3.75:11.25:3.75:1. CA and metal nitrates were dissolved in ethylene glycol at 80 °C and in distilled water at room temperature, respectively. The prepared solutions were mixed together at room temperature under stirring followed by addition of ED. The mixed solution was stirred for 60 min and then heated at 70 °C for 24 h allowing for the gel formation. The gel was calcined at 600 °C for 2 h, then the obtained solid was ground and annealed at 900 °C.
:EG:ED:Me was 3.75:11.25:3.75:1. CA and metal nitrates were dissolved in ethylene glycol at 80 °C and in distilled water at room temperature, respectively. The prepared solutions were mixed together at room temperature under stirring followed by addition of ED. The mixed solution was stirred for 60 min and then heated at 70 °C for 24 h allowing for the gel formation. The gel was calcined at 600 °C for 2 h, then the obtained solid was ground and annealed at 900 °C.
The samples of perovskites were characterized by XRD, BET, TEM with EDX, H2 and CH4 temperature-programmed reduction. X-ray diffraction patterns were obtained with an ARLX′TRA diffractometer (Thermo, Switzerland) using CuKα radiation in the 2θ scanning range of 15–80° with a step of 0.05°. The structural refinement has been carried out using PCW software (version 2.4). Qualitative phase analysis has been carried out by using PDF-2–ICDD files and the ICSD/retrieve database. BET specific surface area (SSA, m2 g−1) was determined from the data on Ar thermodesorption. The TEM micrographs were obtained with a JEM-2010 instrument (lattice resolution 1.4 Å and acceleration voltage 200 kV). Local elemental analysis was performed with an EDX method (a Phoenix Spectrometer). The X-ray photoelectron spectra (XPS) were recorded on a photoelectron spectrometer, KRATOS ES 300, with Mg Kα radiation (1253.6 eV) at pressure 1 × 10−8 Torr. Preliminarily all samples were ground in an agate mortar and deposited on conductive carbon tape.
The redox properties of perovskites were studied using temperature-programmed reduction (TPR) by H2 (10% H2 in Ar, feed rate 2.5 L h−1, temperature ramp from 25 to 900 °C at 10 °C min−1) or CH4 (1% CH4 in He, feed rate 10 L h−1, temperature ramp from 25 to 880 °C at 5 °C min−1). The experiments were carried out in kinetic installations equipped with GC Tcvet-500 and a PEM-2M analyzer (IR absorbance and electrochemical gas sensors). In the case of repeated red–ox cycles, samples were reoxidized in the flow of O2 for 1 h at 500 °C.
Methane dry reforming (MDR) was studied in the temperature-programmed mode (1% CH4 + 1% CO2 in He, contact time 0.005 s) as well as in steady-state conditions (10% CH4 + 10% CO2 in He, contact time 0.01–0.015 s) at temperatures up to 800–850 °C in a flow installation. The long-term testing was performed in the reaction mixture 45% CO2 + 45% natural gas +10% N2 at 800 °C and contact time 0.24 s. Prior to testing, the catalysts were activated in the reaction media at 850 °C for 2 hours. The concentrations of reactants and products were analyzed by using GC Tcvet-500. Temperature-programmed oxidation (TPO) in the feed of 1% O2 in He was used to evaluate the amount of carbon formed over catalysts during long-term testing.
3. Results and discussion
3.1. Structural and textural features
The X-ray diffraction patterns for the initial samples are shown in Fig. 1 and 2. While samples without Ru (PrFe0.7Ni0.3O3−δ and LaFe0.7Ni0.3O3−δ) are single-phase orthorhombic perovskites,27 the partial substitution of Fe with Ru in LnFe0.7−xNi0.3RuxO3−δ (x = 0.05, 0.1) leads to appearance of NiO and the praseodymium or lanthanum oxychloride [JCPDF 88-0064] phase and distortion of perovskite structure (Table 1). The formation of oxychlorides is caused by using Ru(OH)Cl3 as a source of Ru, which is confirmed by the increase in the oxychloride reflections intensity with increase in Ru concentration, being more pronounced for La-containing samples. Simultaneously, especially in the case of LaFe0.7−xNi0.3RuxO3−δ samples, the noticeable growth of NiO reflections is observed (Fig. 1). The shift of all perovskite reflections towards lower angles shows the unit cell expansion due to partial segregation of nickel from perovskite and insertion of larger Ru3+ cations into the structure (0.068 nm versus 0.06 and 0.0645 nm for Ni3+and Fe3+30). | ||
Fig. 1 XRD patterns of LaFe0.7−xRuxNi0.3O3−δ: as prepared (1–3); reduced in H2 (4–6); x = 0 (1, 4); x = 0.05 (2, 5); x = 0.1 (3, 6). NiO ( ), LaOCl (▼), La2O3 ( ), LaOCl (▼), La2O3 ( ), Ni–Fe alloy (◆), Ni–Fe–Ru alloy (*), Ru (+), Ru–Ni alloy (●). Reduction conditions: 5% H2 in He, 800 °C, 2 h. ), Ni–Fe alloy (◆), Ni–Fe–Ru alloy (*), Ru (+), Ru–Ni alloy (●). Reduction conditions: 5% H2 in He, 800 °C, 2 h. | ||
 | ||
Fig. 2 XRD patterns of PrFe0.7Ni0.3O3−δ (1, 3, 5) and PrFe0.6Ru0.1Ni0.3O3−δ (2, 4, 6): as prepared (1, 2), reduced in H2 (3, 4) and used in MDR (5, 6). NiO ( ), PrOCl (▼), Pr2O3 (v), PrxOy (#), Ni–Fe alloy (◆). Reduction conditions: 5% H2 in He, 800 °C, 2 h. MDR conditions: 800 °C in the feed 50% CO2 + 50% natural gas, contact time 0.2 s. ), PrOCl (▼), Pr2O3 (v), PrxOy (#), Ni–Fe alloy (◆). Reduction conditions: 5% H2 in He, 800 °C, 2 h. MDR conditions: 800 °C in the feed 50% CO2 + 50% natural gas, contact time 0.2 s. | ||
| Perovskite | a (Å) | b (Å) | c (Å) | V (Å3) | Secondary phase | SSA, m2 g−1 |
|---|---|---|---|---|---|---|
| a The structural refinement has been carried out using PCW software (version 2.4). Qualitative phase analysis has been carried out by using PDF-2–ICDD files and the ICSD/retrieve database. b From the data on Ar thermodesorption. | ||||||
| LaFN | 5.511 | 7.808 | 5.537 | 238.25 | — | 7 |
| LaFNRu0.05 | 5.529 | 7.817 | 5.547 | 239.74 | LaOCl, NiO | 7 |
| PrFN | 5.522 | 7.742 | 5.462 | 233.54 | — | 4 |
| PrFNRu0.05 | 5.574 | 7.779 | 5.480 | 237.61 | PrOCl, NiO | 7 |
| PrFNRu0.1 | 5.584 | 7.777 | 5.478 | 237.89 | PrOCl, NiO | 4 |
XRD patterns of perovskites reduced in H2 at 800 °C (Fig. 1 and 2) show the reflections in the range of 2θ ∼ 44–44.5° corresponding to the Ni–Fe(Ru)-alloy27 and metal Ru,23 as well as those of orthorhombic perovskite, rare-earth oxides (La2O3 [JCPDF 83-1344] or Pr2O3, PrxOy) and lanthanum or praseodymium oxychloride. The position and intensity of perovskite reflections are practically the same as in the initial and reduced samples showing, in agreement with literature data,22–24 that reduction of Ru-containing perovskites is hampered as compared with Ru-free samples.27 However, the presence of La2O3(Pr2O3, PrxOy) reflections indicates some decomposition of perovskites considering the invariance of La(OH)Cl2 reflections and only some decrease in Pr(OH)Cl2 reflection intensity after reduction. The reflections of the metal phase are broad and of low intensity, which makes their analysis difficult. However, some information on the composition of the metal phase could be deduced. The reflections of the Ni–Fe alloy shift to higher angles with the increase in Ru concentration due to its incorporation into the alloy. Thus, in the case of the LaFe0.65Ni0.3Ru0.05O3−δ sample, the position of alloy reflection is rather close to that of the Ru-free Ni–Fe alloy (2θ = 44°) showing only a small amount of Ru in the alloy. In the XRD pattern of the LaFe0.6Ni0.3Ru0.1O3−δ sample, the reflection at 2θ = 44.2° indicates the presence of the Ni–Fe alloy enriched with Ru, the reflection at 44.35° corresponds to pure metal Ru and the reflection at 44.5° corresponds to the Ni–Ru alloy. The presence of metal Ru in this sample implies that a part of ruthenium in the oxidized sample is not incorporated into the perovskite structure, though in the XRD pattern of the initial LaFe0.6Ni0.3Ru0.1O3−δ perovskite no reflections of individual ruthenium oxide are detected.
3.2. H2 TPR
H2 TPR patterns for reference samples (LaNiO3 and NiO) and perovskites are presented in Fig. 3 and 4. An Asymmetric Gauss Decomposition method (AGDM) has been applied for the analysis of complex H2-TPR profiles of perovskites. A single peak with a maximum at 400 °C is observed in the H2 TPR pattern of NiO whereas LaNiO3 shows two peaks: the first one centered near 337 °C is assigned to the reduction of Ni3+ to Ni2+ and the second peak at 495 °C corresponds to the reduction of Ni2+ to metallic nickel (Fig. 3).17,21,24 For LaFe0.7Ni0.3O3−δ, in agreement with known data,19–21,27 the first peak shifts to lower temperatures and the second one (corresponding to a partial reduction of perovskite) shifts to higher temperatures (Fig. 3). Furthermore, the data of AGDM show the presence of a broad low-intensity peak in the range of 300–600° which could be assigned to the reduction of NiO traces not detected by XRD and the partial reduction of Fe4+ to Fe3+.20,21,27 For LaFe0.7−xNi0.3RuxO3−δ samples (Fig. 3), in the low-temperature region, three peaks of hydrogen consumption are observed: the low intensity peak at 260–270 °C and two overlapping peaks with the maximum at 315–370 °C and 400–410 °C. The first peak and the peak at 315–370 °C could be assigned to the reduction of Ni3+ to Ni2+ and Ru3+ to Ru2+ in perovskite.24,31 The peak at 400–410 °C corresponds to the reduction of NiO: the increase in its intensity at x = 0.1 agrees with XRD data evidencing a higher NiO content in this sample (Fig. 1). The low-intensity peaks in the 500–700 °C region could be assigned to the reduction of Fe4+ cations to Fe3+ within the perovskite lattice.20,21,27 An unresolved, intense, high-temperature peak above 700 °C is attributed to the partial destruction of perovskite structure with the formation of a metallic phase and La2O3 as confirmed by XRD data (Fig. 1). In the case of PrFe0.7−xNi0.3RuxO3−δ series, the TPR profiles are similar (Fig. 4), however, as compared with La-based perovskites, the low-temperature peaks are somewhat shifted to higher temperatures while the high-temperature peaks are shifted to lower temperatures. The higher temperature of the partial reduction of Ni3+ (Ru3+) cations in Pr-based perovskites agrees with the data for Ru-free systems.27,32,33 The shift of the high-temperature peak to lower temperatures is determined by the lower lattice stability of Pr-containing perovskites.33 | ||
| Fig. 3 H2 TPR profiles for: 1, NiO; 2, LaNiO3; 3, LaFe0.7Ni0.3O3−δ; 4, LaFe0.65Ru0.05 Ni0.3O3−δ; 5, LaFe0.6Ru0.1Ni0.3O3−δ. | ||
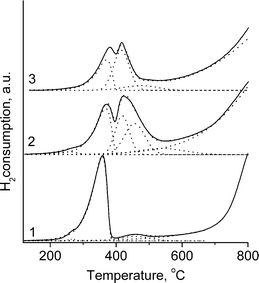 | ||
| Fig. 4 H2 TPR profiles for PrFe0.7−xRuxNi0.3O3−δ: 1, x = 0; 2, x = 0.05; 3, x = 0.1. | ||
In general, when ruthenium is incorporated into perovskite, the peak of the partial reduction of Ni3+ (Ru3+) is noticeably shifted to higher temperatures (from 315 to 370 °C and from 354 to 380 °C for LaFe0.7−xNi0.3RuxO3−δ and PrFe0.7−xNi0.3RuxO3−δ, respectively (Fig. 3 and 4)). Higher stability of both nickel and ruthenium in the perovskite lattice suggests strong inter-cations interaction which agrees with published data.24 Some poisoning effect of Cl− anions in the surface layer is possible as well.
Repeated reduction–reoxidation experiments revealed some evolution of reactivity of perovskites apparently reflecting variation in their real structure/microstructure. In the case of perovskites without the Ru reoxidized after the 1st TPR run, the first peak at ∼315 °C remains only as a shoulder of a new intense asymmetric peak with the main maximum at ∼420 °C. Since the amount of hydrogen consumed in these low-temperature peaks corresponds to complete reduction of all Ni in perovskite to the Ni0 state, this clearly indicates the increase in sample reactivity after reoxidation (Fig. 5a). Nearly identical H2 TPR spectra for the second and the third runs demonstrate reproducibility of sample reactivity after reoxidation. Since reduction of both NiO and Ni-containing perovskites to the metallic phase is a topochemical process including generation of Ni0 nuclei followed by their growth,35 observed variation in perovskites reactivity after red–ox cycles can be explained by microheterogeneity of reoxidized perovskites caused by preferential location of Ni cations in the surface layer and within domain boundaries of defect La ferrite. For the LaFe0.6Ni0.3Ru0.1O3−δ sample, TPR spectra change from run to run reflecting the increase in reactivity (Fig. 5b). In the third run, spectra correspond to practically complete reduction of all nickel and ruthenium cations contained in the sample to the metal state. Since the position of the main peak at ∼400 °C is the same as that for the LaFe0.7Ni0.3O3−δ sample, it clearly corresponds to the topochemical process of NiOx species reduction. Peaks at 180–230 °C are due to reduction of RuOx species formed as a result of ruthenium segregation from the perovskite during the first reduction,23,24 while peaks at 340–390 °C can be assigned to reduction of mixed Ni–Ru oxide species.34 The driving force for the increase of Ru-doped sample reactivity in red–ox cycles is clearly segregation of Ni and Ru in the surface layers as well as Cl removal from the surface.
 | ||
| Fig. 5 Repeated H2 TPR after oxidation for LaFe0.7Ni0.3O3−δ (a) and LaFe0.6Ru0.1Ni0.3O3−δ (b). | ||
3.3 CH4 TPR
Temperature-programmed reduction of perovskites by methane usually occurs at higher temperatures as compared with the reduction by H2 due to a strong C–H bond in CH4 molecules.27,35 The profiles of CH4 and products concentration during TPR for studied perovskites are presented in Fig. 6. The results show that incorporation of Ru in the perovskite strongly affects dynamics of its reduction. Thus, for LaFe0.7Ni0.3O3−δ (Fig. 6a) and PrFe0.7Ni0.3O3−δ27 reduction starts by formation of deep oxidation products (H2O or/and CO2) at 480–540 °C. CO and H2 evolution begins at 620–700 °C and is completed during TPR run resulting in segregation of the Fe–Ni alloy and the corresponding Ln oxide within the remaining perovskite.27 | ||
| Fig. 6 CH4 TPR for perovskites LnFe0.7−xRuxNi0.3O3−δ: (a) Ln = La, x = 0; (b) Ln = La, x = 0.05; (c) Ln = Pr, x = 0.05. | ||
For all Ru-containing perovskites, the start of reduction product evolution shifts to 730–810 °C, and concentration of CO and H2 passed through the maximum and slowly declined with time (Fig. 6b and c). In agreement with H2 TPR data, this evidences hampering of their reduction as compared with Ru-free perovskites. This could be caused by two reasons: inter-cation interaction stabilizing both nickel and ruthenium cations in the perovskite lattice24 and the presence of oxychlorides which could block the surface of perovskite hampering the active metal formation required for methane activation. Further, the dynamics of perovskite reduction and composition of products are affected by the nature of Ln cations. Thus, for PrFe0.65Ru0.05Ni0.3O3−δ, though the reduction products appear at higher temperature as compared with La-based perovskite (775 and 730 °C, correspondingly), the maximum rate of CO and H2 formation is achieved at somewhat lower temperature (820 °C vs. 830 °C), which could be due to a lower stability of Pr(OH)Cl2 basic salt due to a lower basicity of Pr cations. In addition, in the case of Pr-based perovskite, along with CO and H2, products of deep oxidation which are absent for La-containing perovskite are observed. This could be due to combustion of activated CHx species by highly reactive oxygen of PrOx.16,27
3.4. Methane dry reforming
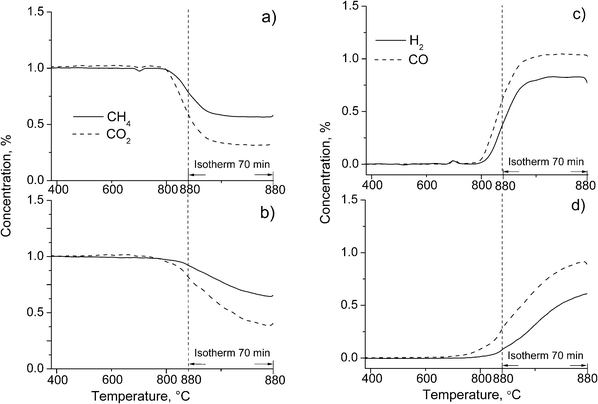 | ||
| Fig. 7 Typical curves for reagents (a, b) and products (c, d) concentration variation in the temperature-programmed reaction. The feed: 1% CH4 + 1% CO2 in He, contact time 0.001 s. | ||
 | ||
| Fig. 8 Temperature-programmed MDR: reaction starting temperature. Reaction conditions: 1% CH4 + 1% CO2 in He, contact time 0.001 s. | ||
Along with Ln nature, the activation process depends on the Ru content in the perovskites. The variation in methane and CO2 conversion during activation of PrFe1−xRuxNi0.3O3−δ at 850 °C in the reaction mixture 10% CH4 + 10% CO2 in He (Fig. 9) evidences that it is retarding with increasing Ru content in perovskite from 0.05 to 0.1% mol. This is conditioned by a bigger amount of oxychloride in the samples with higher Ru concentration as revealed by XRD data (Fig. 1). Thus, dynamics of catalyst activation during MDR reaction is clearly controlled by the same factors controlling their reduction with methane, namely, nickel and ruthenium inter-cation coupling in the perovskite lattice as well as the amount and stability of corresponding oxychloride.
 | ||
| Fig. 9 Time dependence of CH4 and CO2 conversion in MDR over oxidized PrFe0.7−xRuxNi0.3O3−δ (1, x = 0.05, 2, x = 0.1) at 850 °C, the feed 10% CH4 + 10% CO2, He – balance, contact time 0.015 s. | ||
 | ||
| Fig. 10 Temperature dependence of methane (squares) and CO2 (circles) conversion (a) and H2/CO ratio (b). Open symbols – equilibrium values, solid symbols – the catalyst PrFe0.65Ni0.3Ru0.05O3−δ activated in the reaction mixture at 850 °C. The feed 10% CH4 + 10% CO2, He – balance, contact time 0.015 s. | ||
To analyze the effect of perovskites chemical composition and their pretreatment on the catalytic activity the specific effective first-order rate constant of the methane transformation in MDR was estimated using the model of a plug-flow reactor.40 The values of specific rate constants of MDR at 800 °C for all catalysts activated in the reaction mixture and in the flow of H2 at 800 °C are shown in Fig. 11. These data show that, as in the case of Ru-free samples,27 the highest performance has been demonstrated by Pr-based catalysts, moreover, the activity of the catalysts increases with Ru concentration independently of the activation mode. However, the activity of catalysts activated in the reaction mixture is slightly lower due to the presence of PrOCl as detected by XRD (Fig. 2). The pretreatment of catalysts in hydrogen leads to segregation of active metal alloy particles and partial decomposition of PrOCl blocking active centers that increases the catalyst activity. Note that the values of specific rate constants for the samples activated in the reaction mixture and reduced samples are close due to comparatively easy decomposition of PrOCl which is completely destroyed during long term testing in the real reaction mixture (Fig. 2).
 | ||
| Fig. 11 Specific rate constant of CH4 dry reforming over perovskites reduced at 800 °C in H2 or activated in the reaction mixture. The feed 10% CH4 + 10% CO2, He – balance, 800 °C, contact time 0.01 s. | ||
In contrast to Pr-containing samples, for LaFe1−xRuxNi0.3O3−δ catalysts, the activity order strongly depends on the activation mode. Thus, if the catalysts are activated in the reaction media, the next row of activity is observed: LFN > LFNRu0.05 > LFNRu0.1 (Fig. 11). In this case, the decrease of activity with increasing Ru concentration is caused by inter-cation coupling stabilizing both nickel and ruthenium cations in the perovskite lattice24 as well as the rising content of stable LaOCl as shown by XRD data (Fig. 1). The particles of LaOCl blocking the perovskite precursor surface hinder the formation of active metal centers. The results evidencing the poor activation of Ru-containing LaFe1−xRuxNi0.3O3−δ samples in the reaction feed 10% CH4 + 10% CO2 in He agree with CH4-TPR and MDR-TPR results (Fig. 6 and 7). The reversed activity order of catalysts is observed after the reductive activation in hydrogen. According to XRD data (Fig. 1), the perovskite reduction in H2 at 800 °C leads to segregation of metal alloy particles which are the active centers for methane activation, and the activity of catalysts increases with Ru content.
 | ||
| Fig. 12 Methane conversion and H2/CO ratio in long-term tests of PrFe0.7Ni0.3O3 (■), PrFe0.65Ru0.05Ni0.3O3 (▲) and LaFe0.65Ru0.05Ni0.3O3 (O). Dot line – equilibrium value. Reaction conditions: the feed 45% CO2 + 45% natural gas + 10% N2 at 800 °C, pretreatment in reaction media 2 h at 850 °C, contact time 0.24 s. | ||
The amount of carbon deposits on Ru-containing catalysts during long-term testing has been determined using TPO. The results (not presented for brevity) show no CO2 evolution up to 880 °C evidencing no coke formation over the catalysts during the reaction. The absence of any type of coke on the used catalysts has been confirmed by TEM study of PrFe1−xRuxNi0.3O3 samples (Fig. 13).

In agreement with XRD, TEM data show that the PrFe0.6Ru0.1Ni0.3O3 catalyst is comprised of Ni–Ru [JCPDF 06-663] nanoparticles (marked by 1 in Fig. 13a, lattice spacing 2.08 Å) strongly interacting with the surface of Pr ferrite PrFeO3 [JCPDF 47-0065] domains (marked by 2 in Fig. 13a, (021) lattice spacing 2.66 Å). In the case of PrFe0.65Ru0.05Ni0.3O3, small alloy clusters are observed with a good epitaxy with the oxide particle containing both Pr and Fe (Fig. 13b), thus corresponding to the perovskite phase ((020) lattice spacing 2.79 Å). This certainly provides strong metal–support interaction required for preventing sintering and coking. Indeed, no carbon deposits or fibers were detected by TEM in catalysts discharged after reaction.
The XPS study of PrFe0.7−xRuxNi0.3O3−δ perovskites shows that the surface composition of catalysts does not vary appreciably after contact with the reaction feed (Table 2). The initial perovskite contains oxidized forms of ruthenium (III, IV) according to the well-defined Ru3d5/2 peak observed at 282.6 eV (Fig. 14). The low-intense component at 280.5 eV could be assigned to very small (<2 nm) clusters characterized by noticeable relaxation processes in the photoemission that leads to shift of Ru3d peaks. The samples tested in MDR have mainly metal Ru in the surface as indicated by the well-defined Ru3d5/2 peak observed at 280.0 and 280.3 eV for PrRu0.1 and PrRu0.05 samples, respectively. Mathematical analysis with Doniach–Sunjic and Gauss–Lorentz function revealed the presence of a low intensity component at 282.6 eV, which can be assigned to the Ru (III, IV) state. Since samples were transferred through air after discharging from the reactor, this feature can be explained by their partial oxidation due to this manipulation.
 | ||
| Fig. 14 The profile of Ru 3d core-level spectra of PrFe0.6Ru0.1Ni0.3O3−δ perovskites: initial (a) and used (b). | ||
The oxidation state of Fe is identified by the Fe2p3/2 core-level peak as corresponding to Fe(III). It remained almost unchanged after treatment in the reaction media followed by transfer through air. The state of Ni in the initial perovskite is characterized mainly by Ni2p3/2 peaks at 854.1 and 856.1 eV corresponding to Ni(II) and Ni(III) states (Fig. 15). The additional component at 852.5–852.7 eV, corresponding to the Ni0 state, appears in spectra of samples after MDR reaction. With due regard to the presence of mainly metal Ru as well as XRD and TEM data, this Ni metal state can correspond to the Ni–Ru alloy. In addition, the main part of Ni is present in Ni(II) and Ni(III) states as indicated by components at 854.5 and 856.3 eV respectively. This feature can be again caused by the partial reoxidation of Ni metal under contact of the discharged catalyst with air, pure Ni0 particles (generated by reduction of the NiO admixture) being oxidized much easier as compared with Ni–Ru alloy nanoparticles.
 | ||
| Fig. 15 The profile of Ni 2p3/2 core-level spectra of PrFe0.6Ru0.1Ni0.3O3−δ perovskites: initial (a) and used (b). | ||
Table 3 presents methane conversion, H2/CO ratio and amount of carbon deposits obtained at different GHSV and temperatures during MDR using an undiluted feed with CH4/CO2 = 1 for Pr(La)Fe0.65Ru0.05Ni0.3O3 catalysts and various catalysts reported in the literature.16,23,24,41–46 Note that the different conditions of catalysts testing make difficult the comparison of the catalysts activity, however, some conclusions could be drawn. The data show that performance of only Ni-based catalysts depends on the support nature thus the amount of coking changes from 1.2 (4.5%Ni/SiO2)41 to 67% (6.6%Ni/Al2O3)42 while it is 0.06% in the case of 1.5%Ru/CeO2–ZrO2.45 It is known that activity of Ni-containing catalysts and carbon formation over them are linked with the ensemble effect when, for reforming reaction, the ensemble of Ni atoms is smaller than that for nucleation of carbon whiskers,47 thus, active and stable catalysts for MDR must contain nickel particles of high dispersion.3–6,42 However, for catalysts based on pure nickel supported on different substrates sintering is a main problem which could be avoided by using alloys of nickel with other metals (e.g. Fe, Co, Ru).3–12 Highly dispersed active metal alloy species of various composition fixed in the oxide matrix are formed under MDR reaction conditions as a result of perovskite reduction.13–25 LaNi1−xRux (x = 0–0.2),24 Ln1−xCaxRu0.8Ni0.2O3 (Ln = La, Pr, Sm)23,25 have been used as precursors for bimetallic Ni–Ru catalysts containing metal particles distributed in the matrix of corresponding oxides (e.g. CaO–La2O3) which are formed after reductive treatment. Though such systems are sufficiently active and stable in MDR, the formation of filamentous coke is observed in the case of LaNi1−xRux catalysts with high Ni concentrations.24 As concerning Ln1−xCaxRu0.8Ni0.2O3, the high concentration of Ru makes this catalyst rather expensive.
| Sample | Methane conversion, % | H2/CO ratio | Carbon deposits, wt% | GHSV, L g−1 h−1 | T, °C | Ref. |
|---|---|---|---|---|---|---|
| a Carbon balance. | ||||||
| 6.6%Ni/Al2O3 | 68 | — | 67 | 22 | 700 | 41 |
| 4.5%Ni/SiO2 | 50 | — | 1.2 | 21 | 750 | 42 |
| 10%Ni/3% CeO2/3%La2O3/γ-Al2O3 | 77 | 1.09 | 17.5 | 12 | 800 | 43 |
| 10%Ni/8%Y2O3–ZrO2 | 70 | 0.7 | Coke | 36 | 800 | 44 |
| 1.5%Ru/CeO2–ZrO2 | 80 | 0.97 | 0.06 | 12 | 800 | 45 |
| La2NiO4/ZSM-5 | 63 | — | 20 | 48 | 700 | 46 |
| La0.9Pr0.1NiO3 | 48 | 0.81 | 0.0 | 60 | 700 | 16 |
| LaNi0.9Ru0.1O3 | 85 | 0.62 | 20.3 | 72 | 750 | 24 |
| La3.5Ru4O3 | 76 | 0.62 | 0.938 | 72 | 750 | 24 |
| LaRu0.8Ni0.2O3 | 79 | 0.94 | 98.2a | 48 | 700 | 23 |
| PrFe0.65Ru0.05Ni0.3 O3 | 90 | 0.76 | 0.0 | 22 | 800 | This work |
| LaFe0.65Ru0.05Ni0.3 O3 | 87 | 0.72 | 0.0 | 22 | 800 | |
The use of LnFe1−xNi0.3RuxO3−δ (Ln = La, Pr, x = 0–0.1) precursors with low nickel and ruthenium concentrations provides a high dispersion of Ni–Fe–Ru alloy species epitaxially bound with the oxide matrix and, hence, high activity and stability to coking and sintering. Furthermore, if activation of methane is determined by the metallic phase, activation of CO2 occurs with the participation of the oxide matrix and could be controlled by its red–ox properties.27,35 In contrast to ref. 23–25, where the perovskite phase completely decomposes under reaction conditions, the nickelate-ferrite phase used as a precursor in this work is only partly destroyed even after prolonged contact of catalysts with a concentrated feed at high temperatures. A stable perovskite phase along with alloy clusters appear to be the most important factors providing a high and stable performance. This can be explained by a high reactivity and mobility of the surface/lattice oxygen of these ferrite-type perovskites disordered due to reductive segregation of Ni and Ru.27,35 This helps to provide an efficient route of CO2 activation on the surface of perovskites and fast transfer of oxygen atoms or oxygen-containing species (hydroxyls, hydroxocarbonates, etc.) to the interface with segregated Ni–Fe–Ru alloy particles where they consume activated CHx fragments – coke precursors.
4. Conclusions
LnFeNi(Ru)O3 perovskites are promising precursors of active and stable-to-coking catalysts of CO2 reforming of methane. Under reaction conditions, perovskites are transformed into nanocomposites comprised of Ni–Fe(Ru) alloy particles and LnOx epitaxially bound with Ln–Fe–O perovskite particles. Partial substitution of iron by ruthenium gives rise to a synergetic effect between nickel and ruthenium, which is reflected in a higher stability of mixed perovskites to reduction as well as in a higher catalytic activity and stability to coking in MDR. These catalysts provide a high syngas yield and demonstrate resistance to coking for more than 20 hours in dry reforming of the real natural gas. The study of discharged catalysts showed that neither coke deposits nor fibers have been detected.Acknowledgements
Support by OCMOL FP7 Project, RFBR–CNRS 09-03-93112 Project and Russian Federal Innovation Agency via the program “Scientific and Educational cadres” is gratefully acknowledged.Notes and references
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1 CrossRef CAS.
- D. G. Vlachos and S. Caratzoulas, Chem. Eng. Sci., 2010, 65, 18 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev. Sci. Eng., 1999, 41(1), 1 CAS.
- J. J. Spivey and T. Inui, Catalysis, The Royal Society of Chemistry, 2002, vol. 16, p. 133 Search PubMed.
- Y. H. Hu and E. Ruckenstein, Adv. Catal., 2004, 48, 297 CrossRef CAS.
- A. P. E. York, T. Xiao, M. L. H. Green and J. Claridge, Catal. Rev. Sci. Eng., 2007, 49, 511 CAS.
- J. Ma, N. Sun, X. Zhang, N. Zhao, F. Xiao, W. Wei and Y. Sun, Catal. Today, 2009, 148, 221 CrossRef CAS.
- V. M. Gonzalez-Delacruz, F. Ternero, R. Pereniguez, A. Caballero and J. P. Holgado, Appl. Catal., A, 2010, 384, 1 CrossRef CAS.
- C. E. Daza, A. Kiennemann, S. Moreno and R. Molina, Appl. Catal., A, 2009, 364, 65 CrossRef CAS.
- M. M. V. M. Souza, D. A. G. Aranda and M. Schmal, J. Catal., 2001, 204, 498 CrossRef CAS.
- M. Safariamin, L. H. Tidahy, E. Abi-Aad, S. Siffert and A. Aboukais, C. R. Chim., 2009, 12, 748 CrossRef CAS.
- C. Carrara, A. Roa, L. Cornaglia, E. A. Lombardo, C. Mateos-Pedrero and P. Ruiz, Catal. Today, 2008, 133–135, 344 CrossRef CAS.
- J. Woo Nam, H. Chae, S. Ho Lee, H. Jung and K.-Y. Lee, Stud. Surf. Sci. Catal., 1998, 119, 843 CrossRef.
- T. Hayakawa, S. Suzuki and J. Nakamura, Appl. Catal., A, 1999, 183, 273 CrossRef CAS.
- G. Valderrama, M. R. Goldwasser, C. Urbina de Navarro, J. M. Tatibouet, J. Barrault, C. Batiot-Dupeyrat and F. Martinez, Catal. Today, 2005, 107–108, 785 CrossRef CAS.
- G. S. Gallego, J. G. Marín, C. Batiot-Dupeyrat, J. Barrault and F. Mondragon, Appl. Catal., A, 2009, 369, 97 CrossRef CAS.
- G. S. Gallego, C. Batiot-Dupeyrat, J. Barrault, E. Florez and F. Mondragon, Appl. Catal., A, 2008, 334, 251 CrossRef CAS.
- G. Valderrama, A. Kienneman and M. R. Goldwasser, Catal. Today, 2008, 133–135, 142 CrossRef CAS.
- H. Provendier, C. Petit, C. Estournes and A. Kiennemann, Stud. Surf. Sci. Catal., 1998, 119, 741 CrossRef CAS.
- H. Provendier, C. Petit, C. Estournes and A. Kiennemann, Chemistry, 2001, 4, 57 CAS.
- H. Falcon, J. Baranda, J. M. Campos-Martin, M. A. Pena and J. L. G. Fierro, Stud. Surf. Sci. Catal., 2000, 130, 2195 CrossRef.
- M. R. Goldwasser, M. E. Rivas, E. Pietri, M. J. Perez-Zurita, M. L. Cubeiro, L. Gingembre, L. Leclercq and G. Leclercq, Appl. Catal., A, 2003, 255, 45 CrossRef CAS.
- M. R. Goldwasser, M. E. Rivas, E. Pietri, M. J. Perez-Zurita, M. L. Cubeiro, A. Grivobal-Constant and G. Leclercq, J. Mol. Catal. A: Chem., 2005, 228, 325 CrossRef CAS.
- G. C. Araujo, S. M. Lima, J. M. Assaf, M. A. Pena, J. L. Garcia Fierro and M. D. C. Rangel, Catal. Today, 2008, 133–135, 129 CrossRef.
- M. E. Rivas, J. L. G. Fierro, M. R. Goldwasser, E. Pietri, M. J. Perez-Zurita, A. Griboval-Constant and G. Leclercq, Appl. Catal., A, 2008, 344, 10 CrossRef CAS.
- N. K. Labhsetwar, A. Watanabe, T. Mitsuhashi and H. Haneda, J. Mol. Catal. A: Chem., 2004, 223, 217 CrossRef CAS.
- L. Kapokova, S. Pavlova, R. Bunina, G. Alikina, T. Krieger, A. Ishchenko, V. Rogov and V. Sadykov, Catal. Today, 2011, 164, 227 CrossRef CAS.
-
M. P. Pechini, US Patent, 3330697, 1967 Search PubMed.
- T. G. Kuznetsova, V. A. Sadykov, E. M. Moroz, S. N. Trukhan, E. A. Paukshtis, V. N. Kolomiichuk, E. B. Burgina, V. I. Zaikovskii, M. A. Fedotov, V. V. Lunin and E. Kemnitz, Stud. Surf. Sci. Catal., 2002, 143, 659 CrossRef CAS.
- Electronic sources http://abulafia.mt.ic.ac.uk/shannon/ptable.php.
- N. W. Hurst, S. J. Gentry, A. Jones and B. D. McNicol, Catal. Rev. Sci. Eng., 1982, 24(2), 233 CAS.
- P. Ciambelli, S. Cimino, S. De Rossi, L. Lisi, G. Minelli, P. Porta and G. Russo, Appl. Catal., B, 2001, 29, 239 CrossRef CAS.
- P. Porta, S. Cimino, S. De Rossi, M. Faticanti, G. Minelli and I. Pettiti, Mater. Chem. Phys., 2001, 71, 165 CrossRef CAS.
- C. Crisafulli, S. Scire, S. Minico and L. Solarino, Appl. Catal., A, 2002, 225, 1 CrossRef CAS.
- V. A. Sadykov, S. N. Pavlova, T. S. Kharlamova, V. S. Muzykantov, N. F. Uvarov, Yu. S. Okhlupin, A. V. Ishchenko, A. S. Bobin, N. V. Mezentseva, G. M. Alikina, A. I. Lukashevich, T. A. Krieger, T. V. Larina, N. N. Bulgakov, V. M. Tapilin, V. D. Belyaev, E. M. Sadovskaya, A. I. Boronin, V. A. Sobyanin, O. F. Bobrenok, A. L. Smirnova, O. L. Smorygo and J. A. Kilner, Perovskites: Structure, Properties and Uses, Nova Science Publishers Inc, 2010, p. 67 Search PubMed.
- J. Juan-Juan, M. C. Roman-Martinez and M. J. Illan-Gomez, Appl. Catal., A, 2009, 355, 27 CrossRef CAS.
- D. Liu, R. Lau, A. Borgna and Y. Yang, Appl. Catal., A, 2009, 358, 110–118 CrossRef CAS.
- J. Chen, C. Yao, Y. Zhao and P. Jia, Int. J. Hydrogen Energy, 2010, 35, 1630 CrossRef CAS.
- H. M. Swaan, V. C. H. Kroll, G. A. Martin and C. Mirodatos, Catal. Today, 1994, 21, 571 CrossRef CAS.
- N. N. Sazonova, V. A. Sadykov, A. S. Bobin, S. A. Pokrovskaya, E. L. Gubanova and C. Mirodatos, React. Kinet. Catal. Lett., 2009, 98, 35 CrossRef CAS.
- J. Juan-Juan, M. C. Roman-Martınez and M. J. Illan-Gomez, Appl. Catal., A, 2006, 301, 9 CrossRef CAS.
- A. Effendi, K. Hellgard, Z.-G. Zhang and T. Yoshida, Catal. Commun., 2003, 4, 203 CrossRef CAS.
- R. Yang, C. Xing, C. Lv, L. Shi and N. Tsubaki, Appl. Catal., A, 2010, 385, 92 CrossRef CAS.
- J. D. A. Bellido and E. M. Assaf, Appl. Catal., A, 2009, 352, 179 CrossRef CAS.
- J. Chen, C. Yao, Y. Zhao and P. Jia, Int. J. Hydrogen Energy, 2010, 35, 1630 CrossRef CAS.
- W. D. Zhang, B. S. Liu, C. Zhu and Y. L. Tian, Appl. Catal., A, 2005, 292, 138 CrossRef CAS.
- J. R. Rostrup Nielsen, Stud. Surf. Sci. Catal., 1991, 68, 85 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |