Catalytic activity and X-ray photoelectron spectroscopy performance of Bi2MoxW(1−x)O6 solid-solutions†
R.
Rangel
*a,
P.
Bartolo-Pérez
b,
E.
Martínez
c,
X. A.
Trejo-Cruz
a,
G.
Díaz
e and
D. H.
Galván
d
aUniversidad Michoacana de S.N.H., Morelia, Mich, ., Mexico. E-mail: rrangel@umich.mx; Fax: +52 443 3273584; Tel: +52 443 3273584
bDepartamento de Física Aplicada, Centro de Investigación y de Estudios Avanzados-IPN, Mérida, Yucatán, México. E-mail: pascual@mda.cinvestav.mx
cCentro de Investigación en Materiales Avanzados, S.C., Parque de Investigación e Innovación Tecnológica (PIIT), Nueva Carretera Aeropuerto Km. 10, Apodaca, Nuevo León, Mexico. E-mail: eduardo.martinez@cimav.edu.mx
dCentro de Nanociencias y Nanotecnología, Universidad Nacional Autónoma de México, Apdo. Postal 2681, 22800 Ensenada, B. C., México. E-mail: donald@cnyn.unam.mx
eInstituto de Física, Universidad Nacional Autónoma de México, A.P. 20-364, México DF 01000, México. E-mail: diaz@fisica.unam.mx
First published on 20th January 2012
Abstract
The present paper describes the results of the study of solid solutions of Bi2MoxW(1−x)O6, which were prepared by the route of solid-state reaction and tested for catalytic activity in the oxidation of CO into CO2. X-Ray photoelectron spectroscopy (XPS) was conducted to obtain information on the chemical composition, and with the use of Bi2MoO6 and Bi2WO6 synthesized standards, quantitative measures were obtained. Catalytic studies of the samples show that they possess high activity, as a change in the temperature of activation as a function of the sintering temperature with the Mo/W ratio was detected. Bi–Mo mixed oxides have synergistic effects on the oxidation of CO, making possible the full conversion at lower temperatures than compounds that contain only single phases. The work described here demonstrates that a solid solution can be more efficient in oxidation, compared with single oxides studied separately.
1. Introduction
Metal-catalyzed CO oxidation has been widely studied in some detail for decades. Classical catalysts are those that are considered to contain mainly Pt (single elemental or conveniently combined with Pd or Rh) supported on Al2O3.1,2 Other materials, including Au/MOx/Al2O3 (where M = Mg, Mn),3 Pd/CeO2/TiO24 and Ir/TiO25 and Au/TiO2,6 have been reported to exhibit a high catalytic activity for CO oxidation. In this context, Bi–Mo based catalysts have demonstrated versatility, high efficiency and stability.7,8 α-Bi2Mo3O12, β-Bi2Mo2O9, and γ-Bi2MoO6 are known for their excellent catalytic activity for one of the most important industrial processes: the oxidation/ammoxidation of lower olefins.9 There is still wide room for further improvement due to the importance of the process in industry. Especially, the topic of controlling the stoichiometry of the various bismuth molybdates remains of interest because they constitute model catalysts.10 As an example, Williams et al.11 studied the propene oxidation to acrolein using Bi–Mo mixed oxides. They found that the nominal composition of Bi2MOxMe (x ≤ 0.25, Me = V or Ni) produced efficient catalysts to oxidize propene, with 90% conversion. Also, extremely dense single-phase Bi2Mo1−xWxO6 mixed oxides, prepared at 950 °C, with interesting ferroelectric properties have been reported, addressed mainly to studies of X-ray diffraction considering the composition range 0.5 < x < 1.0 focusing on the characterization of the Bi2Mo0.25W0.75O6 compound.Researchers established that ferroelectricity and dielectric constant values are dependant on the Mo/W ratio, unfortunately failing to fully analyze the composition range of the system.12 Another approach has been made when studying the reactivity of propylene or acrolein by testing different proportions of the α-Bi2Mo3O12, β-Bi2Mo2O9, γ-Bi2MoO6 phases, stating that the synergy is observed only in mixtures that include γ-Bi2MoO6 phase.13 The catalytic properties of the α, β and γ, Bi2MoO6 and Bi2WO6 compounds for CO oxidation have also been reported.7,8 Studies conclude that mobile oxygen ions in the three bismuth molybdate (and equivalently in bismuth tungstate) crystal phases are different. The mobile oxygen ions are O(1), O(11) and O(12) in the alpha phase; O(3), O(11), O(16) and O(18) in the beta phase; O(1) and O(5) in the gamma phase. The mobile lattice oxygen ions are proposed to be the source of the oxidizing oxygen responsible for the selective oxidation. One important finding is that mobile oxygen ions are all related to molybdenum ions rather than bismuth ions in the lattice.
The latter is very important because it means that by modifying the physical and chemical environment of the molybdenum oxide polyhedra it is possible to modify the catalyst selectivity or activity. Likewise data on the properties of Bi–Mo–W solid solutions over the entire composition range are still missing.
The present work reports the study of Bi2MoxW(1−x)O6 mixed oxide solid-solutions, which were successfully tested in the process of CO oxidation to CO2. Besides, chemical composition and oxidation states were obtained from X-ray photoelectron spectroscopy (XPS) measurements. It is pertinent to note that significant differences exist between this work and others reported since our approach includes a comprehensive study of compositions. Published reports consider the study of MoO3–WO3 or Bi2MoO6–Bi2WO6 mixtures, which are not truly solid solutions and thus the physicochemical properties are different from those reported here.
2. Materials and methods
Stoichiometric amounts of high purity Bi2O3, WO3 and MoO3 compounds, 99.99% purity from Alfa-Aesar, were mixed and made to react through a solid-state reaction to obtain Bi2MoxW(1−x)O6 compounds. Samples were ground in an agate mortar and shaped into disks of 10 mm diameter and 3 mm thickness using a pressure of 6 ton cm−2. In the next stage, samples were annealed at 700 and 900 °C.Specific surface area measurements were carried out by means of the BET method and measured in a Micrometrics Gemini 2060 RIG-100 model at 77 K. Previously, the samples were degassed for one hour at 200 °C.
X-Ray diffraction (XRD) analysis was performed with a Philips X'pert using CuKα (λ = 0.154 nm) radiation from 10–80°(2θ) operating at 30 keV and 20 mA. XPS analyses were performed in a Perkin-Elmer PHI 560 ESCA-SAM system, with a base pressure of 1 × 10−9 Torr. XPS spectra were obtained after 5 min of Ar+ sputtering. Samples were excited with 1486.6 eV using an Al anode. The spectrometer was calibrated using the Cu 2p3/2 (932.3 eV) and Cu 3p3/2 (74.9 eV) lines. Spectrum deconvolution was carried out using the spectra of X-ray satellite peak subtraction and background correction. For charge-up correction, the C 1s (284.6 eV) spectrum line was used. The reaction was carried out in the range of 150–600 °C, in a micro-flow reaction system under atmospheric pressure. The CO to O2 ratio was adjusted to CO/O2 = 5. The catalyst weight was 400 mg. Catalysts were pretreated in O2 at 60 mL min−1 for 1 h at 150 °C. The reaction products were analyzed using a thermal conductivity gas chromatograph.
3. Results and discussion
Quantitative XPS analyses of the Bi2MoxW1−xO6 catalysts were performed, with the typical survey and high-resolution spectra shown in Fig. 1. XPS spectra for Bi2MoxW1−xO6 catalysts annealed at 700 °C are presented to determine the oxidation state and elemental composition for each member of the solid solution. The Bi 4f5/2 and Bi 4f7/2 spin–orbital splitting photoelectrons for Bi2O3 were located at binding energies of 164.4 and 159.2 eV, respectively, and assigned to the presence of typical Bi3+ in the catalysts annealed at 700 °C over the entire range of solid solution. The characteristic binding energy (BE) values for Bi 4f7/2 (159.2 eV), Mo 3d5/2 (236 eV) and W 4f7/2 (37.5 eV) indicate a trivalent oxidation state for bismuth and a six-valent oxidation state for Mo6+ and W6+.15 Also, the O 1s, Mo 3d3/2, Bi 4f5/2 and W 4f5/2 core-level peaks were observed in coincidence with reported BE values.15,16 Relative atomic concentrations were calculated using the integrated area under the curve of O 1s, Mo 3d, W 4f and Bi 4f transition peaks through the relative atomic concentration formula.14–16 In order to avoid miscalculations typical for complex solid solutions when relative sensitivity factors (RSF) are used,17–21 more accurate qualitative XPS analyses were performed using RSFs obtained from our prepared Bi2MoO6 (x = 1) and Bi2WO6 (x = 0) standards. In this case: SO = 1, SBi = 9.44, SMo = 6.58 and SW = 6.30. Calculated relative atomic concentrations for samples sintered at 700 °C are summarized in Table S1 (see ESI†). For Bi2MoO6 (x = 1) and Bi2WO6 (x = 0), it was found that their atomic concentration was very close to the stoichiometric proportion. For samples with compositions between 0.95 < x < 0.05, the oxygen content was higher than the stoichiometric proportion and segregation of Bi at the surface level was detected. It is likely that Bi is mainly at surface as a consequence of the evaporation of bismuth even at 700 °C for intermediate compositions. It is also observed that the tungsten XPS signal near to Mo 3d5/2 peak at 236 eV becomes more evident when x increases as can be observed from x = 0.5. The Mo 3d5/2 peak is not evident for x = 0.1, x = 0.05 and x = 0 for which the W contribution is dominant. The same is observed at lower binding energy (37.5 eV) where the characteristic splitting for W is the main contribution when x decreases.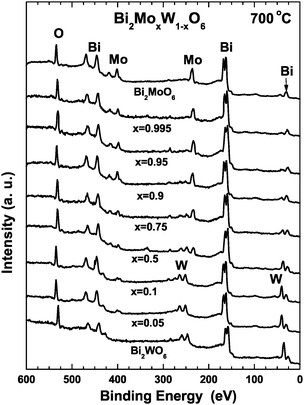 | ||
| Fig. 1 XPS spectra for samples sintered at 700 °C. | ||
In Fig. 2, XPS survey spectra of catalyst and standards of Bi2MoO6 and Bi2WO6 annealed at 900 °C are included. At 900 °C, Bi2MoO6 (x = 1) and Bi2WO6 (x = 0) samples were found to be similar to those annealed at 700 °C with atomic concentration very close to the stoichiometric proportion (see Table SII, ESI†). The thermal treatment at 900 °C for left samples induces some changes in binding energy values and chemical composition when compared with those annealed at 700 °C. The bands were a little broader and shifted slightly to a higher binding energy of 164.60 and 159.3 eV for Bi 4f5/2 and Bi 4f7/2, respectively, which might be assigned to more oxidized Bi ions. The differences in binding energies between Bi 4f5/2 and Bi 4f7/2 were distributed in the range of 5.25–5.40 eV in the entire solid solution. The W 4f7/2 and W 4f5/2 spin–orbital splitting photoelectrons for WO3 were located at binding energies of 35.40 and 37.54 eV, respectively, for samples sintered at 700 °C. When samples were treated at 900 °C, the bands shifted slightly to a lower binding energy of 35.20 and 37.36 eV for W 4f7/2 and W 4f5/2, respectively, which might be assigned to less oxidized W ions. There is no apparent dependence of shifting on molybdenum content. To calculate atomic concentration for the samples prepared at 900 °C, sensitivity factors SO = 1, SBi = 12.66, SMo = 5.08 and SW = 5.64 were employed. Calculations of atomic concentrations are included in Table SII (provided as ESI†). To determine oxidation states of the different elements of the catalysts, the lines were recorded with a narrow sweep in the range of 8–16 eV. It is noteworthy that some samples prepared at 900 °C show Bi deficiencies due to an increase in temperature that leads to a structural transformation to form more stable phases as it will be discussed later. The atomic concentration for Mo and W was very near to the expected composition over the entire range of the solid solution.
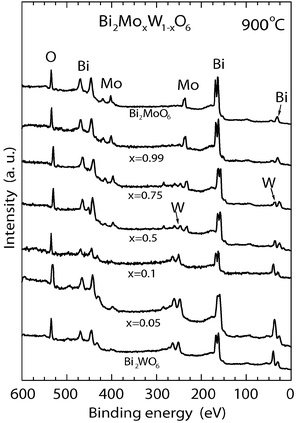 | ||
| Fig. 2 XPS spectra for samples sintered at 900 °C. | ||
It is known that XPS is one of the powerful tools for measuring oxygen mobility of catalysts;22 for that reason the binding energies of O 1s were measured in order to determine the oxygen mobility of the catalysts, see Fig. 3(a) and (b).
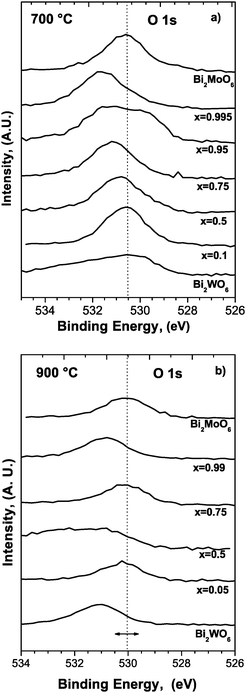 | ||
| Fig. 3 High resolution XPS spectra for oxygen in samples sintered at (a) 700 °C and (b) 900 °C. | ||
XPS spectra in Fig. 3(a) show a shifted and broadened O 1s peak that can be considered as the convolution of some oxygen species. Three types of oxygen species have been reported: type I oxygen with the lowest binding energy (530.3 eV) which corresponds to the oxygen species strongly bonded to the metal component (Mo/W); type II oxygen with the highest binding energy (533.1 eV) which is believed to be the oxygen weakly bonded onto the catalyst surface, and type III oxygen with an intermediate binding energy. Type III oxygen has been considered as an index for the oxygen mobility of the multicomponent bismuth molybdate catalyst. The binding energy of O 1s increases with decreasing valence electron density of lattice oxygen. This indicates that Mo/W–oxygen bonds in the catalyst would be weakened with increasing binding energy of O 1s, making the lattice oxygen more active and mobile. In other words, the higher binding energy of O 1s corresponds to the higher oxygen mobility.
As shown in Fig. 3(a) for 700 °C, the O 1s peak appears at the binding energy of 530.67 eV for Bi2MoO6 corresponding to the type I oxygen. For x = 0.995, the O 1s peak is broadened and shifted to a higher energy, this can be an indirect evidence of type III lattice oxygen existence, which promotes a higher oxygen mobility. For x = 0.95, this behavior is even more evident due to the broadening of the O 1s peak. Broadening and shifting is less evident for x = 0.5 and not observable for x = 0.1 and Bi2WO6, for which the O 1s is similar to that obtained for Bi2MoO6. We propose the increase in lattice oxygen mobility up to x = 0.95 or x = 0.75, probably as a consequence of a reticular expansion, to be mainly responsible for catalytic activity. Also a binding energy shift to higher energy values is presented as an evidence of the effective doping in solid solution at intermediate compositions. The O 1s peak appears at the same value for x = 0.1, Bi2MoO6 and Bi2WO6 as a result of the equivalent chemical vicinity for oxygen.
For 900 °C (Fig. 3(b)) the Bi2MoO6 composition showed that the Bi compound has the same shape of the peak and binding energy as those sintered at 700 °C, which indicates that for this composition it remained with the same structure. On the other hand it can be seen that the oxygen associated with the composite W is moved to the left, implying a possible transition of this compound explained in terms of Bi deficiency due to the higher sintering temperature. Comparing, for example, the compound Bi2Mo0.75W0.25O6, prepared at both 700 and 900 °C, it is shown that the oxygen of the compound is centered for 900 °C, differently from the case of the samples prepared at 700 °C, which implies that for the latter oxygen is more closely linked to the structure of the compound and therefore, its mobility could be lower. Also, as will be discussed in the X-ray diffraction section, the flat shape of this O 1s peak compared to the peak found at 700 °C could be indicative of the presence of both gamma and beta phases in the compound, and that behavior was not found for the compound prepared at 700 °C, for which the gamma phase was the only one found.
An issue of particular concern is to know the oxidation state for the equimolar Bi2MoO6–Bi2WO6 catalyst. Fig. 4 shows the XPS measurements of the Bi 4f7/2 core levels for the sample sintered at 700 °C. In Fig. 4(a), the appearance of Bi 4f5/2 and Bi 4f7/2 peaks is observed. The Bi 4f7/2 peak at the binding energy of 159.2 eV belongs to the Bi2MoO6 compound,18 while the Bi 4f7/2 peak at the binding energy of 157 eV agrees with the Bi2WO6 compound. Then it is possible to ascertain that Bi presents an oxidation state of +3 in both systems. In Fig. 4(b), the W 4f5/2 and W 4f7/2 signals displayed at the binding energies of 37.1 and 35 eV can be assigned to W6+, corresponding to Bi2WO6 in agreement with reported results.19 The above findings show that for a nominal composition Bi2Mo0.5W0.5O6 it is not possible to form a single phase because the formation of Bi2MoO6 and Bi2WO6 compounds was established.
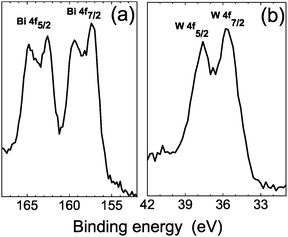 | ||
| Fig. 4 XPS spectra of Bi (a) and W (b) for the Bi2Mo0.5W0.5O6 sample sintered at 700 °C. | ||
The surface area measurements show relatively small values of the order of 1.3 to 4.5 m2 g−1 for samples sintered at 700 °C while for samples prepared at 900 °C they range from 1.0 to 2.2. These values are characteristic of such compounds. The differences found for the compounds prepared at low and high temperature are due to a natural sintering process promoted by the increase in temperature. On the other hand, by comparing different values obtained at a specific temperature, say 700 °C, we consider that it is not possible to establish a specific trend because the differences in the area values as a function of molybdenum or tungsten content are within the experimental error.
Results of the catalytic activity for Bi2MoO6 and Bi2WO6 samples sintered at 700 °C and 900 °C are displayed in Fig. 5. The Bi2MoO6 compound starts its activation at 200 °C, attaining 90% of conversion at 320 °C, see Fig. 5(a). Afterwards, the system continues increasing its CO conversion at a slower rate, from 90% to 95%. For the Bi2WO6 compound, conversion starts at 275 °C, achieving 85% of CO conversion at 375 °C. Later, the activity decreases to 70% at 450 °C. These results suggest that the Bi2MoO6 compound is more stable.
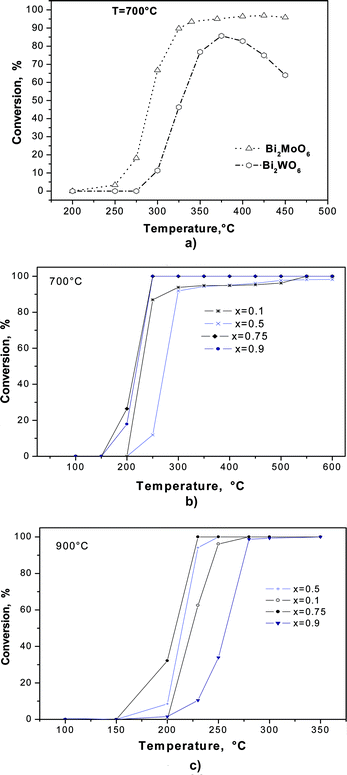 | ||
| Fig. 5 Catalytic activity for (a) Bi2MoO6 and Bi2WO6 sintered at 700 °C, (b) Bi2Mo(x)W(1−x)O6 samples sintered at 700 °C and Bi2Mo(x)W(1−x)O6 samples sintered at (c) 900 °C. | ||
The results on catalytic activity for the solid solutions Bi2MoxW1−xO6 are displayed in Fig. 5(b) and (c), where it can be observed that samples sintered at 700 °C with high Mo contents, say x = 0.9 and 0.75, are more reactive than those containing W.
Samples calcined at 900 °C do not follow the same tendency, since in that case, catalysts including W with x = 0.75 are more reactive than samples with higher content of Mo. Sintering at 900 °C promotes structural changes that modify catalytic activity when compared with samples sintered at 700 °C. This change was also suggested by XPS spectra in which the broadening and shifting exhibits a different behavior than for samples sintered at 700 °C.
Besides, the doping element provides additional electronic states that positively affect the catalytic activity. In such a case, this result is related to the synergistic effect of Mo and W content, where the solid solutions are more reactive than either Bi2MoO6 or Bi2WO6 alone. Also, the variations found in the CO oxidation, comparing the solid solutions with respect to the pure phases, are attributed to the occurrence, in small proportion, of Bi2Mo2−xWxO9 monoclinic phase.
This must be related to the broadened and shifted O 1s XPS peak that was explained in terms of type III lattice oxygen as a consequence of a crystallographic expansion that facilitates the migration of active oxygen species on the catalyst surface, leading to an enhanced oxygen mobility of multicomponent bismuth molybdate catalysts.
Regarding the correlation between the XPS measurements and catalytic activity, it was observed that adding W into Bi2MoxW(1−x)O6 promotes an energy shift in the Mo 3d5/2 transition.23 Thus it is reasonable to consider that W is chemically bound into the Bi2MoO6 structure. As a result, catalytic activity is improved; see Fig. 5(b). For the equimolar Mo/W ratio (Bi2Mo0.5W0.5O6) it was found that Bi2MoO6 and Bi2WO6 coexist as separate phases, which was confirmed through the appearance of Bi 4f7/2 transitions at 159.2 and 157.0 eV, corresponding to Bi2MoO6 and Bi2WO6, respectively. In such a case, the high values of catalytic activity must be recognized as an effect of the mixture of both oxides for this particular value.
In short, the use of Bi2MoxW(1−x)O6 solid solutions provides better conversion values and stability; moreover, deactivation is not observed in the investigated range, compared to either Bi2MoO6 or Bi2WO6 alone.
An important issue is to ensure that we have prepared solid solutions, which are different from mixed oxides. To do that we performed X-ray diffraction analyses, noting that for x = 0.5 the structure is a mixture of oxides while for concentrations x ≠ 0.5 it is possible to form solid solutions, see Fig. 6 and 7. Also in ref. 13 discussed earlier, authors consider that Mo for W substitution on Bi2MoO6 keeps the structural framework of the Aurivillius-type structures, however for x = 0.5 they report to obtain a mixture of γ-Bi2MoO6 and Bi2WO6 phases which reinforces our observations.
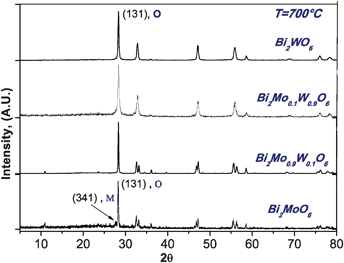 | ||
| Fig. 6 X-Ray diffraction patterns of Bi2MoO6, Bi2WO6, Bi2Mo0.1W0.9O6 and Bi2Mo0.9W0.1O6 synthesized at 700 °C. | ||
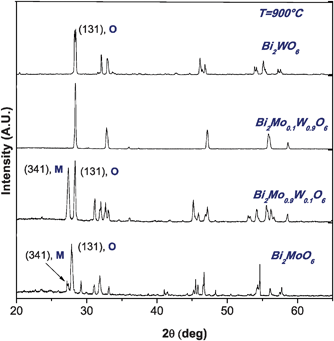 | ||
| Fig. 7 X-Ray diffraction patterns of catalysts Bi2MoO6, Bi2WO6, Bi2Mo0.1W0.9O6 and Bi2Mo0.9W0.1O6 synthesized at 900 °C. | ||
Linking the structural and the catalytic activity results it was noticed that in some solid solutions for example Mo, x = 0.9, the monoclinic phase appears in different proportions, mixed with the orthorhombic phase, see Fig. 6 and 7. However, as seen in the compound Bi2Mo0.25W0.75O6 its reactivity is higher than that shown by the individual oxides. This implies that the addition of molybdenum has been beneficial and improves the activity of bismuth tungstate.
In other words, the γ-Bi2Mo0.75W0.25O6 and β-Bi2Mo1.25W0.25O9 compounds are present in those systems (see Fig. 8) while for compounds containing higher proportion of tungsten, say W = 0.75, there is a single solid solution which indicates that the Bi2WO6 compounds tolerate a higher proportion of doping. We observe that solid solutions are more reactive than single phases. As discussed in the introductory part, for some reactions it has been found that the γ/β ratio is a determining factor to enhance the reactivity in comparison to either γ-Bi2MoO6 or Bi2WO6.13 We observed a similar behavior in our solid solutions containing γ and β phases.
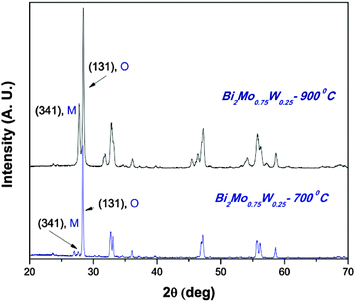 | ||
| Fig. 8 X-Ray diffraction patterns of Bi2Mo0.75W0.25O6 synthesized at 700 °C and 900 °C. | ||
Although studies on Bi–Mo–W–O oxides as powders or thin films have been the subject of numerous reports; the synthesis of single crystals of Bi–Mo–W solid-solutions has not already been published, for which it seems necessary to clarify its physical and chemical properties.
Conclusively we establish that mixed systems have greater reactivity than the pure phases. It was found that compounds having the composition x = 0.75 (Bi2Mo0.75W0.25O6) showed the greatest conversion of CO to CO2. That behavior is attributed to the appearance of γ-Bi2Mo0.75W0.25O6 and β-Bi2Mo1.25W0.25O9 phases. This mixture of phases provides an easy path for the movement of oxygen from the bulk (as discussed earlier, O(18) in the beta phase; O(1) and O(5) in the gamma phase) to the surface of the catalyst. It is therefore clear that the mobility of oxygen in the Bi2MoO6 matrix, achieved through a mechanism of vacancy diffusion, is the most important parameter to improve the catalytic activity and that morphological parameters (surface area for example), in the investigated range of values, are less relevant in this respect.
4. Conclusions
Our results have demonstrated that the Bi2MoxW(1−x)O6 compounds are highly effective in oxidizing CO to CO2. The temperature to start the CO oxidation is lower for the solid solutions than for either Bi2MoO6 or Bi2WO6 compounds. Conversion of about 100% is reached only when using the solid solutions and not with Bi2MoO6 or Bi2WO6 catalysts. From the XPS analysis it was made evident that the chemical environment created by the different W/Mo ratios affects CO conversion. It was observed that the Bi2Mo0.5W0.5O6 sample promotes the formation of Bi2MoO6–BiWO6 mixtures instead of one-single solid solution. The work described here is an important contribution because it demonstrates that a solid solution may be more efficient in the CO oxidation compared to the oxides studied individually. It also shows the combined effect of the different compositions and temperature.Acknowledgements
P. Bartolo-Pérez acknowledges grant 59998 from CONACYT. The authors thank W. Cauich for technical support. Authors thank the help provided by A. Gómez-Cortés from Instituto de Física, Universidad Nacional Autónoma de México. Also to E. Aparicio from CNyN, UNAM for the X-ray determinations.Notes and references
- G. Pérez-Osorio, V. Petranovskii and A. Simakov, PdO/Al2O3–(Ce1−xZrx)O2 catalysts: effect of the sol–gel support composition, Catal. Lett., 2006, 110(1–2), 53–60 CrossRef.
- D. Tibiletti, E. A. Bart de Graaf, S. P. Teh, G. Rothenberg, D. Farrusseng and C. Mirodatos, J. Catal., 2004, 225, 489–497 CrossRef CAS.
- A. Boulahouache, G. Kons, H. G. Lintz and P. Schulz, Appl. Catal., A, 1992, 91, 115–123 CrossRef CAS.
- E. D. Park and J. S. Lee, Catal. Today, 1998, 180, 123–131 CAS.
- S. Castillo, M. Morán-Pineda and R. Gómez, Catal. Commun., 2001, 2, 295–300 CrossRef CAS.
- A. K. Chakraborty, J. Sol-Gel Sci. Technol., 2003, 28, 87–95 CrossRef CAS.
- R. Rangel, P. Bartolo-Pérez, G. Díaz and D. H. Galván, Surf. Rev. Lett., 2002, 9, 1779–1783 CrossRef CAS.
- R. Rangel, P. Bartolo-Pérez, A. Gómez-Cortés, G. Díaz and D. H. Galván, J. Mater. Synth. Process., 2001, 9, 207–221 CrossRef CAS.
- S. Pudar, J. Oxgaard, K. Chenoweth, A. C. T. van Duin and W. A. Goddard III, Mechanism of Selective Oxidation of Propene to Acrolein on Bismuth Molybdates from Quantum Mechanical Calculations, J. Phys. Chem. C, 2007, 111, 16405–16415 CAS.
- J. SiphoMotshweni, Synthesis of Mixed Metal Oxides for use as Selective Oxidation Catalysts, Dissertation Thesis, University of Stellenbosch, US, 2007 Search PubMed.
- S. Williams, M. Puri, A. J. Jacobson and C. A. Mims, Catal. Today, 1997, 37, 43–49 CrossRef CAS.
- A. Castro, P. Bégué, B. Jiménez, J. Ricote, R. Jiménez and J. Galy, Chem. Mater., 2003, 15, 3395–3401 CrossRef CAS.
- M. T. Le, W. J. M. Van Well, P. Stoltze, I. Van Driessche and S. Hoste, Appl. Catal., A, 2005, 282, 189–194 CrossRef CAS.
- A. M. Venezia, Catal. Today, 2003, 77, 359–370 CrossRef CAS.
- T. L. Barr, Modern ESCA: The principles and practice of X-ray photoelectron spectroscopy, CRC Press, Boca Raton, 1994, p. 204 Search PubMed.
- D. Briggs and M. P. Seah, Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, John Wiley, Chichester, 1983, p. 182 Search PubMed.
- P. Bartolo-Pérez, J. L. Peña and M. H. Farías, Rev. Mex. Fis., 1998, 44, 9–23 Search PubMed.
- L. E. Davis, N. C. MacDonald, P. W. Palmberg, G. E. Riach and R. E. Weber, Handbook of Auger Electron Spectroscopy, Physical Electronics, Eden Prairie, 1978 Search PubMed.
- Handbook of X-ray Photoelectron Spectroscopy, ed. J. F. Mouder, W. F. Stickle, P. E. Sobol, K. D. Bomben and J. Chastain, Physical Electronics, Eden Prairie, Minnesota, 1992 Search PubMed.
- P. Bartolo-Pérez, J. L. Peña and M. H. Farías, Superficies Vacio, 1999, 8, 59–63 Search PubMed.
- C. Jenks and T. E. Booler, Appl. Surf. Sci., 2001, 180, 57–64 CrossRef CAS.
- J. Ch. Jung, H. Lee, H. Kim, Y.-M. Chung, T. J. Kim, S. J. Lee, S.-H. Oh, Y. S. Kim and I. K. Song, Catal. Lett., 2008, 124, 262–267 CrossRef CAS.
- A. Ayame, M. Iwata, K. Uchida, N. Igrashi and M. Miyamoto, Jpn. J. Appl. Phys., 2000, 39, 4335–4339 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00506a |
| This journal is © The Royal Society of Chemistry 2012 |