Tetraethylammonium 2-(N-hydroxycarbamoyl)benzoate: a powerful bifunctional metal-free catalyst for efficient and rapid cyanosilylation of carbonyl compounds under mild conditions†
Mohammad G.
Dekamin
*,
Zahra
Karimi
and
Mehdi
Farahmand
Pharmaceutical and Biologically-Active Compounds Research Laboratory, Department of Chemistry, Iran University of Science and Technology, Tehran 16846-13114, Iran. E-mail: mdekamin@iust.ac.ir; Fax: +98-21-7322 7703
First published on 20th March 2012
Abstract
In contrast to tetrabutylammonium hydroxide, its tetraethylammonium counterpart reacts with N-hydroxyphthalimide to afford tetraethylammonium 2-(N-hydroxycarbamoyl)benzoate (TEAHCB) rather than tetraethylammonium phthalimide-N-oxyl (TEAPINO). TEAHCB contains at the same time benzoate and hydroxycarbamoyl functionalities in a proper geometry to activate reaction components as Lewis basic and Lewis acidic centers, respectively. TEAHCB was found to be able to efficiently catalyze very rapid cyanosilylation of a wide variety of carbonyl compounds at 0.15 mol% catalyst loading under solvent-free conditions at room temperature as a simple and readily available bifunctional organocatalyst.
Introduction
In recent years, organocatalytic (metal-free) protocols have received great attention from both practical and environmental standpoints. The rapid-growing interest in the development of organocatalytic protocols is due to their ability to perform organic reactions in wet solvents or under solvent-free conditions in an aerobic atmosphere and to avoid the possibility of heavy metals contamination of the products that may occur with the traditional metallic catalytic systems.1 Hence, organocatalytic protocols seem to be especially attractive for the preparation of compounds that do not tolerate metal contamination, especially pharmaceuticals. Therefore, the use of organocatalysts is a valuable tool to address green chemistry principles.1,2 More recently, bifunctional organocatalysts have received considerable attention for different organic transformations such as Michael addition,3 Biginelli reaction,4 Strecker reaction5 or cyanosilylation of carbonyl compounds.6 Indeed, bifunctional and multifunctional catalysts present high potential for both catalysis and asymmetric synthesis.7,8 These catalysts contain both Lewis base and Lewis acid sites that activate nucleophilic and electrophilic substrates, respectively. The proper geometry of the catalytic centers is very important to incorporate reaction partners effectively in the catalytic cycle. As a result, such catalytic system assemblies promote reactions similar to the natural catalysis by enzymes. In addition, dual coordination by bifunctional organocatalysts further accelerates the reaction by simultaneously enhancing the electrophilic character of one substrate and the nucleophilic character of the other.6b,8On the other hand, hydrocyanation and cyanosilylation of carbonyl compounds are among the most important strategies for C–C bond-forming reactions in organic synthesis. The adduct products provide versatile intermediates such as cyanohydrins and cyanohydrin trialkylsilyl ethers, respectively. In particular, cyanohydrin trimethylsilyl ethers are industrially valuable and important intermediates for the synthesis of α-hydroxy acids and esters, α-amino acids, acyloins, vicinal diols, β-amino alcohols and other biologically active compounds.6,9 For instance, cyanosilylation is the key step in the manufacture of (+)-biotin,10a Ditropan or its analogues,10b and insecticides such as cypermethrin and fluvalinate.11a Cyanohydrin trialkylsilyl ethers are generally prepared by the addition of trimethylsilyl cyanide (TMSCN), a safe and easily handled reagent compared to HCN, NaCN or KCN,6,9 to carbonyl compounds in the presence of Lewis acids,11 Lewis bases10a,12 and double activating9b,13 or bifunctional10b,14 catalytic systems. Therefore, a large body of work has been devoted to the development of cyanohydrin trimethylsilyl ethers synthesis. However, many of these protocols often involve heavy or expensive transition metal catalysts, poor yield of the products or prolonged reaction times, inert atmosphere or anhydrous solvents, the use of hygroscopic catalysts and tedious work-up procedures.10–14
Traditional Lewis acid catalytic systems promote cyanosilylation through direct complexation of the metal species to both the carbonyl group of aldehydes or ketones and the cyano group of TMSCN.9,11 On the other hand, Lewis basic catalysts including the majority of organocatalytic systems often react with the silicon atom of TMSCN to produce more reactive hypercoordinated silicon intermediates.12a,15–21 Hence, a few Lewis acid organocatalytic systems have been introduced for cyanosilylation of carbonyl compounds. Although catalysts such as N-iodosuccinimide22 or different alkyl triphenylphosphonium containing soft halide ions including iodide or bromide23 have been introduced in recent years, the use of a hydrogen bond donor moiety is more feasible and practical in organocatalysis. Interestingly, hydrogen bonding has demonstrated significant effects on the catalytic activity of bifunctional organocatalysts.6,24–27 The use of different thio-urea derivatives,6,24 tetramethyl guanidine,25 alkali-metal salts of α-aminoacids26 and tetraethylammonium 2-(carbamoyl)benzoate27 has proved the influence of hydrogen bonding on the catalytic activity of bifunctional organocatalysts for cyanosilylation of carbonyl compounds.
In continuation of our interest to develop more efficient organocatalysts for cyanosilylation of carbonyl compounds,16c,18b–d,19a,27 we wish herein to report our recent findings on the improved and smooth addition of TMSCN to carbonyl compounds using tetraethylammonium 2-(N-hydroxycarbamoyl)benzoate (TEAHCB, 1), as a new bifunctional organocatalyst, under solvent-free conditions (Scheme 1).
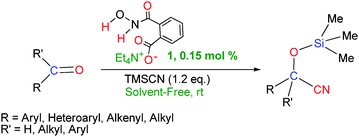 | ||
| Scheme 1 Cyanosilylation of carbonyl compounds using tetraethylammonium 2-(N-hydroxycarbamoyl)benzoate (TEAHCB, 1). | ||
Results and discussion
In our preceding papers,18b,c,28 we have reported that the hydroxide ion in tetrabutylammonium hydroxide (TBAOH, 3a) or alkali metal hydroxides 3b–d reacts preferentially with the more acidic hydroxyl proton of N-hydroxyphthalimide (NHPI, 2). This pattern of reactivity produces the corresponding salts of the phthalimide-N-oxyl (PINO) nucleophilic catalyst 4 which were successfully applied by our research group to facile cyanosilylation of carbonyl compounds,18b,c protection of alcohols and phenols with hexamethyldisilazane,29and cyclotrimerization of isocyanates28,30 (Scheme 2).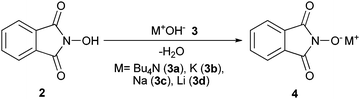 | ||
| Scheme 2 Different phthalimide-N-oxyl salts 4 produced by the reaction of NHPI (2) with TBAOH or alkali metal hydroxides 3a–d. | ||
Interestingly, in our previous research, we found that tetraethylammonium hydroxide (TEAOH, 3e) reacts with the carbonyl groups of imide functionality of the phthalimide instead of the more acidic N−H proton to afford tetraethylammonium 2-(carbamoyl)benzoate (TEACB) as a bifunctional organocatalyst.27 In our hands, TEACB demonstrated higher catalytic activity in both cyanosilylation of carbonyl compounds and cyclotrimerization of isocyanates compared to the PINO nucleophile 4 in the typical catalytic systems.27,30 Hence, we decided to investigate the reactivity of TEAOH toward NHPI (2). It was found that although the imide moiety in the structure of 2 survived through the reaction with 3a–d,18b,c the situation is completely altered in the reaction with 3e to afford TEAHCB (1) as a new bifunctional organocatalyst (Scheme 3).
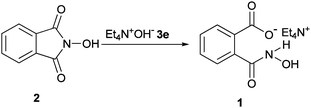 | ||
| Scheme 3 Unusual reaction pathway of 2 and 3e to produce TEAHCB (1). | ||
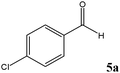
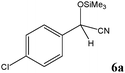
At first, a mixture of 4-chlorobenzaldehyde (5a) and TMSCN was treated with 0.3 mol% of TEAHCB (1) under solvent-free conditions at room temperature (Table 1). It was found that the cyanide addition occurred almost instantaneously to afford the desired trimethylsilylated cyanohydrin 6a with 99% conversion in 5 min (entry 1). By employing lower catalyst loading, 0.15 mol%, the desired product was obtained in quantitative yield. Furthermore, the reaction was studied on a 10 mmol scale, and we were delighted to find that only a minute amount of 1 (0.08 mol%) was required to catalyze the cyanosilylation of 5a at room temperature (entries 2 and 3). It is obvious that the best result in terms of turnover number (TON) and turnover frequency (TOF) could be achieved using 0.15 mol% catalyst loading of 1 (entry 2). On the other hand, no reaction was observed in the absence of 1 under similar reaction conditions (entry 4). Therefore, the lower catalyst loading required for this transformation compared to many organocatalytic systems having oxygen as their sole nucleophilic site18 and even TEACB27 demonstrates the simultaneous role of benzoate functionality of catalyst 1 and hydrogen bonding formed by its N-hydroxycarbamoyl moiety in a proper geometry in activating both TMSCN and carbonyl group for smooth cyanosilylation of diverse carbonyl compounds (Scheme 4, Table 2). It is expected that nucleophilic attack from the benzoate functionality of catalyst 1 on TMSCN produces pentavalent silicon intermediate 7 as the first reaction intermediate. The high reactivity of the pentavalent or hexavalent silicon intermediates to promote many organic transformations mediated by silicon reagents is well known.9c,12a,27,31 Therefore, the cyanide transfer from the pentavalent silicon atom to the activated carbonyl group by hydrogen bonding in complex 8 forms intermediate 9. Subsequent decomposition of pentavalent intermediate 9 gives product 6 and regenerates catalyst 1.
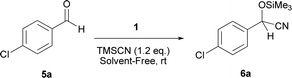
| Entry | Catalyst | Mol% | Time/min | Conversionb (%) | TONc | TOFd/h−1 |
|---|---|---|---|---|---|---|
| a TMSCN (3 mmol) was added to a mixture of 4-chlorobenzaldehyde (5a, 2.5 mmol) and TEAHCB (1) under solvent-free conditions at room temperature except for entry 3 where the reaction was performed on a 10 mmol scale. b Determined by GC analysis. c Turnover number. d Turnover frequency. e Tetraethylammonium 2-(carbamoyl)benzoate as catalyst.27 | ||||||
| 1 | TEAHCB (1) | 0.3 | 5 | 100 | 333 | 4000 |
| 2 | TEAHCB (1) | 0.15 | 8 | 100 | 667 | 5000 |
| 3 | TEAHCB (1) | 0.08 | 25 | 99 | 990 | 2376 |
| 4 | — | — | 120 | 0 | — | — |
| 5 | TEACBe | 0.5 | 15 | 98 | 196 | 784 |
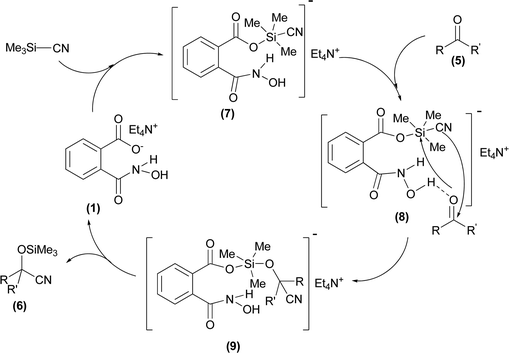 | ||
| Scheme 4 Plausible mechanism for cyanosilylation of carbonyl compounds 5 catalyzed by TEAHCB (1). | ||
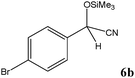
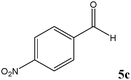
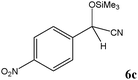
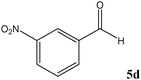
| Entry | Carbonyl compound (5) | Time (min) | Product (6)b | Conversion (%)c | Ref. |
|---|---|---|---|---|---|
| a TMSCN (3 mmol) was added to a mixture of carbonyl compound 5 (2.5 mmol) and TEAHCB 1 (1.1 mg) under solvent-free conditions at room temperature. b All products are known and were well-characterized by IR and NMR spectral data as compared with those obtained from authentic samples or reported in the literature.6,11,12,16,19,20 c Determined by GC analysis. | |||||
| 1 |
|
8 |
|
100 | 11g |
| 2 |
|
10 |
|
100 | 11h |
| 3 |
|
2 |
|
100 | 11h |
| 4 |
|
5 |
|
97 | 11n |
| 5 |
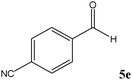
|
3 |
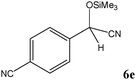
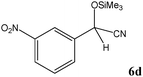
|
99 | 16b |
| 6 |
|
10 |
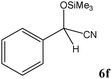
|
100 | 20a |
| 7 |
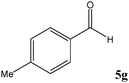
|
15 |
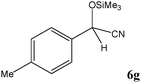
|
97 | 11g |
| 8 |

|
15 |
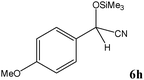
|
100 | 11g |
| 9 |
|
20 |
|
99 | 11h |
| 10 |
|
15 |
|
99 | 11g |
| 11 |
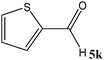
|
15 |
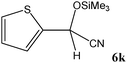
|
100 | 11m |
| 12 |
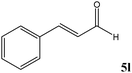
|
15 |
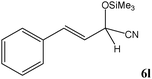
|
99 | 11g |
| 13 |
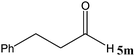
|
15 |
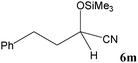
|
98 | 20a |
| 14 |
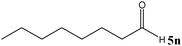
|
15 |
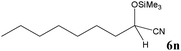
|
100 | 11g |
| 15 |
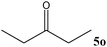
|
20 |
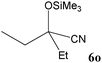
|
98 | 11m |
| 16 |
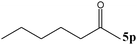
|
20 |
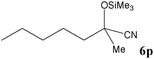
|
95 | 12b |
| 17 |
|
20 |
|
96 | 20a |
| 18 |
|
25 |
|
90 | 20a |
| 19 |
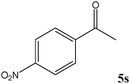
|
20 |
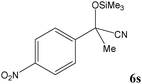
|
100 | 20a |
| 20 |
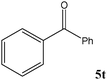
|
30 |
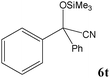
|
70 | 20a |
| 21 |
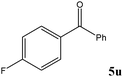
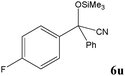
|
30 |
|
92 | 6d |
| 22 |
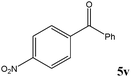
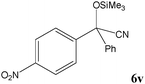
|
30 |
|
95 | 19a |
Encouraged by these results, other carbonyl compounds were subjected to cyanosilylation under optimal reaction conditions (TEAHCB 0.15 mol%, 1.2 equiv. of TMSCN, room temperature, solvent-free conditions). Table 2 shows that aromatic, heterocyclic and aliphatic aldehydes or ketones 5 have been effectively converted to the corresponding products 6. The reactions proceed very cleanly at room temperature under mild conditions. After completion of the reaction (monitored by TLC), the catalyst could be easily separated from the reaction mixture by aqueous extraction. Therefore, a simple work-up affords the desired products without any chromatographic purification. In general, the reaction conditions are very mild and high to quantitative yields of the products could be obtained within very short reaction time.
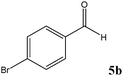
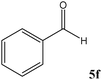
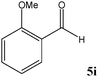
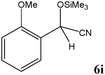
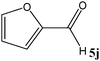
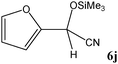
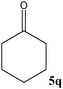
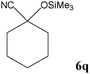
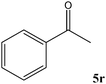
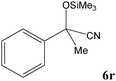
No benzoin condensation or desilylation by-products were observed. Acid-sensitive aldehydes such as furfural (5j), thiophene-2-carbaldehyde (5k) and cinnamaldehyde (5l) produced the desired cyanohydrin trimethylsilyl ethers rather than products of polymerization or Michael addition in quantitative yields (Table 2, entries 10–12). This may indicate that the catalytic system selectively activates the carbonyl function and keeps the double bonds of these acid sensitive substrates intact.27 As shown in Table 2, electronic effects and the nature of the substituents on the aromatic ring showed relatively strong effects on the required reaction time for the complete conversion of aldehydes. As a consequence of proposed bifunctional organocatalysis, substrates containing electron-withdrawing groups (entries 1 and 3–5) react much faster than ones bearing electron-donating groups (entries 7–11). This can be interpreted by considering that the cyanide transfer from the pentavalent silicon atom in complex 8 is the slowest step of the reaction mechanism (Scheme 4). However, 4-bromobenzaldehyde (5b) did not demonstrate any substantial substituent effect and its reaction required similar reaction time to benzaldehyde (5f). On the other hand, 4-methoxybenzaldehyde (5h) required shorter reaction time compared to 2-methoxybenzaldehyde (5i) due to proximity of the methoxy group to the carbonyl group in 5i which impedes hydrogen bonding between the catalyst and the carbonyl moiety of the substrate. Furthermore, due to the steric bulk, ketones such as cyclohexanone (5q), acetophenone (5r), benzophenone (5t), and their derivatives required longer reaction times than aldehydes (entries 16–22).11d,f The reaction of acyclic and cyclic aliphatic carbonyl compounds also afforded the corresponding products in very good yields in longer time compared to aromatic and heterocyclic carbonyl compounds (entries 13–17). Finally, this new catalytic system is far superior in terms of low catalyst loading and shorter reaction time as compared to other protocols for the addition of TMSCN to ketones (entries 16–22).11d,f,k,16,17,27
The comparison of the present method with respect to the catalyst loading, required reaction time, and simplicity of performance with those reported in the literature reveals that a practical and very fast method for the cyanosilylation of carbonyl compounds has been established using TEAHCB as a new bifunctional organocatalyst with unique geometry. To illustrate the efficiency of the present method, Table 3 is shown to compare our results with some of organocatalytic protocols reported in the literature recently.
| Entry | Substrate | Method [equivalent of TMSCN/ TOF (h−1)] | ||||
|---|---|---|---|---|---|---|
| 1a | 220c![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
317ab |
419bc |
527a |
||
| a Solvent-free conditions. b THF as solvent. c CH2Cl2 as solvent. | ||||||
| 1 | Benzaldehyde | 1.2/4000 | — | 1.2/97.0 | 2/0.62 | 1.2/552.0 |
| 2 | 4-Chlorobenzaldehyde | 1.2/5000 | 1.1/8.0 | 1.2/39.6 | - | 1.2/784.0 |
| 3 | 4-Methoxylbenzaldehyde | 1.2/2667 | 1.1/27.6 | 1.2/32.7 | 2/0.15 | 1.2/446.4 |
| 4 | Cinnamaldehyde | 1.2/2640 | 1.1/30.0 | — | — | 1.2/258.7 |
| 5 | 3-Phenylpropanal | 1.2/2613 | — | 1.2/32.7 | — | 1.2/198.0 |
| 6 | 4-Nitroacetophenone | 1.2/2000 | — | — | — | 1.2/99.0 |
Conclusions
In summary, we have developed a highly efficient catalytic cyanosilylation reaction with broad substrate generality using TEAHCB as a new bifunctional organocatalyst in relatively short reaction time at room temperature. This can be the first example of use of TEAHCB as a simple and readily available bifunctional organocatalyst in organic synthesis. The important features of our method are: mild reaction conditions, very low catalyst loading, simple work-up, wide substrate scope, high to quantitative yield and simple preparation of the catalyst from inexpensive precursors.Experimental section
Materials and instruments
FT IR spectra were recorded as KBr pellets on a Shimadzu FT IR-8400S spectrometer. 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were obtained using a Bruker DRX-500 AVANCE spectrometer. All NMR spectra were determined in CDCl3 or D2O at ambient temperature. GC chromatograms were recorded on a Perkin-Elmer 8420 instrument. Microanalysis of catalyst 1 was carried out using a CHNS/O analyzer (Perkin-Elmer Series II, 2400). Melting points were determined using an Electrothermal 9100 apparatus and are uncorrected. All chemicals were purchased from Merck or Aldrich and used as received except for benzaldehyde, for which a fresh distilled sample was used. TEAHCB (1) was ground to a fine powder and then dried at 70 °C under reduced pressure for 1 h. All reactions were protected from air moisture using a CaCl2 guard tube. Analytical TLC was carried out using Merck 0.2 mm silica gel 60 F-254 Al-plates. All compounds were well characterized by GC analysis, IR and NMR spectral data as compared with those obtained from authentic samples or reported in the literature.6,11,12,16,19,20Preparation of tetraethylammonium 2-(N-hydroxycarbamoyl)benzoate (TEAHCB, 1)
To a 25 mL round bottom flask equipped with a magnetic stirrer and a condenser were added 4.23 mmol of N-hydroxyphthalimide 2 (MW = 163.13 g mol−1, 0.69 g) and 4.23 mmol of tetraethylammonium hydroxide 3e (FW = 147.26 g mol−1, 20% w/w in water, d = 1.04 g mL−1, 5.0 mL). The mixture was stirred at room temperature for 5 min and then refluxed for 1 h. After cooling to room temperature, the reaction mixture was evaporated to dryness under reduced pressure to give a deep red viscous liquid and then dissolved in absolute EtOH (5 mL). EtOAc was added dropwise until a turbid solution was obtained and the mixture was heated until the disappearance of turbidity. After cooling, the white crystals of TEAHCB were filtered on a Büchner funnel; yield: 87%; mp = 140–142 °C; 1H NMR (500 MHz, D2O): δ = 1.15–1.19 (t, J = 7.3 Hz, 12H), 3.14–3.18 (q, J = 7.3 Hz, 8H), 7.36–7.38 (d, J = 7.4 Hz, 1H), 7.41–7.44 (t, J = 7.2 Hz, 1H), 7.46–7.50 (t, J = 7.50 Hz, 1H), 7.57–7.58 (d, J = 7.4 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3): δ = 6.9, 52.3, 128.1, 128.7, 129.9, 131.2, 131.8, 138.3, 170.4, 175.6 ppm; Anal. calcd. for C16H26N2O4: C, 61.91; H, 8.44; N, 9.03%; found: C, 62.02; H, 8.46; N, 9.01%.General procedure for cyanosilylation of carbonyl compounds
TMSCN (3.0 mmol, 0.38 mL) was added to a mixture of 2.5 mmol of a carbonyl compound and TEAHCB (1.1 mg). The obtained mixture was stirred at room temperature for 2–30 min. The reaction was monitored by TLC. After completion, the reaction mixture was quenched with water (2.5 mL) and the organic materials were extracted with EtOAc (2 × 2.5 mL). The obtained organic phase was washed with brine followed by water (2.5 mL) and dried over MgSO4. The solvent was evaporated on a rotary evaporator to afford the desired products which in many cases were essentially pure cyanohydrin TMS ethers. Further purification of the products was performed by silica gel column chromatography (EtOAc–Hexane, 1:10). The isolated yields were in good agreement with those obtained by GC Analysis.
Acknowledgements
We warmly thank The Research Council of Iran University of Science and Technology (IUST), Iran, for the financial support of this work (Grant No. 160/5295).Notes and references
- (a) C. Zhong and X. D. Shi, Eur. J. Org. Chem., 2010, 2999 CrossRef CAS; (b) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (c) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS; (d) T. Mitsudome, K. Nose, T. Mizugaki, K. Jitsukawa and K. Kaneda, Tetrahedron Lett., 2008, 49, 5464 CrossRef CAS.
- (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (b) K. Yamaguchi, T. Imago, Y. Ogasawara, J. Kasai, M. Kotani and N. Mizuno, Adv. Synth. Catal., 2006, 348, 1516 CrossRef CAS.
- (a) C. L. Wu, W. J. Li, J. J. Yang, X. M. Liang and J. X. Ye, Org. Biomol. Chem., 2010, 8, 3244 RSC; (b) C. G. Oliva, A. M. S. Silva, D. Resende, F. A. A. Paz and J. A. S. Cavaleiro, Eur. J. Org. Chem., 2010, 3449 CrossRef CAS; (c) O. Andrey, A. Alexakis, A. Tomassini and G. Bernardinelli, Adv. Synth. Catal., 2004, 346, 1147 CrossRef CAS; (d) D. Enders and A. Seki, Synlett, 2002, 26 CrossRef CAS; (e) B. List, P. Pojarliev and H. Martin, Org. Lett., 2001, 3, 2423 CrossRef CAS; (f) P. Bernal, R. Fernandez and J. M. Lassaletta, Chem.–Eur. J., 2010, 16, 7714 CrossRef CAS.
- (a) Y. Wang, H. Yang, J. Yu, Z. Miao and R. Chen, Adv. Synth. Catal., 2009, 351, 3057 CrossRef CAS; (b) D. Ding and C. G. Zhao, Eur. J. Org. Chem., 2010, 3802 CrossRef CAS.
- (a) X. Huang, J. L. Huang, Y. H. Wen and X. M. Feng, Adv. Synth. Catal., 2006, 348, 2579 CrossRef CAS; (b) P. Vachal and E. N. Jacobsen, Org. Lett., 2000, 2, 867 CrossRef CAS; (c) P. Vachal and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 10012 CrossRef CAS.
- (a) S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 15872 CrossRef CAS; (b) Y. Shen, X. Feng, Y. Li, G. Zhang and Y. Jiang, Eur. J. Org. Chem., 2004, 129 CrossRef CAS; (c) X. Liu, B. Oin, X. Zhou, B. He and X. Feng, J. Am. Chem. Soc., 2005, 127, 12224 CrossRef CAS.
- (a) M. Shibasaki, H. Sasai and T. Arai, Angew. Chem., Int. Ed., 1997, 36, 1236 CrossRef; (b) G. J. Rowlands, Tetrahedron, 2001, 57, 1865 CrossRef CAS; (c) M. Shibasaki and N. Yoshikawa, Chem. Rev., 2002, 102, 2187 CrossRef CAS.
- (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (b) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS; (c) D. Cahard and J. A. Ma, Angew. Chem., Int. Ed., 2004, 43, 4566 CrossRef; (d) A. Hamza, G. b. Schubert, T. Soós and I. Pápai, J. Am. Chem. Soc., 2006, 128, 13151 CrossRef CAS.
- (a) R. J. H. Gregory, Chem. Rev., 1999, 99, 3649 CrossRef CAS; (b) N. H. Khan, R. I. Kureshy, S. H. R. Abdi, S. Agrawal and R. V. Jasra, Coord. Chem. Rev., 2008, 252, 593 CrossRef CAS; (c) J. Gawronski, N. Wascinska and J. Gajewy, Chem. Rev., 2008, 108, 5227 CrossRef CAS; (d) M. Sakulsombat, P. Vongvilai and O. Ramstrom, Org. Biomol. Chem., 2011, 9, 1112 CAS; (e) M. G. Dekamin and Z. Mokhtari, Tetrahedron, 2012, 68, 922 CrossRef CAS; (f) D. A. Evans, L. K. Truesdale and G. L. Carroll, J. Chem. Soc., Chem. Commun., 1973, 55 RSC; (g) K. Higuchi, M. Onaka and Y. Izumi, J. Chem. Soc., Chem. Commun., 1991, 1035 RSC.
- (a) M. Seki, M. Hatsuda, Y. Mori and S. Yamada, Tetrahedron Lett., 2002, 43, 3269 CrossRef CAS; (b) S. Masumoto, M. Suzuki, M. Kanaia and M. Shibasaki, Tetrahedron Lett., 2002, 43, 8647 CrossRef CAS.
- (a) H. Firouzabadi, N. Iranpoor and A. A. Jafari, J. Organomet. Chem., 2005, 690, 1556 CrossRef CAS; (b) K. Iwanami, J.-C. Choi, B. Lu, T. Sakakura and H. Yasuda, Chem. Commun., 2008, 1002 RSC; (c) J. Wang, Y. Masui, K. Watanabe and M. Onaka, Adv. Synth. Catal., 2009, 351, 553 CrossRef CAS; (d) M. L. Kantam, S. Laha, J. Yadav, B. M. Choudary and B. Sreedhar, Adv. Synth. Catal., 2006, 348, 867 CrossRef CAS; (e) H. Yang, L. Zhang, P. Wang, Q. Yang and C. Li, Green Chem., 2009, 11, 257–264 RSC; (f) S. Kataoka, A. Endo, A. Harada, Y. Inagi and T. Ohmori, Appl. Catal., A, 2008, 342, 107 CrossRef CAS; (g) C. Baleizão, B. Gigante, H. García and A. Corma, Tetrahedron, 2004, 60, 10461 CrossRef; (h) S. T. Kadam and S. S. Kim, Appl. Organomet. Chem., 2009, 23, 119 CrossRef CAS; (i) A. Heydari and L. Ma’Mani, Appl. Organomet. Chem., 2008, 22, 12 CrossRef CAS; (j) L. Mei, S. W. Wong, H. K. Liang and W. S. Xuan, Appl. Organomet. Chem., 2008, 22, 181 CrossRef CAS; (k) A. Majhi, S. S. Kim and H. S. Kim, Appl. Organomet. Chem., 2008, 22, 407 CrossRef CAS; (l) W. K. Cho, S. M. Kang, A. K. Medda, J. K. Lee, I. S. Choi and H. S. Lee, Synthesis, 2007, 507 Search PubMed; (m) S. K. De and R. A. Gibbs, J. Mol. Catal. A: Chem., 2005, 232, 123 CrossRef CAS; (n) T. Schareina, A. Zapf, A. Cotte, M. Gotta and M. Bellera, Adv. Synth. Catal., 2011, 353, 777 CrossRef CAS; (o) N. Azizi and M. R. Saidi, J. Organomet. Chem., 2003, 688, 283 CrossRef CAS; (p) B. Karimi and L. Ma’Mani, Org. Lett., 2004, 6, 4813 CrossRef CAS.
- (a) G. K. S. Prakash, H. Vaghoo, C. Panja, V. Surampudi, R. Kultyshev, T. Mathew and G. A. Olah, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3026 CrossRef; (b) P. Phuengphai, S. Youngme, P. Gamez and J. Reedijk, Dalton Trans., 2010, 39, 7936 RSC; (c) S. S. Kim, J. Rajagopal and D. H. Song, J. Organomet. Chem., 2004, 689, 1734 CrossRef CAS; (d) B. He, Y. Li, X. Feng and G. Zhang, Synlett, 2004, 1776 CAS.
- (a) D. H. Ryu and E. J. Corey, J. Am. Chem. Soc., 2005, 127, 5384 CrossRef CAS; (b) B. M. Trost and S. Martinez-Sanchez, Synlett, 2005, 627 CrossRef CAS; (c) Z. Zeng, G. Zhao, P. Gao, H. Tang, B. Chen, Z. Zhou and C. Tang, Catal. Commun., 2007, 8, 1443 CrossRef CAS; (d) A. Alaaeddine, T. Roisnel, C. M. Thomas and J. Carpentiera, Adv. Synth. Catal., 2008, 350, 731 CrossRef CAS.
- (a) Y. Q. Wen, W. M. Ren and X. B. Lu, Org. Biomol. Chem., 2011, 9, 6323 RSC; (b) Y. Hamashima, D. Sawada, H. Nogami, M. Kanai and M. Shibasaki, Tetrahedron, 2001, 57, 805 CrossRef CAS; (c) Y. Hamashima, M. Kanai and M. Shibasaki, Tetrahedron Lett., 2001, 42, 691 CrossRef CAS; (d) G. Rajagopal, S. S. Kim and S. C. George, Appl. Organomet. Chem., 2007, 21, 798 CrossRef; (e) Y. Xiong, X. Huang, S. Gou, J. Huang, Y. Wen and X. Feng, Adv. Synth. Catal., 2006, 348, 538 CrossRef CAS.
- (a) For a recent review on Lewis base catalysis, see: S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS; (b) For a review on selective catalysis using hard–soft acid and base (HSAB) principles, see: S. Woodward, Tetrahedron, 2002, 58, 1017 CrossRef CAS.
- (a) S. E. Denmark and W. Chung, J. Org. Chem., 2006, 71, 4002 CrossRef CAS; (b) I. V. P. Raj, G. Suryavanshi and A. Sudalai, Tetrahedron Lett., 2007, 48, 7211 CrossRef CAS; (c) M. G. Dekamin and Z. Karimi, J. Organomet. Chem., 2009, 694, 1789 CrossRef CAS; (d) M. L. Kantam, P. Sreekanth and P. L. Santhi, Green Chem., 2000, 2, 47 RSC.
- (a) B. M. Fetterly and J. G. Verkade, Tetrahedron Lett., 2005, 46, 8061 CrossRef CAS; (b) S. Matsukawa, I. Sekine and A. Iitsuka, Molecules, 2009, 14, 3353 CrossRef CAS.
- (a) H. Zhou, F. X. Chen, B. Qin and X. Feng, Synlett, 2004, 1077 CAS; (b) M. G. Dekamin, J. Mokhtari and M. R. Naimi-Jamal, Catal. Commun., 2009, 10, 582 CrossRef CAS; (c) M. G. Dekamin, S. Javanshir, M. R. Naimi-Jamal, R. Hekmatshoar and J. Mokhtari, J. Mol. Catal. A: Chem., 2008, 283, 29 CrossRef CAS; (d) M. G. Dekamin, M. Farahmand, S. Javanshir and M. R. Naimi-Jamal, Catal. Commun., 2008, 9, 1352 CrossRef CAS.
- (a) M. G. Dekamin, R. Alizadeh and M. R. Naimi-Jamal, Appl. Organomet. Chem., 2010, 24, 229 CrossRef CAS; (b) A. Blanrue and R. Wilhelm, Synlett, 2004, 2621 CAS.
- (a) M. J. Fuchter, Chem.–Eur. J., 2010, 16, 12286 CrossRef CAS; (b) J. J. Song, F. Gallou, J. T. Reeves, Z. Tan, N. K. Yee and C. H. Senanyake, J. Org. Chem., 2006, 71, 1273 CrossRef CAS; (c) Y. Suzuki, M. D. Abu Bakar, K. Muramatsu and M. Sato, Tetrahedron, 2006, 62, 4227 CrossRef CAS; (d) Y. Fukuda, Y. Maeda, S. Ishii, K. Kondo and T. Aoyama, Synthesis, 2006, 589 CAS.
- X. Wang and S. K. Tian, Synlett, 2007, 1416 CAS.
- S. T. Kadam and S. S. Kim, Catal. Commun., 2008, 9, 1342 CrossRef CAS.
- (a) X. Wang and S. K. Tian, Tetrahedron Lett., 2007, 48, 6010 CrossRef CAS; (b) Y. Teng and P. H. Toy, Synlett, 2011, 551 CAS.
- (a) S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 15872 CrossRef CAS; (b) S. Hun, H. Chen, J. W. Wiench, M. Pruski and V. S. Lin, Angew. Chem., Int. Ed., 2005, 44, 1826 CrossRef.
- (a) L. Wang, X. Huang, J. Jiang, X. Liu and X. Feng, Tetrahedron Lett., 2006, 47, 1581 CrossRef CAS; (b) S. Matsukawa and S. Fujikawa, Tetrahedron Lett., 2012, 53, 1075 CrossRef CAS.
- X. Liu, B. Oin, X. Zhou, B. He and X. Feng, J. Am. Chem. Soc., 2005, 127, 12224 CrossRef CAS.
- M. G. Dekamin, S. Sagheb-Asl and M. R. Naimi-Jamal, Tetrahedron Lett., 2009, 50, 4063 CrossRef CAS.
- M. G. Dekamin, F. M. Moghaddam, H. Saeidian and S. Mallakpour, Monatsh. Chem., 2006, 137, 1591 CrossRef CAS.
- M. G. Dekamin, N. Yazdaninia, J. Mokhtari and M. R. Naimi-Jamal, J. Iran. Chem. Soc., 2011, 8, 537 CAS.
- M. G. Dekamin, K. Varmira, M. Farahmand, S. Sagheb-Asl and Z. Karimi, Catal. Commun., 2010, 12, 226 CrossRef CAS.
- (a) G. K. S. Prakash, C. Panja, H. Vaghoo, V. Surampudi, R. Kultyshev, M. Mandal, G. Rasul, T. Mathew and G. A. Olah, J. Org. Chem., 2006, 71, 6806 CrossRef CAS; (b) Y. Orito and M. Nakajima, Synthesis, 2006, 1391 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra for TEAHCB (1) and 1H NMR for selected products 6a and 6s. See DOI: 10.1039/c2cy20037f |
| This journal is © The Royal Society of Chemistry 2012 |