E/Z selectivity in ruthenium-mediated cross metathesis†
Cristina
Luján
and
Steven P.
Nolan
*
EaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, United Kingdom. E-mail: snolan@st-andrews.ac.uk
First published on 13th January 2012
Abstract
The influence of ancillary ligand on the activity of diverse NHC-ruthenium indenylidene complexes in cross metathesis (CM) have been examined. The phospine/NHC ancillary ligands tested lead to varied E/Z selectivities as a function of catalyst ancillary ligands. CM reaction between olefins leads to good initial Z-selectivity. However, secondary metathesis is responsible for the isomerisation of the product mixture leading to the thermodynamically favored E-isomer. Very active Ru catalysts (that activate easily under mild conditions) achieved very good conversions in short reaction times but isomerisation of the initially formed product was rapidly achieved as well. When less labile leaving groups (NHC) were attached to the Ru center, initiation time proved much longer but up to 1.3/1 E/Z ratio was achieved even at long reaction times. The effects of ancillary ligand on initiation and E/Z selectivity are discussed.
Introduction
Olefin metathesis represents a versatile tool for the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds which are present in a vast variety of molecules. The interest for metal-catalyzed olefin metathesis has exponentially risen1 given the high impact of this reaction on the syntheses of natural products and of various functionalized materials and polymers.2 Many variations of olefin metathesis exist such as ring closing metathesis (RCM), ring opening metathesis polymerization (ROMP), ring opening cross metathesis (ROM), alkyne metathesis (AM). Among these, cross metathesis (CM) (Scheme 1) represents the most challenging version, as it lacks the entropic driving force of RCM and the ring-strain release of ROMP.3 In CM, there are concerns regarding selective formation of heterolefins over homocoupled products, and in spite of these issues, the major CM limitation is still poor Z-selectivity at high conversions.4 Grubbs has recently addressed some of these issues using a modified NHC ligand to achieve improved Z-selectivity.5
C double bonds which are present in a vast variety of molecules. The interest for metal-catalyzed olefin metathesis has exponentially risen1 given the high impact of this reaction on the syntheses of natural products and of various functionalized materials and polymers.2 Many variations of olefin metathesis exist such as ring closing metathesis (RCM), ring opening metathesis polymerization (ROMP), ring opening cross metathesis (ROM), alkyne metathesis (AM). Among these, cross metathesis (CM) (Scheme 1) represents the most challenging version, as it lacks the entropic driving force of RCM and the ring-strain release of ROMP.3 In CM, there are concerns regarding selective formation of heterolefins over homocoupled products, and in spite of these issues, the major CM limitation is still poor Z-selectivity at high conversions.4 Grubbs has recently addressed some of these issues using a modified NHC ligand to achieve improved Z-selectivity.5
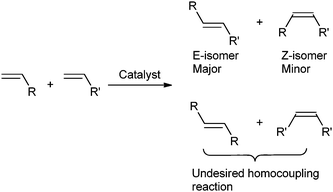 | ||
| Scheme 1 General CM reaction involving two terminal olefins. | ||
One of the main drawbacks of CM is the existence of a competitive secondary metathesis of the desired products6–8 causing poor Z-selectivity. Isomerisation of the desired and more reactive Z-product is promoted to the thermodynamically more stable E-isomer (Scheme 2).9,10
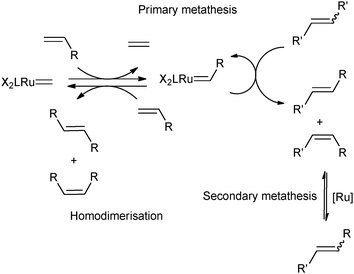 | ||
| Scheme 2 Secondary metathesis mechanism. | ||
Numerous organometallic Ru complexes have been recently reported as metathesis catalysis.2,11 Currently, incarnations of NHC-based ruthenium pre-catalysts stand at the forefront of the state-of-the-art as alkene metathesis catalysts. The stronger σ-donation12,13 of the NHC ligands, compared to the phosphines used in earlier days, adds increased stability to the Ru metal centre. Additionally, complexes containing mixed NHC/phosphine, while stable, display superior activity and more functional group tolerance than bis-NHC or bis-phosphine analogs in olefin metathesis reactions.9d,10a,b,d,14
Selective Z-olefin metathesis has been successfully achieved in the presence of Mo catalysts15 and their use has been demonstrated in the synthesis of natural products.16 High Z-selectivities have also been reported in substrates containing sp-hybridized substituent such as acrylonitrile or enynes.12b,17 To date, CM with both high efficiency and high Z-selectivity has scarcely been accomplished with ruthenium systems.3,5,18 The first example of Z-selective homodimerization of terminal olefins using a ruthenium-based catalyst has been recently reported by Grubbs.19 It has also been shown that E/Z selectivity can be affected by both catalyst and olefin isomer stabilities along with ligand and olefin sterics. Even though a large variety of catalysts have been tested in CM, until now no clear trends have been identified. The only established trend so far identified is that secondary metathesis leading to greater amounts of the more thermodynamically stable E-isomer is related to more thermally stable Ru complexes. The formation of the more thermodynamically stable E-isomer is favored for second generation catalysts.5,20 For example, CM of 1,4-diacetoxy-2-butene (2) with allylbenzene (1) gave an E/Z ratio of ∼3/1 to 4/1 with a diphosphine Ru catalyst while a NHC-containing Ru catalyst afforded an E/Z ratio of ∼6/1 to 10/1 (Scheme 3).5,18d
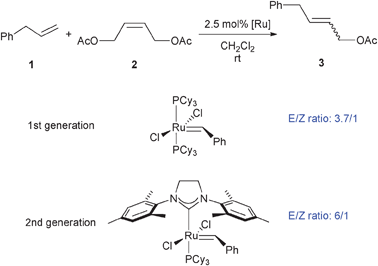 | ||
| Scheme 3 Cross Metathesis involving an internal olefin. | ||
It has been observed that the difference between the reactivity of different 14-electron Ru catalysts intermediates is due to electronic factors.21 According to the accepted reaction mechanism,16 the formation of the active 14-electron Ru species in a diphosphine system is very favorable but the catalyst carries out few catalytic turnovers before being “quenched” by the free phosphine. On the contrary, pre-catalysts containing a NHC ligand dissociate phosphine relatively inefficiently. However, once the phosphine is dislodge, coordination to the olefin is facile compared to re-binding of the free PR3.17,22 The leaving group influence the catalytic properties and the fast and thermodynamically facile formation of the active 14 electron Ru catalyst species is a crucial factor.23 The question as to the origins of the selectivity still remains greatly unanswered. The efficiency and selectivity of a CM process may very well depend on the ancillary ligand and/or on the nature and coordination of the alkene. Therefore the capability of the catalyst to perform secondary metathesis and consequently affect the product isomeric distribution could be directly dependent of the lability of the ancillary ligand. We have recently studied the possible electronic and steric influence of N-aryl group and backbone substitution in Z-olefin isomer synthesis.24 The exact role of these is still unclear and requires further study.
The choice of the ancillary ligand remains crucial in finding an adequate compromise between lability of the dissociating ligand to initiate the reaction and lifetime of the active catalyst. For these reasons, the E/Z-selectivity of a CM reaction was studied in the presence of various Ru pre-catalysts containing either NHC/phosphine or bis-NHC ligands.
Results and discussion
Initially, CM of alkenes 1 and 2 in the presence of pre-catalysts bearing SIMes and phosphine ligands were investigated. CM in the presence of pre-catalyst 425 containing PCy3 afforded a satisfactory 1.1/1 E/Z ratio within one hour, although, only 50% conversion was observed (entry 1, Table 1). On the other hand, the replacement of the PCy3 ligand with the more labile PPh3 ligand9d drastically improved the conversion to 96% but at the expense of the Z-selectivity (entry 2, Table 1). We envisaged that tuning the electron properties of PPh3, by introducing different electron-withdrawing/donating substituents, which does not significantly change the sterics, (see σ in entries 3 and 4, Table 1) could influence the phosphine dissociation/re-binding rates without greatly affecting conversions. Unfortunately, the fast initiation rates of those three Ru catalysts as well as their stability favored E-alkene formation (from 9/1 to 10/1 E/Z ratio, entries 3 and 4, Table 1) by a possible rapid and efficient isomerisation of the product.23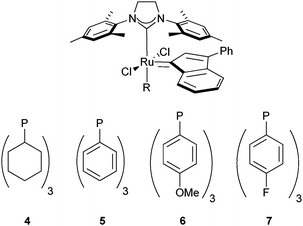
Challenged to establish how the initiation rate and the stability of the catalyst translate into selectivity and conversion of the CM reaction, the behaviour of two SIMes ruthenium–indenylidene complexes, 4 and 5, were studied at shorter reaction times.26 The CM reaction of alkenes 1 and 2 in the presence of 4 and 5 was measured at 10 min intervals. In both cases, better Z-selectivity was observed at low conversions and short reaction times for the first 50 min (Fig. 1 and 2). However, as the reaction proceeded with time a different catalytic behaviour is observed, possibly depending on the lifetime of the catalytically active ruthenium species promoting (more or less) the isomerisation of the Z-product (Fig. 1 and 2).27 The rapid initiation of catalyst 5 affords higher conversions over the same time period as 4. Unfortunately, isomerisation to the more stable E-isomer was also vastly promoted within the first 60 min. It is already known that faster phosphine exchange is observed with PPh3 ligand than with PCy3.20 The more labile nature of PPh3 and lack of its bulkiness has proved to translate into a decreased stability of the corresponding Ru complex,9d resulting in shorter catalyst lifetime with fast initial conversion. Therefore, it was not surprising that no isomerisation was observed at long reaction times. On the other hand, 4 does not show any conversion within the first 50 min but isomerisation of the Z-product is still observed after 5 h (Fig. 1).
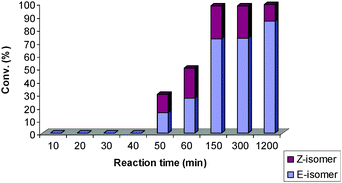 | ||
| Fig. 1 The reactivity of 4 (SIMes/PCy3 bearing) with time.33 | ||
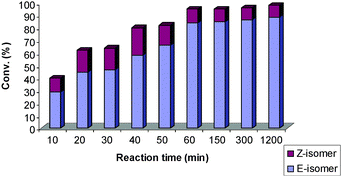 | ||
| Fig. 2 The reactivity of 5 (bearing SIMes/PPh3) with time.27 | ||
In an attempt to examine the generality of our proposal, two different SIPr-phosphine ruthenium pre-catalysts were also tested under the standard CM conditions. Contrary to the SIMes results, CM in the presence of the SIPr/PCy328 (8) leads to high conversions within one hour at room temperature with poor Z-selectivity. Once again, the carbene-PPh3 Ru pre-catalyst29 (9) promoted fast conversion in shorter times, still with disappointing Z-selectivity. Considering that SIPr is bulkier than SIMes and that PCy3 is bulkier than PPh3, the leaving group re-binding rate could logically be affected by steric factors. Re-attachment of the bulky ligands to the catalyst could be more challenging, potentially leading to poorer Z-selectivity.
At shorter reaction times, 8 bearing PCy3 is initially more prone towards Z-selectivity than 9 (Fig. 3 and 4). Both 8 and 9 have very fast initiation, achieving high conversions quickly. For 8, secondary metathesis plays an important role. After achieving full conversion within the first 10 min, the catalyst is still highly active leading to isomerisation of the Z-product (Fig. 4). On the other hand, 9 did not show any important catalytic activity after 20 min at room temperature (Fig. 3).
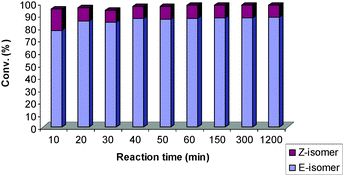 | ||
| Fig. 3 The reactivity of 8 (bearing SIPr/PCy3) with time.27 | ||
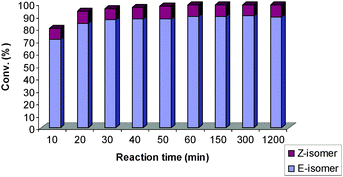 | ||
| Fig. 4 The reactivity of 9 (bearing SIPr/PPh3) with time.27 | ||
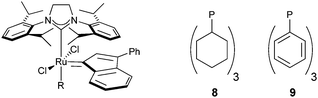
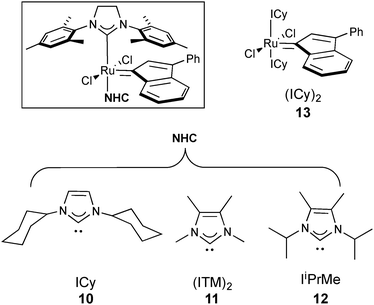
Comparing the different Ru complexes so far tested the ancillary ligand is a stable species in solution. We wondered that if the free leaving group had a stronger binding energy to the metal and was not as stable as a free species, less isomerisation of the alkene could take place. For that reason, various Ru complexes bearing mixed carbenes ligands featuring one sterically small NHC: ICy, ITM, IiPrMe, and one larger congener SIMes (10, 11 and 12 respectively) or two small bis-ICy 13 were tested.28
The stability observed for those catalysts is due to the strongly bonded NHC ligand.30 Their TEPs30,31 are reported in Table 3, the weakest NHC-Ru bond will be the leaving group in complexes 10, 11 and 12 (Table 3). It has been previously reported that those complexes posses high reactivity towards RCM at low catalyst loading.30 CM in the presence of ICy 10 or IiPrMe 12 pre-catalysts, with similar electron-donating properties bearing the same leaving group (SIMes), showed similar good Z-selectivity and conversion at room temperature (Table 3, entries 1 and 3). On the other hand, complex 11 containing two carbenes with similar electron properties and less electron donating than 10 and 12 showed a 3/1 E/Z ratio. It is significant that in all cases NHC/NHC catalysts had both slow initiation rates and conversion. On the other hand, the best Z-selectivity and conversion were observed only after 5 h when two ICy ligands were bound to Ru (Table 3, entry 4).
| Entry | Pre-catalyst | Ligand | TEPa (cm−1) | t (h) | Conv.b,c (%) | E/Zb |
|---|---|---|---|---|---|---|
| a Determined in Ni(CO)3(NHC).32 b Reaction conditions: 1,4-diacetoxy -2-butene (2 equiv.), [Ru] pre-catalyst (1 mol%), CH2Cl2, rt, N2. c Determined by 1H NMR. | ||||||
| 1 | 10 | ICy | 2051.7 | 20 | >99 | 1.8/1 |
| 2 | 11 | ITM | 2046.6 | 20 | 67 | 3.1/1 |
| 3 | 12 | IiPrMe | 2049.7 | 20 | 55 | 1.9/1 |
| 4 | 13 | ICy | 2051.7 | 5 | 89 | 1.3/1 |
Although we were pleased to see that good Z-selectivity had been achieved, these gains were mitigated by sluggish conversions requiring long reaction times. Reactions using pre-catalysts 10–13 were repeated at higher temperatures in an attempt to promote higher and faster conversions. After 1 h at 80 °C nearly full conversion was achieved with complexes 10, 11 and 12 (Table 4, entries 1, 3 and 5). The general trend observed with these complexes at 80 °C was an important isomerization of the product at long reaction times 12 (Table 4, entries 2, 4 and 6). In contrast, Z-selectivity between 1 and 2 in the presence of 13 gave full conversion after 5 h. Surprisingly, the E/Z ratio obtained was the same as the reaction at room temperature with a considerable conversion increase (E/Z ratio 2.2/1; Table 4, entry 7). To our delight, isomerisation of the product and long reaction times did not occurred, the E/Z ratio remained the same (Table 4, entry 8). Albeit 13 afforded a great E/Z selectivity at 80 °C and generally better conversions were achieved for the other catalysts tested, concurrent increased in most of the cases in thermodynamic product of 7/1 E/Z ratio made this method less desirable (Table 4, entries 2, 4 and 6).
| Entry | Pre-catalyst | Ligands | t (h) | Conv.a,b (%) | E/Zb |
|---|---|---|---|---|---|
| a Reaction conditions: 1,4-diacetoxy-2-butene (2 equiv.), [Ru] pre-catalyst (1 mol%), toluene, 80 °C, N2. b Determined by 1H NMR. | |||||
| 1 | 10 | SIMes/ICy | 1 | 98 | 5.8/1 |
| 2 | 10 | SIMes/ICy | 20 | 99 | 7/1 |
| 3 | 11 | SIMes/ITM | 1 | 98 | 2.9/1 |
| 4 | 11 | SIMes/ITM | 20 | 99 | 5.2/1 |
| 5 | 12 | SIMes/IiPrMe | 1 | >99 | 3.3/1 |
| 6 | 12 | SIMes/IiPrMe | 20 | 99 | 5.5/1 |
| 7 | 13 | ICy/ICy | 5 | >99 | 2.2/1 |
| 8 | 13 | ICy/ICy | 20 | >99 | 2.4/1 |
The significance of the effect of the secondary metathesis to the E/Z final ratio was tested using alkene 3 with a known 2.9/1 E/Z ratio and one Ru complex with a low and one with a fast isomerisation rate, 4 and 12 respectively, in CH2Cl2 at room temperature (Scheme 4). As expected, the more active pre-catalyst 4 isomerized the olefin within 1 h into the more thermodynamically stable E-olefin (10/1 E/Z ratio). As anticipated, the ratio obtained did not increased at longer reaction times possibly due to the catalysts short lifetime. Conversely, no isomerisation was observed in the presence of 12 over 16 hours at room temperature.
 | ||
| Scheme 4 Secondary metathesis studies. | ||
All the results so far have shown the tendency of Ru complexes to promote initial good Z-selectivity. However, isomerisation towards the more thermally stable E-isomer takes place in the case of very active Ru catalysts. Following those results we were very interested to see the impact of accelerating the termination step by the addition of excess ligand to the reaction mixture. The amount of free catalyst, therefore, should decrease and, hopefully, reduce the isomerisation of the metathesis product.
When an excess of PPh3 was added to the reaction mixture in the presence of complex 5 the conversion rate remained the same (80% conversion within an hour), along with improved Z-selectivity, from 7.3/1 (Table 1, entry 2) to 5.3/1. On the other hand, an excess of PCy3 to the reaction catalyzed by 4 lowered the conversion rate, but again, also promoted Z-selectivity during the first hour (Fig. 5). Surprisingly, even though, the catalyst remained active for the next 19 h, significant isomerisation was not observed (Fig. 5). Therefore, an excess of the ligand present in the reaction mixture re-binding to the Ru catalyst disfavors secondary metathesis achieving better Z-selectivity even at long reaction times.
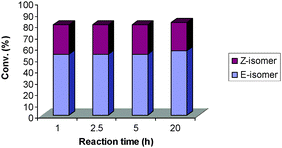 | ||
| Fig. 5 CM reaction in the presence of SIMes/PCy34 and an excess of PCy3.27 | ||
Conclusions
In previous work we tried unsuccessfully to determine if there was any correlation between electronic and steric effects of the NHCs attached to diverse Ru metal centers. Keeping with this trend, the influence of the ancillary ligand in the E/Z selectivity outcome has now been studied. Generally, CM reaction between two olefins achieves good initial Z-selectivity, and secondary metathesis is responsible of isomerisation of the product mixture towards the most stable E-isomer. Therefore CM selectivity is compromised by highly active and long-lived Ru catalysts, such as Grubbs second generation. Very active Ru catalysts, such as 5 and 9, achieved very good conversions at short reaction times, but as we expected the high activity of the catalyst also promoted isomerisation of the product. Ru catalysts with a slow initiation rate, such as 4 achieved better Z-selectivity with less isomerisation. Measuring initial conversion at short reaction gave good E/Z ratios, with isomerisation only occurring later. This demonstrates the importance of olefin isomerisation. When less labile leaving groups (NHC) were attached to the Ru center the initiation time was much longer but as we expected very good Z-selectivity was achieved even at long reaction times. The addition of an excess of the ancillary ligand reduced the amount of active catalytic species reducing the isomerisation of the product.Experimental
General procedure for the CM reactions
A solution of allyl benzene (0.5 mmol), cis-1,4-diacetoxy-2-butene (2, 1.2 mmol) and the Ru catalyst (1 mol%) in solvent (5 mL) was stirred under N2 atmosphere. Reaction conversion was followed by taking 0.5 mL reaction samples, quenched with ethyl vinyl ether (1 mL) and analysed by 1H NMR shifts at 4.57 (E-isomer) and 4.77 (Z-isomer) ppm.34CM at room temperature in CH2Cl2
A solution of allyl benzene (1, 0.5 mmol), cis-1,4-diacetoxy-2-butene (2, 1.2 mmol) and the Ru catalyst (1 mol%) in CH2Cl2 (5 mL) was stirred at room temperature and under N2 atmosphere. Reaction conversion was followed by taking 0.5 mL reaction samples, quenched with ethyl vinyl ether (1 mL) and analysed by 1H NMR shifts.CM at 80 °C in toluene
A solution of allyl benzene (1, 0.5 mmol), cis-1,4-diacetoxy-2-butene (2, 1.2 mmol) and the ancillary ligand (8 mol%) and the Ru catalyst (1 mol%) in toluene (5 mL) was stirred at 80 °C and under N2 atmosphere. Reaction conversion was followed by taking 0.5 mL reaction samples, quenched with ethyl vinyl ether (1 mL) and analysed by 1H NMR shifts.CM in the presence of excess ancillary ligand
A solution of allyl benzene (1, 0.5 mmol), cis-1,4-diacetoxy-2-butene (2, 1.2 mmol) and the ancillary ligand (8 mol%) and the Ru catalyst (1 mol%) in CH2Cl2 (5 mL) was stirred at room temperature and under N2 atmosphere. Reaction conversion was followed by taking 0.5 mL reaction samples, quenched with ethyl vinyl ether (1 mL) and analysed by 1H NMR shifts.Acknowledgements
The EC is gratefully acknowledged for funding through the seventh framework program (CP-FP 211468-2-EUMET). SPN is a Royal Society Wolfson Research Merit Award holder.Notes and references
- For recent reviews on olefin metathesis: (a) F. Boeda, H. Clavier and S. P. Nolan, Chem. Commun., 2008, 2726–2740 RSC; (b) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2009, 110, 1746–1787 CrossRef; (c) A. M. Lozano-Vila, S. Monsaert, A. Bajek and F. Verpoort, Chem. Rev., 2010, 110, 4865–4909 CrossRef CAS.
- (a) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS; (c) T. J. Donohoe, A. J. Orr and M. Bingham, Angew. Chem., Int. Ed., 2006, 45, 2664–2667 ( Angew. Chem. , 2006 , 118 , 2730–2736 ) CrossRef CAS; (d) M. D. McReynolds, J. M. Dougherty and P. R. Hanson, Chem. Rev., 2004, 104, 2239–2258 CrossRef CAS; (e) A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243–251 CrossRef CAS; (f) S. Kotha and K. Lahiri, Synlett, 2007, 2767–2784 CrossRef CAS; (g) W. A. L. van Otterlo and C. B. de Koning, Chem. Rev., 2009, 109, 3743–3782 CrossRef CAS; (h) P. R. Krishna and M. Alivelu, Helv. Chim. Acta, 2011, 94, 1102–1107 CrossRef CAS.
- (a) A. Fürstner, Angew. Chem., 2000, 112, 3013–3043 ( Angew. Chem., Int. Ed. , 2000 , 39 , 3140–3172 ) CrossRef; (b) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS; (c) U. Frenzel and O. Nuyken, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2895–2916 CrossRef CAS.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360–11370 CrossRef CAS.
- K. Endo and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 8525–8527 CrossRef CAS.
- P. Teo and R. H. Grubbs, Organometallics, 2010, 29, 6045–6050 CrossRef CAS.
- Primary metathesis involves the reaction of Ru catalyst with a terminal olefin forming a Ru alkylidene species that will react with another olefin to afford E- or Z-product. Secondary metathesis, on the other hand, involves the reaction of the E-/Z-product with Ru catalysts resulting in the interconversion of the product isomers towards the most thermodynamically stable.
- B. Allaert, N. Ledoux, N. Dieltiens, H. V. Mierde, C. V. Stevens, P. Van Der Voort and F. Verpoort, Catal. Commun., 2008, 9, 1054–1059 CrossRef CAS.
- (a) C. W. Lee and R. H. Grubbs, Org. Lett., 2000, 2, 2145–2147 CrossRef CAS; (b) B. Kang, J. M. Lee, J. Kwak, Y. S. Lee and S. Chang, J. Org. Chem., 2004, 69, 7661–7664 CrossRef CAS.
- D. R. Anderson, T. Ung, G. Mkrtumyan, G. Bertrand, R. H. Grubbs and Y. Schrodi, Organometallics, 2008, 27, 563–566 CrossRef CAS.
- (a) A. Fürstner, J. Grabowski and C. W. Lehmann, J. Org. Chem., 1999, 64, 8275–8280 CrossRef; (b) L. Jafarpour, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 5416–5419 CrossRef CAS; (c) For a review on ruthenium-indenylidene complexes see: F. Boeda, H. Clavier and S. P. Nolan, Chem. Commun., 2008, 2726–2740 RSC.
- (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (b) K. Öfele, J. Organomet. Chem., 1968, 12, 42–43 CrossRef; (c) H. W. Wanzlick and H. J. Schönherr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141–142 CrossRef CAS; (d) J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2674–2678 CrossRef CAS.
- (a) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247–2250 CrossRef CAS; (b) T. Weskamp, W. C. Schattenmann, M. Spiegler and W. A. Herrmann, Angew. Chem., Int. Ed., 1998, 37, 2490–2493 CrossRef CAS; (c) M. S. Sanford, J. A. Love and R. H. Grubbs, Organometallics, 2001, 20, 5314–5318 CrossRef CAS; (d) J. Huang, H.-J. Schanz, E. D. Stevens and S. P. Nolan, Organometallics, 1999, 18, 5375–5380 CrossRef CAS; (e) J. C. Conrad, G. P. A. Yap and D. E. Fogg, Organometallics, 2003, 22, 1986–1988 CrossRef CAS.
- (a) T. Weskamp, F. J. Kohl, W. Hieringer, D. Gleich and W. A. Herrmann, Angew. Chem., Int. Ed., 1999, 38, 2416–2419 CrossRef CAS; (b) L. Ackermann, A. Fürstner, T. Weskamp, F. J. Kohl and W. A. Herrmann, Tetrahedron Lett., 1999, 40, 4787–4790 CrossRef CAS.
- For impressive E/Z selectivity using molybdenum systems, see: (a) I. Ibrahem, M. Yu, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3844–3845 CrossRef CAS; (b) W. E. Crowe and D. R. Goldberg, J. Am. Chem. Soc., 1995, 117, 5162–5163 CrossRef CAS.
- S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461–466 CrossRef CAS.
- E. C. Hansen and D. Lee, Org. Lett., 2004, 6, 2035–2038 CrossRef CAS.
- (a) G. C. Vougioukalakis and R. H. Grubbs, Organometallics, 2007, 26, 2469–2472 CrossRef CAS; (b) G. C. Vougioukalakis and R. H. Grubbs, J. Am. Chem. Soc., 2008, 130, 2234–2245 CrossRef CAS; (c) K. Voigtritter, S. Ghorai and B. H. Lipshutz, J. Org. Chem., 2011, 76, 4697–4702 CrossRef CAS. For a comparative catalytic evaluation of the most commonly used ruthenium-based catalyst: (d) T. Ritter, A. Hejl, A. G. Wenzel, T. W. Funk and R. H. Grubbs, Organometallics, 2006, 25, 5740–5745 CrossRef CAS.
- B. K. Keitz, K. Endo, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 9686–9688 CrossRef CAS.
- (a) M. S. Sanford, M. Ulman and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 749–750 CrossRef CAS; (b) M. S. Sanford, J. A. Love and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 6543–6544 CrossRef CAS.
- B. F. Straub, Angew. Chem., Int. Ed., 2005, 44, 5974–5978 CrossRef CAS.
- J. A. Love, M. S. Sanford, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 10103–10109 CrossRef CAS.
- B. F. Straub, Adv. Synth. Catal., 2007, 349, 204–214 CrossRef CAS.
- C. Luján and S. P. Nolan, J. Organomet. Chem., 2011, 696, 3935 CrossRef.
- J. Broggi, C. A. Urbina-Blanco, H. Clavier, A. Leitgeb, C. Slugovc, A. M. Z. Slawin and S. P. Nolan, Chem.–Eur. J., 2010, 16, 9215–9225 CrossRef CAS.
- For Hammett σp, see: Advanced Organic Chemistry, ed. M. B. Smith and J. March, Wiley Interscience, New York, NY, 5th edn, 2001, ch. 9 Search PubMed.
- (a) N. Bahri-Laleh, R. Credendino and L. Cavallo, Beilstein J. Org. Chem., 2011, 7, 40–45 CrossRef CAS; (b) A. G. Wenzel, G. Blake, D. G. VanderVelde and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 6429–6439 CrossRef CAS.
- X. Bantreil, R. A. M. Randall, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3007–3011 CrossRef CAS.
- Conversions determined by 1H NMR.
- H. Clavier, C. A. Urbina-Blanco and S. P. Nolan, Organometallics, 2009, 28, 2848–2854 CrossRef CAS.
- C. A. Urbina-Blanco, S. Manzini, J. P. Gomes, A. Doppiu and S. P. Nolan, Chem. Commun., 2011, 47, 5022–5024 RSC.
- The Tolman Electronic Parameter (TEP) is employed as a measure of the electron donating or withdrawing ability of a ligand, see: C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS.
- W. H. Henderson, C. T. Check, N. Proust and J. P. Stambuli, Org. Lett., 2010, 12, 824–827 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00457g |
| This journal is © The Royal Society of Chemistry 2012 |