Synthesis of cyclic carbonates using monometallic, and helical bimetallic, aluminium complexes†
Received
13th December 2011
, Accepted 16th January 2012
First published on 17th January 2012
Abstract
The aluminium complexes of a series of bis(pyrazol-1-yl)methane derived ligands were investigated as catalysts for the synthesis of cyclic carbonates from carbon dioxide and epoxides. A bimetallic, helical, heteroscorpionate complex displayed significantly higher catalytic activity than the corresponding monometallic complexes. The optimal reaction conditions were found to involve the use of 5 mol% of the bimetallic complex and 5 mol% of tetrabutylammonium bromide under solvent free conditions in the presence of 10 bar of wet carbon dioxide. Under these conditions, eleven monosubstituted epoxides were converted into the corresponding cyclic carbonates in 51–91% yield. A kinetic study showed that the reactions were first order in epoxide, catalyst and tetrabutylammonium bromide and allowed a catalytic cycle to be proposed.
Introduction
The reaction between epoxides and carbon dioxide can lead to either cyclic1,2 or polycarbonates1,3 (Scheme 1), both of which are of significant commercial importance. Cyclic carbonates are the thermodynamic products of the reaction and have many applications including as electrolytes for lithium ion batteries,4 solvents4,5 and chemical intermediates.6 Interest in this reaction has increased significantly over the last five years in the context of using carbon dioxide as a sustainable building block for the chemical industry and avoiding atmospheric emissions of carbon dioxide.
 |
| | Scheme 1 Synthesis of cyclic and polycarbonates. | |
Over the last four years, the North group has developed bimetallic aluminium(salen) complexes such as 1 as highly effective catalysts for the synthesis of cyclic carbonates from epoxides and carbon dioxide at atmospheric pressure and room temperature.7,8 Mechanistic studies8 showed that the bimetallic nature of the complexes was critical to the exceptionally high catalytic activity of these complexes, as was the presence of a tetraalkylammonium salt which could function as a source of both a nucleophilic halide and a tertiary amine, thus allowing the assembly of the key transition state shown in Fig. 1 in which the carbonate forming reaction occurs intramolecularly.
 |
| | Fig. 1 Catalysts 1 and 2 and the key transition state for cyclic carbonate synthesis. | |
In view of the high level of catalytic activity displayed by bimetallic salen complexes such as 1, it was of interest to investigate the use of other bimetallic aluminium complexes as catalysts for cyclic carbonate synthesis. Thus, recently the North group reported the use of bimetallic aluminium(acen) complex 2 as a catalyst for cyclic carbonate synthesis.9 In recent years, a number of research groups have been interested in the design of “heteroscorpionate” ligands, their coordination chemistry and their catalytic applications.10 In this field, the Otero group has reported the synthesis of acetamidate or thioacetamidate heteroscorpionate ligands based on a bis(pyrazol-1-yl)methane moiety.11 This kind of ligand exhibits high versatility in terms of coordination modes due to the existence of three possible tautomers.11 Recently, helical bimetallic aluminium complex 3 as well as related monometallic complexes 4–7 containing these versatile heteroscorpionate ligands (Fig. 2) have been reported.12 These complexes are active initiators for the ring-opening polymerization of cyclic esters. In view of the potential applications of aluminium complexes in catalysis it was of interest to study their potential as catalysts for the synthesis of cyclic carbonates. In this paper we report the results of a collaborative project on the use of complexes 3–7 as catalysts for the synthesis of cyclic carbonates from epoxides and carbon dioxide.
 |
| | Fig. 2 Structures of complexes 3–7. | |
Results and discussion
An initial screening of complexes 3–7 as catalysts for cyclic carbonate synthesis was carried out using 5 mol% of the catalyst and 5 mol% of tetrabutylammonium bromide at room temperature for 24 hours with styrene oxide 8a as substrate under solvent free conditions and under one bar pressure of carbon dioxide (Scheme 2). The results of this study are shown in Table 1. As can be seen from entries 1–5, bimetallic complex 3 was more active than any of the monometallic complexes 4–7. It is also apparent that thioamide derived complexes 3–4 are significantly more active than imidate derived complexes 5–7. Control experiments (entries 6–8) showed that neither tetrabutylammonium bromide nor complexes 3–4 displayed significant catalytic activity in the absence of the other catalyst component.
Table 1 Synthesis of styrene carbonate 9a using aluminium complexes 3–7
| Entry |
Catalyst |
Bu4NBr (mol%) |
CO2/bar |
Conversiona (%) |
TOFc/h−1 |
|
Determined by GCMS analysis.
Using dry carbon dioxide.
TOF = moles of product/(moles of catalyst × time).
|
| 1 |
3
|
5 |
1 |
69 |
0.6 |
| 2 |
4
|
5 |
1 |
53 |
0.4 |
| 3 |
5
|
5 |
1 |
24 |
0.2 |
| 4 |
6
|
5 |
1 |
0 |
0 |
| 5 |
7
|
5 |
1 |
29 |
0.2 |
| 6 |
|
5 |
1 |
6 |
0.05 |
| 7 |
3
|
0 |
1 |
0 |
0 |
| 8 |
4
|
0 |
1 |
0 |
0 |
| 9b |
3
|
5 |
1 |
23 |
0.2 |
| 10b |
4
|
5 |
1 |
18 |
0.15 |
| 11 |
3
|
5 |
10 |
96 |
0.8 |
| 12 |
4
|
5 |
10 |
44 |
0.4 |
| 13 |
|
5 |
10 |
6 |
0.05 |
| 14 |
3
|
|
10 |
0 |
0 |
| 15 |
4
|
|
10 |
0 |
0 |
The results presented in entries 1–8 of Table 1 were obtained using carbon dioxide generated by the evaporation of cardice pellets. The carbon dioxide atmosphere generated in this way will contain a significant amount of water vapour. Since catalysts 3–7 contain hydrolytically sensitive aluminium-alkyl groups, experiments were also carried out using complexes 3–4 under dry reaction conditions using anhydrous carbon dioxide obtained from a commercial cylinder. As shown in entries 9 and 10 of Table 1, this had a significantly detrimental effect on both catalysts, reducing the catalytic activity to one third of that obtained in the presence of water in both cases. To confirm that water was the cause of this difference in reactivity, reactions were carried out using catalyst 3 and dry carbon dioxide with known amounts of water added to the reaction. As shown in Fig. 3, the presence of small amounts of water did have a beneficial effect on the activity and under optimal conditions (0.75 mol% water), 68% conversion of epoxide 8a into cyclic carbonate 9a could be obtained which compares with the 69% conversion obtained using cardice pellets under the same reaction conditions. However, addition of more than 0.75 mol% of water was detrimental, consistent with over hydrolysis of aluminium complex 3. Thus, it appears that complexes 3–7 are precatalysts of catalytically active species which contain aluminium-oxo units.
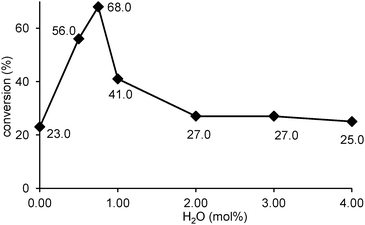 |
| | Fig. 3 Effect of water on the synthesis of styrene carbonate 9a catalysed by complex 3. | |
To increase the conversion of epoxide 8a into cyclic carbonate 9a obtained after a reaction time of 24 hours, the effect of increasing the reaction pressure to 10 bar was investigated. As shown in entry 11 of Table 1, this has a very beneficial effect in the case of reactions catalysed by bimetallic complex 3, increasing the conversion to 96%. However, for monometallic complex 4, a lower conversion was obtained at 10 bar pressure than at 1 bar (entry 12). Control experiments (entries 13–15) showed that even at 10 bar, neither tetrabutylammonium bromide nor complexes 3–4 displayed significant catalytic activity on their own.
The effect of changing the amount of catalyst 3 and tetrabutylammonium bromide whilst keeping the catalyst to cocatalyst ratio constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was investigated in reactions carried out at 10 bar with epoxide 8a as substrate. The results are shown in Fig. 4. It is apparent that reactions carried out using less than 5 mol% of catalyst 3 and tetrabutylammonium bromide gave much lower conversions, and whilst 99–100% conversion could be obtained using more than 5 mol% of 3 and tetrabutylammonium bromide, this was a minimal improvement. The effect of changing the catalyst 3 to tetrabutylammonium bromide ratio was investigated in experiments in which the amount of either 3 or tetrabutylammonium bromide was fixed at 5 mol% and the amount of the other catalyst component was varied. The results are shown in Fig. 5 and indicate that at least 5 mol% of both catalyst components are required and hence that the optimal catalyst 3 to tetrabutylammonium bromide ratio is 1:1. Thus, use of 5 mol% of both catalyst 3 and tetrabutylammonium bromide was shown to be optimal.
:1 was investigated in reactions carried out at 10 bar with epoxide 8a as substrate. The results are shown in Fig. 4. It is apparent that reactions carried out using less than 5 mol% of catalyst 3 and tetrabutylammonium bromide gave much lower conversions, and whilst 99–100% conversion could be obtained using more than 5 mol% of 3 and tetrabutylammonium bromide, this was a minimal improvement. The effect of changing the catalyst 3 to tetrabutylammonium bromide ratio was investigated in experiments in which the amount of either 3 or tetrabutylammonium bromide was fixed at 5 mol% and the amount of the other catalyst component was varied. The results are shown in Fig. 5 and indicate that at least 5 mol% of both catalyst components are required and hence that the optimal catalyst 3 to tetrabutylammonium bromide ratio is 1:1. Thus, use of 5 mol% of both catalyst 3 and tetrabutylammonium bromide was shown to be optimal.
Having determined the optimal reaction conditions to be room temperature, 10 bar pressure of undried carbon dioxide and 5 mol% of catalyst 3 and tetrabutylammonium bromide, the effect of varying the epoxide was investigated. Table 2 shows the results obtained using eleven terminal epoxides under these standard conditions. Good yields of cyclic carbonates† were obtained from epoxides with aliphatic (8b–d,i) or aromatic (8a,j–k) side chains. Glycidol 8e was converted into glycidol carbonate 9e with a relatively low yield of 51%, probably because the alcohol functionality of 8/9e can react with the alkylaluminium groups in catalyst 3 (the 1H NMR spectrum of a 1:1 mixture of glycidol and catalyst 3 showed evidence of such an interaction). In contrast, functionalized epoxides 8f,g gave excellent yields of cyclic carbonates 9f,g. Notably, commercially important ethylene carbonate 9h could be obtained in 80% yield from ethylene oxide 8h. Whilst all the monosubstituted epoxides investigated were substrates for catalyst 3, attempts to extend the chemistry to cyclohexene oxide, cis-2,3-butene oxide or 2-methyl-2,3-butene oxide were unsuccessful even when the pressure was increased to 30 bar.
Table 2 Synthesis of cyclic carbonates 9a–k using bimetallic aluminium complex 3
| Epoxide |
Conversiona (%) |
Yieldb (%) |
TOFe/h−1 |
|
Determined by GCMS analysis in the case of 8/9a and by 1H NMR analysis for 8/9b–k.
Yield of isolated pure cyclic carbonates 9a–k.
Conversion not determined due to the volatility of epoxides 8h,i.
Reaction carried out at 0 °C.
TOF = moles of product/(moles of catalyst × time).
|
|
8a
|
96 |
83 |
0.7 |
|
8b
|
83 |
77 |
0.6 |
|
8c
|
86 |
76 |
0.6 |
|
8d
|
71 |
65 |
0.5 |
|
8e
|
63 |
51 |
0.4 |
|
8f
|
95 |
91 |
0.8 |
|
8g
|
100 |
86 |
0.7 |
|
8h
|
c
|
80 |
0.7 |
|
8i
d
|
c
|
57 |
0.5 |
|
8j
|
89 |
77 |
0.6 |
|
8k
|
81 |
69 |
0.6 |
To investigate the mechanism of the reaction, a kinetics study was undertaken using propylene carbonate 9i as solvent at 27 °C and 1 bar carbon dioxide pressure with styrene oxide 8a as substrate as we have previously described for reactions catalysed by complex 1.8 However, under these conditions, the reactions were rather slow (at most 50% conversion after six hours), so GCMS analysis of samples withdrawn from the reaction was found to be a more sensitive method of monitoring the concentration changes than in situ FTIR. Attempts to monitor reactions over longer time periods were unsuccessful as catalyst decomposition became apparent. The general rate equation for the reaction is given in eqn (1). However, the kinetics study was carried out in the presence of a large excess of carbon dioxide and since complex 3 and tetrabutylammonium bromide are both catalysts, eqn (1) can be simplified to eqn (2a,b).
| | | Rate = k[8a]a[CO2]b[3]c[Bu4NBr]d | (1) |
| | | kobs = k[CO2]b[3]c[Bu4NBr]d | (2b) |
| | | log(kobs) = log(k) + blog[CO2] + clog[3] + dlog[Bu4NBr] | (3) |
Under these reaction conditions, all of the reactions were found to show a good fit to overall first order kinetics indicating that a = 1.† To determine the order with respect to complex 3 and tetrabutylammonium bromide, two series of reactions were carried out at four different concentrations of 3 or tetrabutylammonium bromide. These experiments were carried out in duplicate to show their reproducibility and the average of the two rate constants (kavg) used in subsequent analysis. The rate data are presented in Table 3. Plots of log(kavg) against log[3] or log[Bu4NBr] had gradients of 1.14 and 1.11, respectively, suggesting that c = d = 1 (eqn (3)).† This was confirmed by plots of kavg against [3] or [Bu4NBr] which in both cases showed a linear correlation between the concentration and observed rate constant (Fig. 6 and 7).
| [3] (M) |
[Bu4NBr] (M) |
108kobs1 (s−1) |
108kobs2 (s−1) |
108kavg (s−1) |
|
All reactions carried out at 27 °C in propylene carbonate as solvent with [8a]0 = 1.07 M.
|
| 0.02685 |
0.0537 |
762 |
720 |
741 |
| 0.0537 |
0.0537 |
1792 |
2185 |
1989 |
| 0.08055 |
0.0537 |
2820 |
2952 |
2886 |
| 0.1074 |
0.0537 |
3889 |
3145 |
3517 |
| 0.0537 |
0.02685 |
1213 |
941 |
1077 |
| 0.0537 |
0.08055 |
4388 |
3198 |
3793 |
| 0.0537 |
0.1074 |
5114 |
4826 |
4970 |
![Linear correlation between kavg and [3].](/image/article/2012/CY/c2cy00517d/c2cy00517d-f6.gif) |
| | Fig. 6 Linear correlation between kavg and [3]. | |
![Linear correlation between kavg and [Bu4NBr].](/image/article/2012/CY/c2cy00517d/c2cy00517d-f7.gif) |
| | Fig. 7 Linear correlation between kavg and [Bu4NBr]. | |
Compared to the previously reported kinetic analysis of cyclic carbonate synthesis catalysed by complex 1,8 the main difference in reactions catalysed by complex 3 is the order with respect to tetrabutylammonium bromide concentration. Thus, whilst reactions catalysed by complex 1 were second order in tetrabutylammonium bromide concentration, those involving complex 3 are first order in tetrabutylammonium bromide concentration. This implies that for reactions catalysed by complex 3, only one tetrabutylammonium bromide molecule is involved in the mechanism before or during the rate determining step of the catalytic cycle. It is well known2 that one role of an onium halide cocatalyst is to supply halide which ring-opens the Lewis-acid coordinated epoxide to form a halo-alkoxide. Carbon dioxide can then insert into the metal alkoxide bond to give a metal carbonate which can ring-close to form the cyclic carbonate and regenerate the onium halide as shown in Scheme 3. The catalytic cycle shown in Scheme 3 is also consistent with the observed optimal 1:1 ratio of complex 3 to tetrabutylammonium bromide (Fig. 5). In the case of reactions catalysed by complex 1, the tetrabutylammonium bromide had a second role: to generate tributylamine in situ. The tributylamine then reacted with the carbon dioxide to form a zwitterionic carbamate which was stabilized by coordination to the second aluminium ion of complex 1. This also allowed the key carbonate forming step to occur intramolecularly rather than intermolecularly and thus accounted for the exceptionally high catalytic activity displayed by complex 1.
 |
| | Scheme 3 Catalytic cycle for cyclic carbonate synthesis. | |
In the case of reactions catalysed by complex 3, tributylamine is also formed under the reaction conditions and could be detected by GCMS analysis of the reaction mixture. However, examination of the crystal structure of complex 312 revealed that the two aluminium ions are on opposite faces of the N–C–S unit and hence, any adduct formed between complex 3, tributylamine and carbon dioxide could not react intramolecularly with the ring-opened epoxide. Thus, in this case, tributylamine appears to be solely a cocatalyst decomposition product and the reaction follows the mechanism shown in Scheme 3. This analysis was supported by the results of experiments in which tributylamine was used as a cocatalyst instead of, or as well as, tetrabutylammonium bromide. Thus, when a combination of complex 3 and tributylamine (5 mol% of each) was used to convert epoxide 8a into cyclic carbonate 9a, no conversion was observed and when 5 mol% of catalyst 3, tetrabutylammonium bromide and tributylamine was used, a conversion of just 34% was obtained. When compared to the 69% conversion (Table 1, entry 1) obtained under the same conditions in the absence of tributylamine, it is apparent that for this catalyst system, tributylamine has no catalytic activity and actually acts as an inhibitor of cyclic carbonate synthesis.
Complex 3 is only a precatalyst for cyclic carbonate synthesis. The catalytic activity of the system increases in the presence of water and alkyl aluminium complexes are known to be subject to hydrolysis by water forming Al–O–Al species. The crystal structure of complex 312 also revealed that formation of intramolecular μ–oxo bridges is impossible as the two aluminium ions are too far apart from one another and on opposite faces of the N–C–S unit. Hydrolysis of the aluminium ethyl groups may therefore result in intermolecular μ–oxo bridge formation, leading to the formation of oligomeric species which are catalytically active. However, the first order dependence of the kinetics on the concentration of complex 3 indicates that each monomer unit within the oligomer is independently catalytically active and thus that there is no cooperative catalysis between two aluminium ions whether they are in the same or different complex 3 units. Attempts to confirm the formation of oligomeric species from complex 3 and water by mass spectrometry or NMR spectroscopy were unsuccessful due to the decomposition of complex 3 by protic solvents.
Conclusions
A combination of complex 3 and tetrabutylammonium bromide forms an active catalyst system for the synthesis of cyclic carbonates from carbon dioxide and terminal epoxides at ambient temperature and 10 bar pressure. The differing kinetic behaviour of complexes 1 and 3 provides important mechanistic information which supports the previously proposed catalytic cycle for cyclic carbonate synthesis and shows the importance of the correct juxtaposition of the two metal ions within a bimetallic complex to obtain optimal catalytic activity.
Whilst a large number of catalysts have been reported for the synthesis of cyclic carbonates from terminal epoxides and carbon dioxide, the vast majority of these require pressures above 15 bar and temperatures above 90 °C.2 Only a small number of catalyst systems have been reported to be active at (or below) room temperature and at 1–10 bar carbon dioxide pressure. Of the aluminium based systems that are active under such mild conditions, complex 3 with a TOF of up to 0.8 h−1 is the second most active, outperformed only by aluminium(salen) complexes such as 1 (TOF 13–64 h−1)7,8,13 which have been shown to be catalytically active as bimetallic complexes. Other aluminium based catalysts have TOFs of <0.02 to 0.5 h−1 under similar reaction conditions.14 More active catalysts under mild reaction conditions can be formed by complexes of more toxic metals: bismuth (TOF 31 h−1),15 cobalt (TOF 4–706 h−1)16 and chromium (TOF 3–48 h−1).17 Finally, Kisch et al. showed that the combination of zinc chloride and tetrabutylammonium iodide formed a very active catalyst system (TOF 143 h−1) at room temperature and 1 bar carbon dioxide pressure.18
Experimental
Commercially available chemicals (Alfa, Aldrich, Fluka) were used as received. Complexes 3–7 were prepared as reported previously.10,11 GC-MS were recorded on a Varian CP-800-SATURN 2200 GC-MS ion-trap mass spectrometer using a FactorFour (VF-5 ms) capillary column (30 m × 0.25 mm) with helium as the carrier gas. The conditions used were: initial temperature 60 °C, hold at initial temperature for 3 min then ramp rate 15 °C min−1 to 270 °C; hold at final temperature for 5 min; TR 7.33 min (styrene oxide), TR 12.09 min (styrene carbonate). For the first 3.50 min, the eluent was routed away from the mass detector. Subsequently, the detector was operated in full EI scan mode. 1H and 13C NMR spectra were recorded on a Bruker Avance 300 spectrometer at 300 MHz for 1H and 75 MHz for 13C. All spectra were recorded at ambient temperature and were referenced to the residual solvent peak.
General procedure for cyclic carbonate synthesis at 1 bar pressure
Styrene oxide 8a (199 mg, 1.66 mmol), catalyst 3 (42.1 mg, 0.083 mmol) and Bu4NBr (26.7 mg, 0.083 mmol) were placed in a sample vial fitted with a magnetic stirrer bar and placed in a large conical flask. The conical flask was placed in an oil bath thermostatted at 26 °C. Cardice pellets were added to the conical flask which was fitted with a rubber stopper pierced by a deflated balloon. The reaction mixture was stirred for 24 h, then the conversion of epoxide to cyclic carbonate was determined by analysis of a sample by GCMS.
General procedure for cyclic carbonate synthesis at 10 bar pressure
An epoxide (1.66 mmol), catalyst 3 (42.1 mg, 0.083 mmol) and Bu4NBr (26.7 mg, 0.083 mmol) were placed in a sample vial fitted with a magnetic stirrer bar and placed in a stainless steel pressure reactor. Sufficient cardice pellets were added to the reactor to generate a pressure of 10 bar once they evaporated. The reactor was kept at room temperature (or an ice/water bath at 0 °C when propylene oxide 8i was the substrate). The reaction mixture was stirred for 24 h, then the conversion of epoxide to cyclic carbonate was determined by analysis of a sample by GCMS (for 8/9a) or 1H-NMR spectroscopy (for 8/9b–k). The remaining sample was filtered through a plug of silica, eluting with CH2Cl2 to remove the catalyst. The eluent was evaporated in vacuo to give either the pure cyclic carbonate or a mixture of cyclic carbonate and unreacted epoxide. In the latter case, the mixture was purified by flash chromatography to give the pure cyclic carbonate. Chromatographic and spectroscopic data for cyclic carbonates 9a–k have been reported previously.7,8†
General procedure for kinetic studies
Styrene oxide (0.20 g, 1.66 mmol), catalyst 3 (2.5, 5.0, 7.5 or 10 mol%) and Bu4NBr (2.5, 5.0, 7.5 or 10 mol%) were dissolved in propylene carbonate (1.43 mL), placed in a sample vial fitted with a magnetic stirrer bar and placed in a large conical flask. The conical flask was placed in an oil bath thermostatted at 27 °C. Cardice pellets were added to the conical flask which was fitted with a rubber stopper pierced by a deflated balloon. Samples were taken at convenient intervals (approximately every 30 minutes) and analysed by GC/MS to determine the relative concentrations of styrene oxide and styrene carbonate.
Acknowledgements
We gratefully acknowledge financial support from the Ministerio de Ciencia e Innovación (MICINN) of Spain (Grant No. CTQ2008-00318/BQU, Grant No. EEBB-2011-43839 and Consolider-Ingenio 2010 ORFEO CSD2007-00006) and the Junta de Comunidades de Castilla-La Mancha, Spain (Grant No. PCI08-0010).
Notes and references
- R. Zevenhoven, S. Eloneva and S. Teir, Catal. Today, 2006, 115, 73–79 CrossRef CAS.
- M. Yoshida and M. Ihara, Chem.–Eur. J., 2004, 10, 2886–2893 CrossRef CAS; J. Sun, S.-i. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS; W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2–12 CrossRef CAS; T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC; M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC; A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822–9837 CrossRef CAS.
- H. Sugimoto and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5561–5573 CrossRef CAS; D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS; G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS; D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765–10780 CrossRef CAS; M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS; B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef CAS.
- M. North, F. Pizzato and P. Villuendas, ChemSusChem, 2009, 2, 862–865 CrossRef CAS; M. North and M. Omedes-Pujol, Tetrahedron Lett., 2009, 50, 4452–4454 CrossRef CAS; W. Clegg, R. W. Harrington, M. North, F. Pizzato and P. Villuendas, Tetrahedron: Asymmetry, 2010, 21, 1262–1271 CrossRef CAS; M. North and P. Villuendas, Org. Lett., 2010, 12, 2378–2381 CrossRef CAS; M. North and M. Omedes-Pujol, Beilstein J. Org. Chem., 2010, 6, 1043–1055 Search PubMed; C. Beattie, M. North and P. Villuendas, Molecules, 2011, 16, 3420–3432 Search PubMed; M. Morcillo, M. North and P. Villuendas, Synthesis, 2011, 1918–1925 Search PubMed; P. Lenden, P. M. Ylioja, C. González-Rodríguez, D. A. Entwistle and M. C. Willis, Green Chem., 2011, 13, 1980–1982 RSC.
-
G. J. Harmsen, E. van der Heide and C. L. Vrouwenvelder, Shell international research, WO2004089866(A1) Search PubMed;
W. E. Evans, M. L. Hess, M. A. Matusz and E. M. G. van Kruchten, Shell international research, WO2009140318(A1) Search PubMed;
J. G. J. Beckers, E. van der Heide, G. M. M. van Kessel and J.-P. Lange, Shell international research, WO2005051939(A1) Search PubMed;
T. M. Nisbet and G. G. Vaporciyan, Shell international research, WO2008128956(A1) Search PubMed;
E. van der Heide, G. M. M. van Kessel, T. M. Nisbet and G. G. Vaporciyan, Shell Oil Company, US20070197802(A1) Search PubMed;
R. G. Duranleau, E. C. Y. Nieh and J. F. Knifton, Texaco Inc., US4691041 1986 Search PubMed;
J. F. Knifton, Texaco Inc., US4661609 1987 Search PubMed;
J. F. Knifton, Texaco Inc., US4734518 1988 Search PubMed; J. F. Knifton and R. G. Duranleau, J. Mol. Catal., 1991, 67, 389–399 Search PubMed; S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 Search PubMed.
- J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323–3326 CrossRef CAS; J. Meléndez, M. North and P. Villuendas, Chem. Commun., 2009, 2577–2579 RSC; M. North, P. Villuendas and C. Young, Chem.–Eur. J., 2009, 15, 11454–11457 CrossRef CAS; I. S. Metcalfe, M. North, R. Pasquale and A. Thursfield, Energy Environ. Sci., 2010, 3, 212–215 RSC; J. Meléndez, M. North, P. Villuendas and C. Young, Dalton Trans., 2011, 40, 3885–3902 RSC; M. North, B. Wang and C. Young, Energy Environ. Sci., 2011, 4, 4163–4170 RSC.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS; W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem.–Eur. J., 2010, 16, 6828–6843 CAS.
- M. North and C. Young, Catal. Sci. Technol., 2011, 1, 93–99 RSC.
- See for examples: A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda and A. Lara-Sanchez, Dalton Trans., 2004, 1499–1510 Search PubMed; C. Pettinari and R. Pettinari, Coord. Chem. Rev., 2005, 249, 663–691 RSC; H. R. Bigmore, S. C. Lawrence, P. Mountford and C. S. Tredget, Dalton Trans., 2005, 635–651 CrossRef CAS; R. G. Howe, C. S. Tredget, S. C. Lawrence, S. Subongkoj, A. R. Cowley and P. Mountford, Chem. Commun., 2006, 223–225 RSC; A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sanchez, L. F. Sánchez-Barba, M. Sánchez-Molina, S. Franco, M. I. López-Solera and A. M. Rodríguez, Inorg. Chem., 2007, 46, 8475–8477 RSC; A. Otero, J. Fernández-Baeza, A. Antiñolo, A. Lara-Sanchez, J. Tejeda and L. F. Sánchez-Barba, Eur. J. Inorg. Chem., 2008, 5309–5326 CrossRef CAS; H. Kopf, B. Holzberger, C. Pietraszuk, E. Hübner and N. Burzlaff, Organometallics, 2008, 27, 5894–5905 CrossRef CAS; G. A. Santillan and C. J. Carrano, Dalton Trans., 2009, 6599–6605 CrossRef CAS; A. Otero, J. Fernández-Baeza, J. Tejeda, A. Lara-Sanchez, S. Franco, J. Martínez-Ferrer, M. P. Carrión, L. F. Sánchez-Barba, I. López-Solera and A. M. Rodríguez, Inorg. Chem., 2011, 50, 1826–1839 RSC.
- A. Otero, J. Fernández-Baeza, A. Antiñolo, J. Tejeda, A. Lara-Sanchez, L. Sánchez-Barba, M. Sánchez-Molina, S. Franco, I. López-Solera and A. M. Rodríguez, Eur. J. Inorg. Chem., 2006, 707–710 CrossRef CAS; A. Otero, J. Fernández-Baeza, A. Lara-Sanchez, C. Alonso-Moreno, I. Márquez-Segovia, L. F. Sánchez-Barba and A. M. Rodríguez, Angew. Chem., Int. Ed., 2009, 48, 2176–2179 CrossRef CAS; A. Otero, A. Lara-Sanchez, J. Fernández-Baeza, E. Martínez-Caballero, I. Márquez-Segovia, C. Alonso-Moreno, L. F. Sánchez-Barba, A. M. Rodríguez and I. López-Solera, Dalton Trans., 2010, 39, 930–940 RSC.
- A. Otero, A. Lara-Sanchez, J. Fernández-Baeza, C. Alonso-Moreno, J. Tejeda, J. A. Castro-Osma, I. Márquez-Segovia, L. F. Sánchez-Barba, A. M. Rodríguez and M. V. Gómez, Chem.–Eur. J., 2010, 16, 8615–8619 CrossRef CAS; A. Otero, A. Lara-Sanchez, J. Fernández-Baeza, C. Alonso-Moreno, J. A. Castro-Osma, I. Márquez-Segovia, L. F. Sánchez-Barba, A. M. Rodríguez and J. C. García-Martínez, Organometallics, 2011, 30, 1507–1522 CrossRef CAS.
- X.-B. Lu, Y.-J. Zhang, K. Jin, L.-M. Luo and H. Wang, J. Catal., 2004, 227, 537–541 CrossRef CAS.
- N. Takeda and S. Inoue, Bull. Chem. Soc. Jpn., 1978, 51, 3564–3567 CAS; K. Kasuga, S. Nagao, T. Fukumoto and M. Handa, Polyhedron, 1996, 15, 69–72 CrossRef CAS.
- S.-F. Yin and S. Shimada, Chem. Commun., 2009, 1136–1138 RSC.
- A. Berkessel and M. Brandenburg, Org. Lett., 2006, 8, 4401–4404 CrossRef CAS; S.-W. Chen, R. B. Kawthekar and G.-J. Kim, Tetrahedron Lett., 2007, 48, 297–300 CrossRef CAS; J. Wang, J. Wu and N. Tang, Inorg. Chem. Commun., 2007, 10, 1493–1495 CrossRef CAS; L. Jin, H. Jing, T. Chang, X. Bu, L. Wang and Z. Liu, J. Mol. Catal. A: Chem., 2007, 261, 262–266 CrossRef CAS; T. Chang, H. Jing, L. Jin and W. Qiu, J. Mol. Catal. A: Chem., 2007, 264, 241–247 CrossRef CAS; L. Jin, Y. Huang, H. Jing, T. Chang and P. Yan, Tetrahedron: Asymmetry, 2008, 19, 1947–1953 CrossRef CAS; S. Zhang, Y. Song, H. Jing, P. Yan and Q. Cai, Chin. J. Catal., 2009, 30, 1255–1260 Search PubMed.
- R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS; X. Zhang, Y.-B. Jia, X.-B. Lu, B. Li, H. Wang and L.-C. Sun, Tetrahedron Lett., 2008, 49, 6589–6592 CrossRef CAS.
- H. Kisch, R. Millini and I.-J. Wang, Chem. Ber., 1986, 119, 1090–1094 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 

