Direct arylation of dithienylperfluorocyclopentenes via palladium-catalysed C–H bond activation: a simpler access to photoswitches†‡
Kassem
Beydoun
,
Julien
Boixel
,
Véronique
Guerchais
* and
Henri
Doucet
*
Institut Sciences Chimiques de Rennes, UMR 6226 CNRS-Université de Rennes 1 “Organométalliques, Matériaux et Catalyse”, Campus de Beaulieu, 35042 Rennes, France. E-mail: veronique.guerchais@univ-rennes1.fr; henri.doucet@univ-rennes1.fr; Fax: +33 0223236939
First published on 6th March 2012
Abstract
The palladium-catalysed direct arylation of dithienylperfluorocyclopentene (DTE) derivatives proceeds in moderate to high yields with a variety of aryl bromides in the presence of 5 mol% Pd(OAc)2/dppb as the catalyst, and KOAc as the base. The use of cyclopentyl methyl ether as the solvent was found to be crucial to avoid the decomposition of the reactants and products. The reaction proceeds regioselectively at C5 of thiophenes, and tolerates various substituents such as formyl, acetyl, ester, nitrile or nitro on the aryl bromide. Therefore, this method allows a straightforward modulation of the electron density distribution on DTE derivatives.
Introduction
The palladium-catalysed direct arylation of heteroaromatics has recently emerged as a very powerful method for the preparation of arylated heteroaromatics including thiophenes.1–4 However, there are still limitations for these reactions in terms of substrate scope. The presence of acetyl, amino, bromide, ester, formyl, nitrile, methylalcohol or trimethylsilyl as the functional groups on the thiophenes has largely described.5a–n On the other hand, the arylation of dithienylperfluorocyclopentene (DTE) derivatives has attracted less attention.5oAmong various photochromic systems,6 DTE derivatives have particularly attracted increased interest for their excellent stability and fatigue-resistance properties making them important building blocks for the synthesis of photoswitching materials (Fig. 1).7
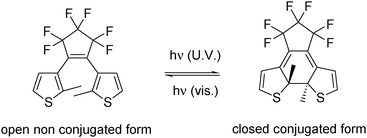 | ||
| Fig. 1 Photochromism of dithienylperfluorocyclopentene. | ||
Arylated DTE can be prepared by Suzuki–Miyaura or Negishi couplings (Scheme 1, top).8,9 However, such syntheses are time consuming and the yields are often modest. Therefore, to overcome these limitations, a straightforward and selective method for the functionalisation of DTE derivatives is highly desirable.
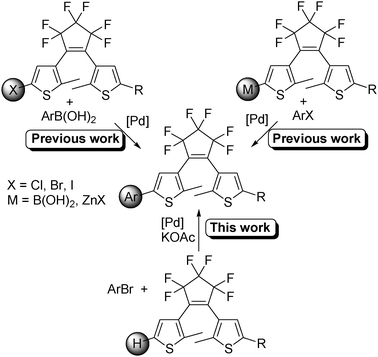 | ||
| Scheme 1 | ||
Very recently, it has been reported that dithienylperfluorocyclopentene 1 can be easily obtained in high yield by Suzuki–Miyaura cross-coupling of 2-methyl-3-thienylboronic acid with 1,2-dichlorohexafluorocyclopentene (Scheme 2).10 It should be noted that this latter compound is commercially available at an affordable cost allowing the synthesis of 1 on a large scale.
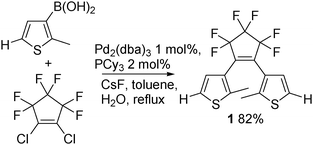 | ||
| Scheme 2 | ||
Therefore, the palladium-catalysed direct arylation of 1 or its derivatives for access to arylated DTE derivatives would present considerable advantages (Scheme 1, bottom). It would allow us to reduce the number of steps to prepare these compounds. Moreover, lower amounts of wastes would be produced. In addition, such couplings are expected to present a better functional group tolerance, which would allow a straightforward modification of the nature of the aryl group and hence of the photochromic properties of the resulting DTE derivatives.
Results and discussion
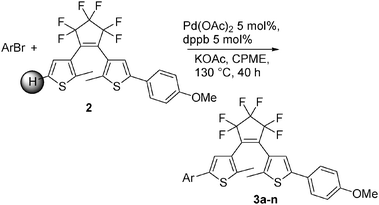
We now report (i) conditions for the palladium-catalysed direct regioselective mono- or diarylation of DTE derivatives with (hetero)aryl bromides using a cheap base and an air stable catalyst, (ii) the formation of a wide variety of diarylated derivatives of DTE with either two different or two identical aryl groups, (iii) that this method tolerates several functional groups.Keeping in mind the conditions used for the direct arylation in our previous works, we examined the influence of the nature of the solvent, catalyst precursor and reaction temperature for the coupling of 4-bromobenzonitrile with 2 (Scheme 3) using KOAc as a base.3,5k–m We observed that, in the course of this reaction, the use of DMAc as the solvent and 2–5 mol% PdCl(C3H5)(dppb) [dppb: 1,4-bis(diphenylphosphino)butane)], Pd(OAc)2 or Pd(OAc)2/dppb as the catalysts at 140 °C did not result in the formation of the target product 3a. Instead, decomposition of 2 was observed. Similar results were obtained at 130 °C. On the other hand, the use of cyclopentyl methyl ether (CPME) as the solvent for this reaction, using 5 mol% Pd(OAc)2 as the palladium source and 5 mol% dppb as the ligand at 140 °C, afforded cleanly the desired arylated product 3a in 69% yield. No formation of other regioisomers or diarylated thiophenes was detected. We had previously observed that cyclopentyl methyl ether promotes the palladium-catalysed direct arylations in high yields with some heteroaromatic derivatives.11 This solvent presents several advantageous features such as limited miscibility in water and low formation of peroxides. Moreover CPME can be manufactured by the addition of MeOH to cyclopentene. This process produces no apparent waste.12 The good performance of KOAc as the base for this coupling is consistent with a concerted metallation deprotonation (CMD) pathway.13
 | ||
| Scheme 3 | ||
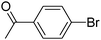
Using the best conditions, the scope of this reaction was examined using para-, meta- or ortho-substituted aryl bromides and also heteroaryl bromides (Scheme 4, Table 1). Other electron-deficient para-substituted aryl bromides also gave the desired 5-arylated DTE products 3b–3e (Table 1, entries 2–5). A good yield of 61% for 3d was obtained for the coupling of 2 with 4-bromoacetophenone; whereas reaction with methyl 4-bromobenzoate gave 3e in only 26% yield. Moderate yields of 3b and 3c were obtained in the presence of 4-bromonitrobenzene and 4-bromobenzaldehyde. As expected, the meta-substituted aryl bromide, 3-bromonitrobenzene, gave 3f in a similar yield to 4-bromonitrobenzene (Table 1, entry 6). A very low yield of 21% for 3g was obtained in the presence of 2-bromonaphthalene (Table 1, entry 7). This is probably due to a slow oxidative addition of this aryl bromide to palladium under these conditions. On the other hand, congested substrate, 2-bromobenzonitrile, was found to be very reactive, and gave 3h in 84% yield (Table 1, entry 8).
 | ||
| Scheme 4 | ||
| Entry | Aryl bromide | Product | Yield (%) |
|---|---|---|---|
| Conditions: Pd(OAc)2 (0.05 equiv.), dppb (0.05 equiv.), aryl bromide (2 equiv.), 1 (1 equiv.), KOAc (3 equiv.), CPME, 40 h, 130 °C.a Aryl bromide (1 equiv.), KOAc (2 equiv.). | |||
| 1 |

|
3a | 69a |
| 2 |

|
3b | 39 |
| 3 |

|
3c | 40 |
| 4 |
|
3d | 61 |
| 5 |

|
3e | 26 |
| 6 |

|
3f | 32 |
| 7 |

|
3g | 21 |
| 8 |

|
3h | 84 |
| 9 |

|
3i | 55 |
| 10 |
|
3j | 51a |
| 11 |

|
3k | 80 |
| 12 |

|
3l | 94 |
| 13 |

|
3m | 63a |
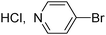
DTE derivatives have been also used as ligands for incorporation into transition metal complexes.7e Therefore, preparative methods for the introduction of N-containing heterocycles such as pyridines,14 quinolines or pyrimidines remain a crucial issue for the elaboration of photoresponsive materials. We observed that the coupling of 3- or 4-bromopyridines with 2 also proceeds nicely to give 3i and 3j in 55% and 51% yields, respectively (Table 1, entries 9 and 10). Very high yields were obtained for the coupling of 2 with 3-bromoquinoline or 4-bromoisoquinoline to produce 3k and 3l in 80% and 94% yields, respectively (Table 1, entries 11 and 12). Finally, the reactivity of 5-bromopyrimidine was examined, and the desired product 3m was obtained in 63% yield (Table 1, entry 13).
A lower yield was obtained in the course of the coupling of 1 equiv. of 2 with 1 equiv. of 4,4′-dibromobiphenyl (Scheme 5). The desired product 3n was only obtained in 29% yield. However, this functionalised highly conjugated structure allows access to multi-DTE systems.15
 | ||
| Scheme 5 | ||
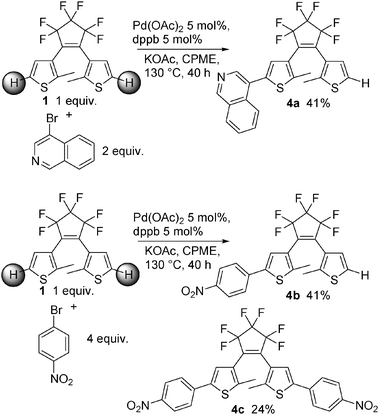
We also performed direct arylation reactions on 1 using two aryl bromides (Scheme 6). From 2 equiv. of 4-bromoisoquinoline and 1 equiv. of 1, the monoarylated product 4a was obtained in 41% yield; whereas the diarylated product could not be isolated. On the other hand, in the presence of 4 equiv. of 3-bromonitrobenzene, a mixture of the monoarylated product 4b and the diarylated product 4c was obtained in a 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24 ratio.
:24 ratio.
 | ||
| Scheme 6 | ||
Conclusion
In summary, we have demonstrated that when appropriate reaction conditions are employed, the palladium-catalysed direct arylation of DTE derivatives proceeds with a variety of aryl bromides. It should be noted that this protocol, which employs a moderate loading of an air stable catalyst and a cheap base, is applicable to a wide range of functions, including reactive ones, such as formyl, acetyl, ester, nitrile or nitro on the aryl bromide. This procedure allowed us to synthesize new families of arylated DTE derivatives, including donor–acceptor (D–A) systems (3a). Such functional group tolerance allows the easy modification of the electronic structure of DTE derivatives, a strategy enabling the tuning of their photochromic properties. For all these reactions, CPME 99+% was used without any purification. As this solvent is hydrophobic, there is no need to dry it before use. The major by-products of these couplings are KBr/AcOH instead of metallic salts with more classical coupling procedures. For these reasons, this process should give a more economically viable and environmentally attractive access to several arylated dithienylperfluorocyclopentenes. In addition, coordination of DTE ligands will open up new perspectives for the design of photoswitchable molecules and materials.Experimental Section
General
All reactions were performed in Schlenck tubes under argon. CPME of analytical grade was not distilled before use. Potassium acetate 99+ was used. Commercial aryl bromides were used without purification. 1H (500 MHz), 13C (125 MHz) spectra were recorded in CDCl3 solutions. Chemical shifts are reported in ppm relative to CDCl3 (1H: 7.29 and 13C: 77.0). Flash chromatography was performed on silica gel (230–400 mesh) using pentane/ether. 2 was prepared according to the reported procedure.7bGeneral procedure
As a typical experiment, the reaction of the aryl bromide (1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) at 130 °C during 40 h in CPME (4 mL) in the presence of Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) under argon affords the corresponding coupling products 3a–n after evaporation of CPME and filtration on silica gel (pentane/ether) or (pentane/dichloromethane).4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-benzonitrile (3a)16
The reaction of 4-bromobenzonitrile (0.091 g, 0.5 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.098 g, 1 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords the corresponding product 3a in 69% (0.197 g) isolated yield as a dark blue solid. 1H NMR (500 MHz, CDCl3): δ 7.69 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 8.6 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H), 7.41 (s, 1H), 7.17 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.03 (s, 3H), 1.98 (s, 3H).4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-nitrobenzene (3b)
The reaction of 4-bromonitrobenzene (0.202 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3b in 39% (0.116 g) isolated yield as a dark green solid (mp: 184 °C). 1H NMR (500 MHz, CDCl3): δ 8.27 (d, J = 8.9 Hz, 2H), 7.70 (d, J = 8.9 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H), 7.46 (s, 1H), 7.17 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.05 (s, 3H), 1.99 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.6, 146.9, 144.1, 142.5, 140.2, 139.4, 139.3, 126.9, 126.7, 126.0, 125.8, 125.5, 125.2, 124.5, 121.1, 114.4, 55.4, 14.7, 14.5. Elemental analysis calcd (%) for C28H19F6NO3S2 (595.58): C, 56.47; H, 3.22; found: C, 56.66; H, 3.32%.4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-benzaldehyde (3c)17
The reaction of 4-bromobenzaldehyde (0.185 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3c in 40% (0.115 g) isolated yield as a light blue solid. 1H NMR (500 MHz, CDCl3): δ 10.03 (s, 1H), 7.92 (d, J = 8.3 Hz, 2H), 7.72 (d, J = 8.3 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H), 7.46 (s, 1H), 7.18 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H).1-[4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-phenyl]-ethanone (3d)
The reaction of 4-bromoacetophenone (0.199 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3d in 61% (0.181 g) isolated yield as a dark blue solid (mp: 161 °C). 1H NMR (500 MHz, CDCl3): δ 7.99 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.5 Hz, 2H), 7.49 (d, J = 8.8 Hz, 2H), 7.42 (s, 1H), 7.18 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.64 (s, 3H), 2.03 (s, 3H), 1.99 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 197.2, 159.6, 143.0, 142.4, 140.7, 140.3, 137.6, 136.1, 129.2, 127.0, 126.4, 126.1, 125.6, 125.4, 124.1, 121.2, 114.4, 55.4, 26.6, 14.7, 14.5. Elemental analysis calcd (%) for C30H22F6O2S2 (592.61): C, 60.80; H, 3.74; found: C, 60.92; H, 3.91%.Methyl 4-(4-{3,3,4,4,5,5-hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-benzoate (3e)
The reaction of methyl 4-bromobenzoate (0.215 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3e in 26% (0.079 g) isolated yield as a blue solid (mp: 170 °C). 1H NMR (500 MHz, CDCl3): δ 8.07 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.8 Hz, 2H), 7.41 (s, 1H), 7.17 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.96 (s, 3H), 3.86 (s, 3H), 2.02 (s, 3H), 1.98 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 166.5, 159.6, 142.8, 142.3, 140.8, 140.3, 137.5, 130.4, 129.2, 127.0, 126.3, 126.1, 125.3, 123.4, 121.2, 114.4, 55.4, 55.2, 14.7, 14.5. Elemental analysis calcd (%) for C30H22F6O3S2 (608.62): C, 59.20; H, 3.64; found: C, 59.43; H, 3.78%.3-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-nitrobenzene (3f)
The reaction of 3-bromonitrobenzene (0.202 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3f in 32% (0.096 g) isolated yield as a light green oil. 1H NMR (500 MHz, CDCl3): δ 8.40 (s, 1H), 8.17 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.59 (t, J = 8.0 Hz, 1H), 7.49 (d, J = 8.8 Hz, 2H), 7.42 (s, 1H), 7.18 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.6, 148.8, 143.0, 142.5, 140.2, 139.3, 135.0, 131.2, 130.0, 127.0, 126.4, 126.1, 125.5, 124.3, 122.3, 121.1, 120.3, 114.4, 55.4, 14.6, 14.5. Elemental analysis calcd (%) for C28H19F6NO3S2 (595.58): C, 56.47; H, 3.22; found: C, 56.54; H, 3.35%.4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(naphth-2-yl)-2-methylthiophen-3-yl]-cyclopent-1-enyl-}5-methylthiophen-2-yl)-methoxyphenyl (3g)
The reaction of 2-bromonaphthalene (0.207 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3g in 21% (0.063 g) isolated yield as a light green oil. 1H NMR (500 MHz, CDCl3): δ 7.99 (s, 1H), 7.90–7.83 (m, 3H), 7.70 (d, J = 8.5 Hz, 1H), 7.55–7.47 (m, 4H), 7.44 (s, 1H), 7.20 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.03 (s, 3H), 2.01 (s, 3H). 13C NMR (125 MHz, CDCl3): 159.5, 142.3, 142.2, 133.5, 132.9, 130.7, 128.7, 128.0, 127.8, 127.0, 126.9, 126.8, 126.5, 126.3, 126.2, 126.1, 125.8, 124.1, 123.8, 122.8, 121.3, 114.4, 55.4, 14.6, 14.5. Elemental analysis calcd (%) for C32H22F6OS2 (600.64): C, 63.99; H, 3.69; found: C, 63.86; H, 3.81%.2-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methyl-thiophen-2-yl)-benzonitrile (3h)
The reaction of 2-bromobenzonitrile (0.182 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3h in 84% (0.241 g) isolated yield as a blue oil. 1H NMR (500 MHz, CDCl3): δ 7.77 (d, J = 7.8 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.59 (d, J = 7.8 Hz, 1H), 7.57 (s, 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.45 (t, J = 7.8 Hz, 1H), 7.19 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.5, 143.6, 142.3, 140.6, 137.2, 136.6, 134.2, 133.0, 129.5, 128.1, 127.4, 126.9, 126.1, 126.0, 125.4, 121.1, 118.3, 114.4, 110.4, 55.4, 14.6, 14.4. Elemental analysis calcd (%) for C29H19F6NOS2 (575.59): C, 60.51; H, 3.33; found: C, 60.72; H, 3.50%.3-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-pyridine (3i)
The reaction of 3-bromopyridine (0.158 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3i in 45% (0.124 g) isolated yield as a blue solid (mp: 146 °C). 1H NMR (500 MHz, CDCl3): δ 8.83 (s, 1H), 8.56 (d, J = 4.8 Hz, 1H), 7.84 (d, J = 7.9 Hz, 1H), 7.49 (d, J = 8.8 Hz, 2H), 7.37–7.32 (m, 2H), 7.17 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.04 (s, 3H), 1.99 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.5, 151.5, 148.9, 146.7, 142.4, 140.3, 138.3, 135.8, 132.7, 129.5, 127.0, 126.3, 126.1, 125.5, 123.7, 121.2, 114.4, 55.4, 14.6, 14.5. Elemental analysis calcd (%) for C27H19F6NOS2 (551.57): C, 58.79; H, 3.47; found: C, 59.16; H, 3.42%.4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-pyridine (3j)18
The reaction of 4-bromopyridine hydrochloride (0.100 g, 0.5 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.098 g, 1 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3j in 51% (0.140 g) isolated yield as a green solid. 1H NMR (500 MHz, CDCl3): δ 8.62 (d, J = 5.9 Hz, 2H), 7.50 (s, 1H), 7.48 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 5.9 Hz, 2H), 7.17 (s, 1H), 6.94 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H), 2.04 (s, 3H), 1.97 (s, 3H).3-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-quinoline (3k)
The reaction of 3-bromoquinoline (0.208 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3k in 80% (0.240 g) isolated yield as a dark green solid (mp: 139 °C). 1H NMR (500 MHz, CDCl3): δ 9.14 (s, 1H), 8.24 (s, 1H), 8.13 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.60 (t, J = 8.0 Hz, 1H), 7.50 (d, J = 8.8 Hz, 2H), 7.48 (s, 1H), 7.20 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.07 (s, 3H), 2.02 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.6, 148.0, 147.4, 142.6, 142.4, 140.3, 138.6, 131.2, 129.6, 129.4, 127.8, 127.7, 127.4, 126.9, 126.5, 126.4, 126.1, 125.6, 123.9, 121.2, 114.4, 55.4, 14.6, 14.5. Elemental analysis calcd (%) for C31H21F6NOS2 (601.63): C, 61.89; H, 3.52; found: C, 61.76; H, 3.49%.4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-isoquinoline (3l)
The reaction of 4-bromoisoquinoline (0.208 g, 1 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3l in 94% (0.283 g) isolated yield as a dark blue solid (mp: 134 °C). 1H NMR (500 MHz, CDCl3): δ 9.26 (s, 1H), 8.59 (s, 1H), 8.10 (d, J = 8.0 Hz, 1H), 8.05 (d, J = 8.0 Hz, 1H), 7.73 (t, J = 8.0 Hz, 1H), 7.67 (t, J = 8.0 Hz, 1H), 7.51 (d, J = 8.8 Hz, 2H), 7.26 (s, 1H), 7.21 (s, 1H), 6.95 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.17 (s, 3H), 2.10 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.5, 152.8, 143.4, 142.9, 142.3, 140.2, 135.9, 133.9, 131.2, 128.4, 128.1, 128.0, 127.6, 126.9, 126.1, 125.7, 125.6, 125.2, 124.0, 121.3, 114.4, 55.4, 14.5, 14.4. Elemental analysis calcd (%) for C31H21F6NOS2 (601.62): C, 61.89; H, 3.52; found: C, 61.77; H, 3.41%.5-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(43-methoxyphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-pyrimidine (3m)
The reaction of 5-bromopyrimidine (0.080 g, 0.5 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.098 g, 1 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 20 h affords product 3m in 63% (0.174 g) isolated yield as a yellow solid (mp: 146 °C). 1H NMR (500 MHz, CDCl3): δ 9.16 (s, 1H), 8.92 (s, 2H), 7.49 (d, J = 8.7 Hz, 2H), 7.41 (s, 1H), 7.16 (s, 1H), 6.95 (d, J = 8.7 Hz, 2H), 3.86 (s, 3H), 2.07 (s, 3H), 1.99 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 159.6, 157.6, 153.2, 143.8, 142.6, 140.2, 134.2, 127.7, 126.9, 126.7, 126.0, 125.4, 124.9, 121.1, 114.4, 55.4, 14.7, 14.5. Elemental analysis calcd (%) for C26H18F6N2OS2 (552.56): C, 56.52; H, 3.28; found: C, 56.75; H, 3.19%.4-(4-{3,3,4,4,5,5-Hexafluoro-2-[5-(4-bromobiphenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-methoxyphenyl (3n)15
The reaction of 4,4′-dibromobiphenyl (0.156 g, 0.5 mmol), 2 (0.237 g, 0.5 mmol) and KOAc (0.098 g, 1 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 3n in 29% (0.051 g) isolated yield as a dark blue solid. 1H NMR (500 MHz, CDCl3): δ 7.63 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 7.50 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.5 Hz, 2H), 7.35 (s, 1H), 7.19 (s, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 2.02 (s, 3H), 1.99 (s, 3H).4-{4-[3,3,4,4,5,5-Hexafluoro-2-(2-methylthiophen-3-yl)-cyclopent-1-enyl]-5-methylthiophen-2-yl}-isoquinoline (4a)
The reaction of 4-bromoisoquinoline (0.208 g, 1 mmol), 1 (0.184 g, 0.5 mmol) and KOAc (0.147 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords product 4a in 41% (0.101 g) isolated yield as a pink solid (mp: 113 °C). 1H NMR (500 MHz, CDCl3): δ 9.26 (s, 1H), 8.57 (s, 1H), 8.12–8.04 (m, 2H), 7.78 (t, J = 6.9 Hz, 1H), 7.70 (t, J = 6.9 Hz, 1H), 7.25 (d, J = 5.3 Hz, 1H), 7.22 (s, 1H), 7.14 (d, J = 5.3 Hz, 1H), 2.09 (s, 3H), 2.07 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 152.8, 143.5, 142.9, 141.7, 135.8, 133.9, 131.2, 128.4, 128.1, 128.0, 127.6, 127.2, 125.7, 125.2, 125.0, 124.0, 123.8, 14.3, 14.2. Elemental analysis calcd (%) for C24H15F6NS2 (495.51): C, 58.17; H, 3.05; found: C, 58.29; H, 3.18%.4-{4-[3,3,4,4,5,5-Hexafluoro-2-(2-methylthiophen-3-yl)-cyclopent-1-enyl]-5-methylthiophen-2-yl}-nitrobenzene (4b) and 4-(4-{3,3,4,4,5,5-hexafluoro-2-[5-(4-nitrophenyl)-2-methylthiophen-3-yl]-cyclopent-1-enyl}-5-methylthiophen-2-yl)-nitrobenzene (4c)
The reaction of 4-bromonitrobenzene (0.404 g, 2 mmol), 1 (0.184 g, 0.5 mmol) and KOAc (0.294 g, 1.5 mmol) with Pd(OAc)2 (0.005 g, 0.025 mmol) and dppb (0.010 g, 0.025 mmol) in CPME (4 mL) at 130 °C during 40 h affords a mixture of product 4b in 41% (0.100 g) isolated yield as a blue solid (mp: 172 °C), and of product 4c in 24% (0.73 g) isolated yield as a dark blue solid (mp: 214 °C).4b 1H NMR (500 MHz, CDCl3): δ 8.27 (d, J = 8.9 Hz, 2H), 7.69 (d, J = 8.9 Hz, 2H), 7.44 (s, 1H), 7.22 (d, J = 5.3 Hz, 1H), 7.10 (d, J = 5.3 Hz, 1H), 2.01 (s, 3H), 1.97 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 146.9, 144.1, 141.8, 139.4, 139.3, 127.1, 126.6, 125.8, 125.2, 124.8, 124.5, 124.0, 14.5, 14.4. Elemental analysis calcd (%) for C21H13F6NO2S2 (489.46): C, 51.53; H, 2.68; found: C, 51.61; H, 2.87%.
4c 1H NMR (500 MHz, CDCl3): δ 8.28 (d, J = 8.9 Hz, 4H), 7.71 (d, J = 8.9 Hz, 4H), 7.47 (s, 2H), 2.06 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 147.0, 144.0, 139.8, 139.2, 128.4, 125.9, 124.6, 124.4, 14.8. Elemental analysis calcd (%) for C27H16F6N2O4S2 (610.55): C, 53.11; H, 2.64; found: C, 53.23; H, 2.74%.
Acknowledgements
K. B. is grateful to CNRS and “Conseil regional de Bretagne” for a grant. We thank the CNRS and “Rennes Metropole” for providing financial support.Notes and references
- (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (b) T. Satoh and M. Miura, Chem. Lett., 2007, 36, 200 CrossRef CAS; (c) I. V. Seregin and V. Gevoryan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (d) B.-J. Li, S.-D. Yang and Z.-J. Shi, Synlett, 2008, 949 CAS; (e) F. Bellina and R. Rossi, Tetrahedron, 2009, 65, 10269 CrossRef CAS; (f) L. Ackermann, R. Vicente and A. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS; (g) J. Roger, A. L. Gottumukkala and H. Doucet, ChemCatChem, 2010, 2, 20 CrossRef CAS; (h) C. Fischmeister and H. Doucet, Green Chem., 2011, 13, 741 RSC.
- A. Ohta, Y. Akita, T. Ohkuwa, M. Chiba, R. Fukunaga, A. Miyafuji, T. Nakata, N. Tani and Y. Aoyagi, Heterocycles, 1990, 31, 1951 CrossRef CAS.
- For recent contributions on direct arylations or vinylations of heteroaromatics from our laboratory: (a) A. L. Gottumukkala, F. Derridj, S. Djebbar and H. Doucet, Tetrahedron Lett., 2008, 49, 2926 CrossRef CAS; (b) Y. Fall, H. Doucet and M. Santelli, ChemSusChem, 2009, 2, 153 CrossRef CAS; (c) J. Roger and H. Doucet, Adv. Synth. Catal., 2009, 351, 1977 CrossRef CAS; (d) J. J. Dong, J. Roger, F. Požgan and H. Doucet, Green Chem., 2009, 11, 1832 RSC; (e) M. Ionita, J. Roger and H. Doucet, ChemSusChem, 2010, 3, 367 CrossRef CAS; (f) L. Chen, J. Roger, C. Bruneau, P. H. Dixneuf and H. Doucet, Chem. Commun., 2011, 47, 1872 RSC.
- For selected recent examples of direct C5 arylations of thiophenes: (a) J. Fournier Dit Chabert, G. Chatelain, S. Pellet-Rostaing, D. Bouchu and M. Lemaire, Tetrahedron Lett., 2006, 47, 1015 CrossRef CAS; (b) M. Nakano, T. Satoh and M. Miura, J. Org. Chem., 2006, 71, 8309 CrossRef CAS; (c) E. David, S. Pellet-Rostaing and M. Lemaire, Tetrahedron, 2007, 63, 8999 CrossRef CAS; (d) G. L. Turner, J. A. Morris and M. F. Greaney, Angew. Chem., Int. Ed., 2007, 46, 7996 CrossRef CAS; (e) P. Amaladass, J. A. Clement and A. K. Mohanakrishnan, Tetrahedron, 2007, 63, 10363 CrossRef CAS; (f) M. Nakano, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1851 CrossRef CAS; (g) C.-Y. Liu, H. Zhao and H.-h. Yu, Org. Lett., 2011, 13, 4068 CrossRef CAS; (h) K. Beydoun, M. Zaarour, J. A. G. Williams, H. Doucet and V. Guerchais, Chem. Commun., 2012, 48, 1260 RSC.
- For selected examples of palladium-catalysed direct C5 arylations using thiophenes bearing formyl-, acetyl-, amino-, nitrile-, halo-, silyl or methylalcohol substituents: (a) S. Pivsa-Art, T. Satoh, Y. Kawamura, M. Miura and M. Nomura, Bull. Chem. Soc. Jpn., 1998, 71, 467 CrossRef CAS; (b) L. Lavenot, C. Gozzi, K. Ilg, I. Orlova, V. Penalva and M. Lemaire, J. Organomet. Chem., 1998, 567, 49 CrossRef CAS; (c) T. Okazawa, T. Satoh, M. Miura and M. Nomura, J. Am. Chem. Soc., 2002, 124, 5286 CrossRef CAS; (d) K. Masui, H. Ikegami and A. Mori, J. Am. Chem. Soc., 2004, 126, 5074 CrossRef CAS; (e) H. A. Chiong and O. Daugulis, Org. Lett., 2007, 9, 1449 CrossRef CAS; (f) K. Kobayashi, A. Sugie, M. Takahashi, K. Masui and A. Mori, Org. Lett., 2005, 7, 5083 CrossRef CAS; (g) B. Liégaut, D. Lapointe, L. Caron, A. Vlassova and K. Fagnou, J. Org. Chem., 2009, 74, 1826 CrossRef; (h) J. J. Dong, J. Roger and H. Doucet, Tetrahedron Lett., 2009, 50, 2778 CrossRef CAS; (i) J. Roger, F. Požgan and H. Doucet, Adv. Synth. Catal., 2010, 352, 696 CrossRef CAS; (j) B. Liégault, I. Petrov, S. I. Gorlesky and K. Fagnou, J. Org. Chem., 2010, 75, 1047 CrossRef; (k) J. Roger and H. Doucet, Eur. J. Org. Chem., 2010, 4412 CAS; (l) F. Derridj, J. Roger, S. Djebbar and H. Doucet, Org. Lett., 2010, 12, 4320 CrossRef CAS; (m) L. Chen, J. Roger, C. Bruneau, P. H. Dixneuf and H. Doucet, Chem. Commun., 2011, 47, 1872 RSC; (n) D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594 CAS; (o) For direct arylations of DTE with aryl iodides: H. Kamiya, S. Yanagisawa, S. Hiroto, K. Itami and H. Shinokubo, Org. Lett., 2011, 13, 6394 CrossRef CAS.
- (a) G. H. Brown, Photochromism, Wiley-Interscience, New York, 1971 Search PubMed; (b) H. Dürr and H. Bouas-Laurent, Photochromism: Molecules and Systems, Elsevier, Amsterdam, 1990 Search PubMed; (c) M. Irie, Photo-reactive Materials for Ultrahigh-density Optical Memory, Elsevier, Amsterdam, 1994 Search PubMed; (d) F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327 RSC; (e) D. Gust, T. A. Moore and A. L. Moore, Chem. Commun., 2006, 1169 RSC.
- (a) R. Hanazawa, R. Sumiya, Y. Horikawa and M. Irie, J. Chem. Soc., Chem. Commun., 1992, 206 RSC; (b) G. M. Tsivgoulis and J.-M. Lehn, Adv. Mater., 1997, 9, 627 CrossRef CAS; (c) M. Irie, Chem. Rev., 2000, 100, 1685 CrossRef CAS; (d) Y. Hasegawa, T. Nakagawa and T. Kawai, Coord. Chem. Rev., 2010, 254, 2643 CrossRef CAS; (e) V. Guerchais, L. Ordronneau and H. Le Bozec, Coord. Chem. Rev., 2010, 254, 2533 CrossRef CAS.
- For Negishi type palladium-catalysed arylation, see for example: M. Irie, K. Sakemura, M. Okinaka and K. Uchida, J. Org. Chem., 1995, 60, 8305 CrossRef CAS.
- For Suzuki–Miyaura palladium-catalysed arylation, see: (a) K. Yagi, C. F. Soong and M. Irie, J. Org. Chem., 2001, 66, 5419 CrossRef CAS; (b) A. Mulder, A. Jukovic, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 1748 RSC; (c) N. Soh, K. Yoshida, H. Nakajima, K. Nakano, T. Imato, T. Fukaminato and M. Irie, Chem. Commun., 2007, 5206 RSC; (d) A. De Meijere, L. Zhao, V. N. Belov, M. Bossi, M. Noltemeyer and S. W. Hell, Chem.–Eur. J., 2007, 13, 2503 CrossRef CAS; (e) S. F. Yan, V. N. Belov, M. L. Bossi and S. W. Hell, Eur. J. Org. Chem., 2008, 2531 CrossRef CAS; (f) S. Hermes, G. Dassa, G. Toso, A. Bianco, C. Bertarelli and G. Zerbi, Tetrahedron Lett., 2009, 50, 1614 CrossRef CAS; (g) B. He and O. S. Wenger, J. Am. Chem. Soc., 2011, 133, 17027 CrossRef CAS.
- S. Hiroto, K. Suzuki, H. Kamiya and H. Shinokubo, Chem. Commun., 2011, 47, 7149 RSC.
- K. Beydoun and H. Doucet, ChemSusChem, 2011, 4, 526 CrossRef CAS.
- K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251 CrossRef CAS.
- (a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS; (b) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
- (a) A. Fernandez-Acebes and J.-M. Lehn, Chem.–Eur. J., 1999, 5, 3285 CrossRef CAS; (b) B. Gorodetsky and N. R. Branda, Adv. Funct. Mater., 2007, 17, 786 CrossRef CAS; (c) H. D. Samachetty, V. Lemieux and N. R. Branda, Tetrahedron, 2008, 64, 8292 CrossRef CAS; (d) J. Kärnbratt, M. Hammarson, S. Li, H. L. Anderson, B. Albinsson and J. Androasson, Angew. Chem., Int. Ed., 2010, 49, 1854 CrossRef.
- J. Areephong, H. Logtenberg, W. R. Browne and B. L. Feringa, Org. Lett., 2010, 12, 2132 CrossRef CAS.
- G. Pariani, A. Bianco, R. Castagna and C. Bertarelli, J. Phys. Chem., 2011, 115, 12184 CrossRef CAS.
- W. Liu, S. Pu, S. Cui, G. Liu and C. Fan, Tetrahedron, 2011, 67, 4236 CrossRef CAS.
- M. Liu, S. Pu, G. Liu, C. Fan and S. Cui, J. Phys.: Conf. Ser., 2011, 276, 0112075 Search PubMed.
Footnotes |
| † Dedicated to Dr Hubert Le Bozec on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00491g |
| This journal is © The Royal Society of Chemistry 2012 |