Decomposition of nitrous oxide over Co-zeolite catalysts: role of zeolite structure and active site†
Xinyan
Zhang
a,
Qun
Shen
a,
Chi
He
a,
Chunyan
Ma
a,
Jie
Cheng
a,
Zhiming
Liu
*b and
Zhengping
Hao
*a
aDepartment of Environmental Nano-materials, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, P. R. China. E-mail: zpinghao@rcees.ac.cn; Fax: +86-10-62923564; Tel: +86-10-62923564
bBeijing University Chemical Technology, State Key Laboratory Chemical Resource Engineering, Beijing 100029, P. R. China. E-mail: liuzm@mail.buct.edu.cn; Fax: +86-10-64427356; Tel: +86-10-64427356
First published on 27th February 2012
Abstract
A series of Co exchanged zeolites with ZSM-5, BEA, MOR and USY structures were prepared and investigated for N2O catalytic decomposition under identical reaction conditions. It is found that Co-zeolites with different structures show dramatically different catalytic activities, which could be attributed to various Co species formed in them. Co-ZSM-5, Co-BEA and Co-MOR exhibit much higher activities than Co-USY catalysts, which is attributed to the predominant formation of active isolated Co2+ ions in the ion exchange positions; while in Co-USY Co mainly exists as less active Co oxides. Moreover, it is observed that the activities of Co2+ ions in ZSM-5, BEA and MOR zeolites are quite different and are related to the specific Co ion sites presented in each zeolite structure. In Co-ZSM-5, the most active sites are the α-type Co ions, which are weakly coordinated to framework oxygens in the straight channel. On the other hand, in Co-BEA and Co-MOR, the most active sites are β-type Co ions, which are coordinated to the framework oxygens of the elongated six-membered ring of BEA and the interconnected small channel of MOR, respectively. The main factors affecting the activities of these individual Co ions are indicated to be their location in the zeolite structure, their chemical coordination and the distances between the Co ions. The highest activity of the α-type Co ions in ZSM-5 could be attributed to their favorite location in the zeolite and weak coordination to framework oxygens, which make them easily accessible and coordinated to reactants. The large number of β-sites and their structural arrangement in MOR allow the formation of two unique adjacent β-Co ions in Co–Co pairs, which cooperate in N2O splitting, consequently yielding the high activity of β-Co ions in MOR.
1. Introduction
Nitrous oxide (N2O) has received more and more attention from the scientists because of its great contribution to the greenhouse effect and ozone layer depletion.1 With regard to protecting the environment, it is of great urgency to develop effective technologies to control N2O emission from its anthropogenic sources.2 Among all N2O elimination technologies the catalytic decomposition of N2O to N2 and O2 is proposed as an economical method.3 In this process, cobalt containing zeolites have gained great concern for their excellent catalytic performances.Co-ZSM-5 zeolites are most extensively studied due to their outstanding catalytic performance in N2O decomposition,4–7 however, it was shown that the cobalt exchanged into different zeolites structure exhibited quite different catalytic performances. Co-BEA and Co-MOR are also demonstrated to be very effective for N2O decomposition.7–10 Conversely, when exchanged in the Y zeolite, cobalt is nearly inactive.11 Moreover, conflicting results appeared regarding the influence of cobalt loading on the catalytic performances of these Co-zeolites for N2O decomposition. Smeets et al.5 reported that the catalytic activity of Co-ZSM-5 increased steadily with the cobalt loading, showing a constant TOF in N2O decomposition (mole of N2O transformed per mole of Co), independent of the Co content in the zeolite. However, the activities of Co-MOR catalysts were found not linearly proportional to the Co content in the catalysts.7 For extensively exchanged catalysts, the TOF values are significantly lower, indicating the presence of cobalt species with different activities. All these opposite behaviors imply that the nature and proportion of Co species formed in the zeolites are an important factor determining their catalytic activities; and it is accepted and convinced by our recent work that Co atoms present in the oxide state contribute little to N2O decomposition, while the isolated Co2+ ions are more active for this reaction.7,10 However, further information about their different activities depending on the Co loading and their different activities varying with zeolite types is lacking and unclear. Recently, some researchers have further identified the cobalt species formed in the zeolites, i.e., various mono-atomic Co2+ ions at the exchange sites (α, β and γ),12–14 Co-oxo clusters in the zeolite channel or Co3O4 nanoparticles outside the zeolite channel.5,7 However, as far as the various mono-atomic Co2+ ions are concerned, their relationship with and contribution to the activity for N2O decomposition are still unclear; while it has been indicated that the Co ions at different cation sites would possess a different coordination environment and reactivity to the reactants, and result in varied activities in NO reduction with CH4.15 And this might be the reason for the conflicting catalytic performance of Co-zeolites reported in different literature above. Besides, Co-zeolites with different zeolite types reported for N2O decomposition are prepared and performed under different reaction conditions in different studies, therefore making the comparison of different studies difficult to conduct.
Therefore, in this current work, the same series of Co ion solutions were exchanged with four most used zeolites with structures of ZSM-5, BEA, MOR and USY, and the catalytic performances of all synthesized catalysts were investigated under identical reaction conditions. We aim to study the key open issues related to the conflicting activities obtained over these Co-zeolites with different zeolite types: which one is the most active, which is the actual active site in them, and how the activities depend on the Co loading. The relationship between the cobalt species status (i.e., nature and distribution), which are determined by the zeolite structure and cobalt exchange level, and the catalytic performance was systematically studied for the first time. The Co ions at different cationic sites of these zeolites were determined by quantitative analysis of Co2+ ions UV-vis spectra and their activities for N2O decomposition were tentatively understood according to their coordination and local framework in the zeolites. We have found that Co-ZSM-5, Co-BEA and Co-MOR exhibit much higher activities than Co-USY catalysts, which is attributed to the predominant formation of active isolated Co2+ ions in the ion exchange positions; while in Co-USY Co mainly exists as less active Co oxides. Moreover, the activities of Co2+ ions in ZSM-5, BEA and MOR zeolites are quite different and are related to the specific Co ion sites presented in each zeolite structure. Three main factors, i.e., their location in the zeolite structure, their chemical coordination and the distances between the Co ions greatly contribute to the activities of these individual Co ions.
2. Experimental
2.1 Catalysts preparation
Commercial H-ZSM-5, H-Beta, H-MOR and H-USY zeolites with similar Si/Al molar ratios (∼12) were purchased from Sinopec Co. and used as parent zeolites. In general, various Co-zeolite catalysts were prepared by wet ion exchange (WIE) using Co(NO3)2·6H2O as the precursor at room temperature for 24 h and subsequently calcinated at 600 °C for 4 h. Typically, 3.0 g of H-ZSM-5 was exchanged with 300 mL of Co(NO3)2·6H2O solution with Co concentrations of 0.001, 0.005, 0.01, 0.05 and 0.1 mol L−1, respectively. The obtained catalysts were denoted as Co-ZSM-5x, where x is the theoretical Co exchange degree (x = 200 Co/Al, mol/mol). In all cases, ion exchange was performed for 24 h at room temperature. After that, the sample was washed thoroughly with deionized water, dried at 80 °C and calcined at 600 °C for 4 h. The same preparation procedure was employed for Co-BEAx, Co-MORx and Co-USYx, as for Co-ZSM-5x. For direct comparison, every series of catalysts with four different zeolite structures (ZSM-5, BEA, MOR and USY) prepared by exchanging with the same cobalt solution was also denoted as Co-zeolitesy, with y being the Co concentration used in the exchange process. For example, Co-zeolites0.005 include Co-ZSM-516, Co-BEA26, Co-MOR52 and Co-USY36, which were obtained by exchanging the zeolites with 0.005 mol L−1 of Co solution.2.2 Materials characterization
The Si, Al and Co contents of the synthesized Co-zeolite catalysts were determined by ICP-OES using an Optima 2000 spectrometer. The X-ray diffraction (XRD) patterns of all samples were measured on a Rigaku powder diffractometer (D/MAX-RB) using Cu Kα radiation (λ = 0.15418 nm) at a scanning rate of 4° min−1 in a 2θ range of 10–50°. UV-vis diffuse reflectance spectra (UV-vis DRS) were recorded in the air against BaSO4 in the region of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–24000 cm−1 on a Hitachi UV-3000 spectrometer.
500–24000 cm−1 on a Hitachi UV-3000 spectrometer.
The FTIR spectra of these Co-zeolite catalysts were collected on the Bruker Tensor27 spectrometer with 256 scans at a resolution of 4 cm−1. A self-supporting catalyst pellet (about 50 mg) was placed in the IR flow cell and the spectrum was taken at room temperature in a vacuum system. Prior to collection, the catalyst was treated at 400 °C in a vacuum for 1 h after calcination in air at 600 °C for 4 h and then cooled to room temperature. In situ FTIR spectra of NO adsorption on these Co-zeolite catalysts were also collected on the Bruker Tensor27 spectrometer. The reference spectrum (i.e. background spectrum) was taken at room temperature (25 °C) in He stream. After the He stream was switched to a gas mixture containing 1% NO in He at a total flow rate of 30 ml min−1, a series of time-dependent FTIR spectra of NO adsorption on the catalysts were sequentially recorded.
H2 temperature-programmed reduction (H2-TPR) experiments were conducted on a Micromeritics Chemisorb 2720 apparatus. Prior to reduction, the catalyst (50 mg) was treated in He stream at 600 °C for 0.5 h and then cooled to room temperature in He stream. Then the sample was heated to 900 °C at a heating rate of 10 °C min−1 in 5 vol% H2/He. The H2 consumption was measured online by a TCD detector.
2.3 Activity evaluation
N2O decomposition experiments were performed in a fixed-bed flow microreactor at atmosphere pressure. In each run, 0.1 g of the catalyst (40–60 mesh) was placed in a quartz reactor (4 mm id) and pretreated in He stream at 600 °C for 1 h. After the reactor was cooled to 200 °C, the reactant gas mixture (5000 ppm of N2O; He balance) was fed into the reactor. The total flow rate of the mixed gases was set at 60 ml min−1 (GHSV = 30000 h−1). The outlet gas composition was analyzed online using a gas chromatograph (Agilent 6820 series) equipped with a TCD detector and two serial columns (a Porapar Q column for the separation of N2O and N2/O2, and a molecular sieve 5 Å column for the separation of N2 and O2). The steady-state N2O conversion at a determined temperature ascending from 200 to 600 °C at 25 °C intervals was calculated based on the GC peak area.
3. Results and discussion
3.1 Catalysts composition
The actual Co loadings and Co/Al molar ratios of various catalysts are listed in Table 1. In general, Co loadings are closely related to zeolite structure and exchange solution concentration. The Co loadings over all studied zeolites increase with the increasing Co concentrations of precursor solution, and for a certain concentration the final Co contents of Co-zeolitey catalysts with different zeolite topologies follow the order of Co-MOR > Co-USY > Co-BEA > Co-ZSM-5. Moreover, all Co-zeolite0.1 catalysts show typical diffraction peaks corresponding to the characteristic parent zeolite structure,16 indicating that the zeolite frameworks are well preserved after ion exchange and subsequent calcinations, as shown in Fig. S1 of the ESI.† In addition, no obvious diffraction peaks corresponding to crystallized Co3O417 can be observed for all Co-zeolite0.1 catalysts, which suggest the good dispersion of cobalt species on the internal/external surface of the zeolites and the limited aggregation of Co3O4 nanoparticles.| Catalyst | Co concentration/mol L−1 | Co/Al | Co (wt.%) | Catalyst | Co concentration/mol L−1 | Co/Al | Co (wt.%) |
|---|---|---|---|---|---|---|---|
| Co-ZSM-512 | 0.001 | 0.06 | 0.26 | Co-MOR44 | 0.001 | 0.22 | 1.88 |
| Co-ZSM-516 | 0.005 | 0.08 | 0.35 | Co-MOR52 | 0.005 | 0.26 | 2.25 |
| Co-ZSM-522 | 0.01 | 0.11 | 0.5 | Co-MOR64 | 0.01 | 0.32 | 2.86 |
| Co-ZSM-520 | 0.05 | 0.1 | 0.47 | Co-MOR66 | 0.05 | 0.33 | 2.85 |
| Co-ZSM-524 | 0.1 | 0.12 | 0.55 | Co-MOR70 | 0.1 | 0.35 | 3.09 |
| Co-BEA22 | 0.001 | 0.11 | 0.53 | Co-USY24 | 0.001 | 0.12 | 2.29 |
| Co-BEA24 | 0.005 | 0.12 | 0.58 | Co-USY36 | 0.005 | 0.18 | 3.32 |
| Co-BEA26 | 0.01 | 0.13 | 0.6 | Co-USY38 | 0.01 | 0.19 | 3.58 |
| Co-BEA28 | 0.05 | 0.14 | 0.63 | Co-USY40 | 0.05 | 0.2 | 3.72 |
| Co-BEA30 | 0.1 | 0.15 | 0.65 | Co-USY42 | 0.1 | 0.21 | 3.86 |
3.2 Cobalt species formed in zeolites

 | ||
| Fig. 2 FTIR spectra of NO adsorption on Co-ZSM-5 (A), Co-BEA (B), Co-MOR (C) and Co-USY (D) at 298 K. From light to dark: NO interacted with samples for 5, 10, 20 and 30 min. | ||
3.3 Co ions at different cationic sites of the zeolites
 | ||
| Fig. 3 FTIR spectra in the T–O–T region of Co-ZSM-5, Co-BEA, Co-MOR and Co-USY. Light for Co-zeolites0.005 and dark for Co-zeolites0.1 (A). Deconvolution of the FTIR spectra of Co-ZSM-524 (B). | ||
Moreover, after deconvolution of the IR spectra two or more bands are identified, which indicates the presence of various Co ions located at different cationic sites. For example, Drozdová et al. have assigned the two bands at 935 and 970 cm−1 for Co-ZSM-5 (Fig. 3B) to α- and β-Co sites in the ZSM-5 channel, respectively.27 These Co ions located at specific sites of the zeolites coordinate differently to the framework oxygens and perturb the framework oxygens to varied extent. These properties may be very important for the catalytic activities as demonstrated by previous researchers.15 Therefore, in order to evaluate the individual Co ion sites in more detail, the UV-vis DRS experiments were further conducted over the Co-zeolites, which are demonstrated to be an effective technique to quantitatively analyze the corresponding Co ions at different cationic sites.25–29
500–24000 cm−1 reveals the existence of various Co2+ species in these ion-exchanged Co-zeolite catalysts. After spectral deconvolution, several characteristic UV-vis adsorption bands are extracted, which are attributed to specific isolated Co2+ ions located at the typical cationic sites (referred to as α, β and γ) in the zeolite channel. Taking the Co-MOR catalysts as an example, the single band at 14800 cm−1 corresponds to the α-type Co ions, which are coordinated to framework oxygens in the main channel of mordenite. Four bands at 15900, 17500, 19200 and 21100 cm−1 belong to the β-type Co ions, which are coordinated to framework oxygens in the twisted eight-membered rings of the MOR cavity. Two bands at 20150 and 22050 cm−1 are assigned to γ-type Co ions located at the so-called “boat-shaped” site of mordenite. Similar absorption spectra are observed and extracted for Co-ZSM-5 and Co-BEA catalysts except that the positions of seven bands were slightly shifted due to the small difference in the local structure of various zeolites.27,28 However, the Co-USY catalysts are characterised by a strong adsorption band at 13700 cm−1, a broad weak band located at 20000–24000 cm−1 and a band at 19600 cm−1. The former two bands are typical for Co3O4 particles and the latter is attributed to CoOx clusters. The distribution of the individual Co2+ ion types and other Co oxides in Co-zeolites0.005 and Co-zeolites0.1 was quantitatively analyzed by calculating the integral intensities of their spectral bands. The UV-vis spectra results (Table S1 of the ESI†) reveal that (i) the bands corresponding to isolated Co2+ are strongest for catalysts Co-MOR, Co-ZSM-5 and Co-BEA, showing high population of isolated Co2+ in these zeolites. Co-USY presents the strongest bands corresponding to Co3O4 and CoOx species, indicating that the predominant Co species are Co3O4 and CoOx species; (ii) as for isolated Co2+ ions in Co-ZSM-5, Co-BEA and Co-MOR, the most populated site is the β-site (50–75%), followed by the α-site (5–20%) and then by the least populated γ-site (0–10%); (iii) the population and distribution of these isolated Co2+ ions in ZSM-5, BEA and MOR zeolites are impacted both by the zeolite type and the Co exchange level.
 | ||
| Fig. 4 Deconvolution of UV-vis/DRS spectra of Co-ZSM-5 (A and B), Co-BEA (C and D), Co-MOR (E and F) and Co-USY (G and H). | ||
Therefore, in order to further clarify the dependence of Co loading on distribution of α, β and γ types of Co2+ ions, the spectral deconvolution of all the series of Co-ZSM-5, Co-BEA and Co-MOR catalysts with different Co exchange levels was conducted and the results are shown in Fig. 5 and 8. With the increase of Co loading, the relative population of α-type Co2+ (relative to the total Co loading in the zeolite) decreases for the ZSM-5, BEA and MOR zeolites (Fig. 5). The relative population of β-type Co2+ ions increases for Co-BEA and Co-MOR catalysts over the whole Co/Al range, while for Co-ZSM-5 it increases when the Co/Al ratio increases from 0.06 to 0.08, then decreases to some extent. Nevertheless, the total concentration of β-type Co2+ also increases with increasing Co/Al ratios in Co-ZSM-5 zeolites over the whole Co/Al range (Fig. 8C). These results reveal that with the increase of Co loading the concentration of β-type Co2+ ions increases at the cost of α-type Co2+ ions. The extent of this effect is more pronounced in Co-BEA and Co-MOR than that in Co-ZSM-5. In contrast, the concentration of the γ-type Co2+ ions is very low and does not significantly change over the whole Co/Al range.
 | ||
| Fig. 5 The effect of Co loading on the relative population of Co2+ ions of the types α, β and γ in Co-ZSM-5 (A), Co-BEA (B) and Co-MOR (C). | ||
3.4 Catalytic performances
 | ||
| Fig. 6 Conversion of N2O over Co-ZSM-5 (A), Co-BEA (B), Co-MOR (C) and Co-USY (D). | ||
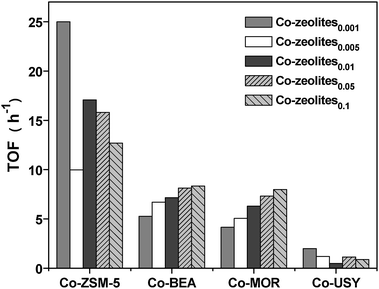
Moreover, for each zeolite type, the TOF does not exhibit a constant value, but depends on the Co/Al ratio of the catalysts. For Co-ZSM-5, the TOF values decrease roughly with increase of the Co/Al ratio, while Co-MOR and Co-BEA catalysts show a constantly increasing trend. This observation clearly evidence that the Co2+ ions in ZSM-5, BEA and MOR zeolites exhibit quite different activities, with respect not only to zeolite structures but also to the specific Co ions presented in zeolites. Therefore, we further related TOF as activity per Co2+ ion, denoted as TOFCo2+, to Co ions at different cationic sites (α, β and γ) to analyze the activity of the individual sites. To simplify the analysis, we have focused on the activity of the cobalt α- and β-sites as low concentration of the γ-type Co ions can be neglected. With Co-ZSM-5 zeolites, the same trend in the dependence of TOFCo2+ values with the α type Co ions on Co2+ concentration is observed, which indicates the α-type Co ions are the most active sites in Co-ZSM-5 zeolites (Fig. 8A). However, with Co-BEA and Co-MOR zeolites, the TOFCo2+ values and concentration of β-type Co ions both increase with increasing Co2+ concentration, suggesting that β-type Co ions are the most active sites in BEA and MOR structures (Fig. 8B and C).
4. Discussion
It is generally accepted that in zeolites exist several forms of cobalt exhibiting different activity, and their distribution greatly depends on the preparation process, cobalt loading and zeolite structures.5–9,12,13,30–32 As proved by characteristic results of H2-TPR, NO-FTIR and UV-vis spectra (Fig. 1–4), Co mainly forms cobalt oxides in Co-USY while Co-ZSM-5, Co-BEA and Co-MOR catalysts possess high population of isolated Co2+ in the zeolites. On the other hand, the activity results show that the activity of Co-zeolites with four zeolite types follows quite a different order of Co-MOR > Co-BEA ≈ Co-ZSM > Co-USY (Fig. 6) from that of Co loading: Co-MOR > Co-USY > Co-BEA > Co-ZSM-5 (Table 1). Besides, the TOF calculation results indicate that the Co-USY catalysts exhibit much lower TOF than the other three zeolites (Fig. 7). These performance activities related to the characteristic results imply that isolated Co2+ ions are the active sites for N2O decomposition and cobalt oxides contribute little to N2O conversion. A similar observation was reported over Co-MFI and Co-MOR zeolites that mono-atomic Co ions in exchange positions exhibit much higher activity than Co oxides in N2O decomposition.7,10On the other hand, for the other three zeolites, ZSM-5, BEA and MOR, which mainly contain mono-atomic Co2+ ions in exchange positions, the Co2+ ions exhibit quite different activities, with respect not only to different zeolite structures but also to the specific Co ions presented in one zeolite structure. This might explain the different catalytic performance obtained in the literature above4–11 and clarify the different dependence effect of these Co-zeolites with the Co loading and zeolite type: although mono-atomic Co2+ ions in exchange positions are generally active for N2O decomposition, however, their activities are highly related to their specific location in different zeolites.
To understand the different activities of these specific Co ions in different zeolite topologies, TOFCo2+ values (activity per Co2+ ion) were further calculated, ruling out the contribution of small amounts of cobalt oxides. The TOFCo2+ values of Co-ZSM-5, Co-BEA and Co-MOR were related to Co ions at different cationic sites (α, β and γ) to analyze the activity of the individual sites. From the analysis of the dependence of TOFCo2+ values and the concentration of α- and β-type Co ions on cobalt content in ZSM-5, BEA and MOR zeolites (Fig. 8), several findings are drawn as follows:
(i) In ZSM-5, the α-type Co ions are indicated to be the most active sites. These ions are coordinated to four framework oxygens in the straight channel of the ZSM-5.26,27 The highly active α-type Co ions are easily accessible to reactants and intermediates and they exhibit the weakest bonding to framework oxygens among the individual Co ions.
(ii) On the other hand, however, in BEA and MOR, the most active sites are suggested to be β-type Co ions. These ions are coordinated to six framework oxygens of the elongated six-membered ring of the hexagonal cage in BEA and the interconnected small channel of the MOR cavity.25,27 The β-type Co ions in MOR are located in the interconnected small channel of mordenite, and consequently they might be diffusionally hindered compared to the α-type Co ions in MOR. Moreover, the β-type Co ions in MOR exhibit stronger perturbation of the framework T–O bonds, which means stronger bonding to framework oxygens than the α-type Co ions in MOR. Thus the electron-donating molecule N2O will less readily coordinate to the cobalt cations in β-sites. The β-type Co ions in BEA are also located in the small channel of BEA, being less accessible to reactants than the α-site. However, in contradiction to the β-sites in MOR, β-type Co ions in BEA exhibit weaker bonding to framework oxygens compared to that of the Co ions in the α-site of BEA, thereby more readily coordinate to N2O.
These observations point out that for the activity of the Co ions in N2O decomposition not only the strength of the Co ions in the framework but also their location in the inner volume of the zeolite might affect their activities. The α-type Co ions in ZSM-5 structure are implied to be located at the most advantageous position with respect to the reactant accessibility and weakest bonding to framework oxygens. With MOR and BEA, the β-type Co ions are also accessible to the reactant N2O, but they are diffusionally hindered compared with the α-type Co ions located in the main channels of MOR and BEA, as they are located in the interconnected small channel of mordenite and inside the small six-membered ring channel of BEA. In addition, the β-type Co ions in MOR exhibit stronger bonding to framework oxygens, thereby less readily coordinated to N2O compared to that of the Co ions in the α-site; while an opposite situation is seen in the BEA. Therefore, it is reasonable to find that TOFCo2+ values of Co-ZSM-5 are much higher than Co-BEA and Co-MOR (Fig. 8). However, for the latter two, although different bonding to framework oxygens is observed, they exhibit almost equal activity. This observation suggests that there must be another factor affecting the rate of the catalytic reaction, i.e., the distance of the Co ions, if the N2O decomposition proceeds with the participation of two neighboring sites. Such a mechanism has been proposed on Fe-FER where there is two iron cations cooperation in the N2O decomposition reaction.33 It can be assumed that, at one site, the Co⋯NNO complex is formed, and the other site attracts the oxygen atom of the Co⋯NNO complex to form Co⋯O species. To attain this N2O splitting by mutual action of two adjacent Co ions, suitable distances would be expected. Therefore, we roughly estimated the distance between these active Co ion sites in ZSM-5, BEA and MOR according to zeolite frameworks and geometrical arrangement of these Co ions. The closest Co sites have been found with the α-type Co ions in ZSM-5 structure (ca. 5.5 Å), while distance of β-type Co ions facing each other across the channel in BEA and MOR is much greater, ca. 7.5 Å and 7.0 Å, respectively. Therefore, a strong attraction between the oxygen atom of the Co⋯NNO complex and the adjacent Co ion (distance O–Co ∼1.5 Å) could be expected in ZSM-5. While for BEA and MOR, the distance between the oxygen atom of the Co⋯NNO complex and the adjacent Co ion would be 3.5 Å and 3.0 Å, respectively. The distance of O–Co below 3.0 Å would produce strong attraction between them while above 3.0 Å it would be too far to form effective attraction.33 Consequently, the ZSM-5 zeolite possesses the most advantageous distance of two adjacent α-type Co ions, which provides potential for cooperation of these two ions in the N2O splitting, followed by β-type Co ions in MOR, while for BEA this distance is too long to form effective Co⋯O.
A direct detection of the existence of the active site containing two close collaborating Co ions accommodated in ZSM-5 and MOR is beyond the present experimental capabilities of the structural techniques. However, a simple evaluation of the probability of the existence of the active sites with such an arrangement of closed Co ions could be employed regarding the occupation of various cationic positions by divalent Co cations in the zeolite. As shown in the UV-vis results, the prevailing Co ions are β-sites in both ZSM-5 and MOR. Furthermore, the concentration of Co ions for MOR is much higher than ZSM-5. Taking these two reasons into account, the formation of collaborating β-type Co ions facing each other across the channel in Co–Co pairs is quite unique and realistic for MOR structure, while in ZSM-5 the chance of α-sites forming Co–Co pairs is rare or could be neglected if they ever exist. Therefore, these two unique close collaborating β-type Co ions in Co–Co pairs in MOR may contribute to its high activity, compensating its difficulty in reactant accessibility and strong bonding to the framework oxygens. Thus, β-type Co ions in MOR exhibit similarly high activity as β-type Co ions in BEA, which have weak bonding to the framework oxygens.
Overall, three factors may determine the activity of the individual Co ions, i.e. their location in the zeolite structure, their coordination and the distances between the Co ions. Their contribution to the activity varies with the zeolite type.
(i) The α-type Co ions in ZSM-5 exhibit the highest activity among the active Co ions of the three zeolites, which could be attributed to their favorite location (straight channel of the ZSM-5) and weak coordination to framework oxygens, which make them easily accessible and coordinated to reactants, and these two factors may play the most important role in determining the activity of the individual Co ions.
(ii) The β-type Co ions in MOR are strongly coordinated to framework oxygens in the interconnected small channel of mordenite and consequently, they are less accessible and coordinated to N2O. However, the large number of β-sites and their structural arrangement in MOR form Co–Co pairs of collaborating β-type Co ions facing each other across the channel (as shown in Fig. 9). These two unique close collaborating Co ions cooperate in the N2O splitting thereby contributing greatly to the superior activity of β-type Co ions in MOR, which is as equally high as the β-type Co ions in BEA. This is consistent with results of the previous literature and our recent works that Co-MOR exhibits high activity for N2O decomposition.7,10 The above results show that Co-MOR shows highest N2O conversion among Co-ZSM-5, Co-BEA and Co-MOR, which could be attributed to its highest exchange level with most abundant active β-type Co ion sites in MOR.
 | ||
| Fig. 8 The dependence of TOFCo2+, concentration of the α- and β-type Co2+ ions on Co2+ content in Co-ZSM-5 (A), Co-BEA (B) and Co-MOR (C). | ||
 | ||
| Fig. 9 Schematic representation of unique structure of Co-MOR including N2O interacting with two close β-type Co ions facing each other across the channel. | ||
5. Conclusions
Zeolite structure has a dramatic effect on Co species formation and their catalytic activities. The activities of catalysts with four zeolite types follow the order of Co-MOR > Co-BEA ≈ Co-ZSM-5 > Co-USY, which could be attributed to various Co species formed in them. In Co-USY catalysts Co atoms mainly form cobalt oxides while in Co-ZSM-5, Co-BEA and Co-MOR catalysts Co atoms mainly exist as isolated Co2+ in ion exchange positions, which are the active sites for N2O decomposition. Moreover, the activities of Co2+ ions in ZSM-5, BEA and MOR zeolites are related to the specific Co ions presented in different zeolite structures. In ZSM-5, the α-type Co ions, weakly coordinated to framework oxygens in the straight channel, are indicated to be the most active sites. In BEA and MOR, the most active sites are proposed as β-type Co ions, which are coordinated to the framework oxygens of the elongated six-membered ring of the hexagonal cage in BEA and the interconnected small channel of the MOR cavity, respectively. Due to their favorite location in the zeolite and coordination to framework oxygens, the α-type Co ions in ZSM-5 are easily accessible and coordinated to reactants, therefore, they exhibit the highest activity among the active sites of the three zeolites. The large number of β-sites and their structural arrangement in MOR allow formation of two unique close β-type Co ions in Co–Co pairs, which cooperate in the N2O splitting, thereby contributing to the high activity of β-type Co ions in MOR. Consequently, Co-MOR yields the highest activity among Co-ZSM-5, Co-BEA and Co-MOR as it possesses the most abundant active β-type Co ion sites.Acknowledgements
This work was financially supported by the Special Co-construction Project of Beijing Municipal Commission of Education, National Natural Science Funds for Distinguished Young Scholar (20725723) and National Basic Research Program of China (2010CB732300).References
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117–151 CrossRef.
- Climate change, the scientific basis. Contribution of the Working Group I to the Third Assessment Report of the IPCC, ed. J. T. Houghton, et al., Cambridge, 2001 Search PubMed.
- J. Pérez-Ramírez, Appl. Catal., B, 2007, 70, 31–35 CrossRef.
- B. M. Abu-Zied and W. Schwieger, Appl. Catal., B, 2009, 85, 120–130 CrossRef CAS.
- P. J. Smeets, Q. G. Meng, S. Corthals, H. Leeman and R. A. Schoonheydt, Appl. Catal., B, 2008, 84, 505–513 CrossRef CAS.
- R. S. da Cruz, A. J. S. Mascarenhas and H. M. C. Andrade, Appl. Catal., B, 1998, 18, 223–231 CrossRef CAS.
- M. C. Campa, V. Indovina and D. Pietrogiacomi, Appl. Catal., B, 2009, 91, 347–354 CrossRef CAS.
- J. A. van Bokhoven, D. C. Koningsberger, P. Kunkeler, H. van Bekkum and A. P. M. Kentgens, J. Am. Chem. Soc., 2000, 122, 12842–12847 CrossRef CAS.
- L. Čapek, J. Dědeček and B. Wichterlová, J. Catal., 2004, 227, 352–366 CrossRef.
- X. Y. Zhang, Q. Shen, C. He, Y. F. Wang, J. Cheng and Z. P. Hao, J. Hazard. Mater., 2011, 192, 1756–1765 CrossRef CAS.
- Y. Li and J. N. Armor, Appl. Catal., B, 1992, 1, L21–L29 CrossRef CAS.
- H.-H. Chen, S.-C. Shen, X. Y. Chen and S. Kawi, Appl. Catal., B, 2004, 50, 37–47 CrossRef CAS.
- X. Wang, H. Chen and W. M. H. Sachtler, Appl. Catal., B, 2001, 29, 47–60 CrossRef CAS.
- J. Q. Zhang, W. B. Fan, Y. Y. Liu and R. F. Li, Appl. Catal., B, 2007, 76, 174–184 CrossRef CAS.
- J. Dědeček and B. Wichterlová, J. Phys. Chem. B, 1999, 103, 1462–1476 CrossRef.
- V. Sebastian, S. Irusta, R. Mallada and J. Santamaría, Appl. Catal., A, 2009, 366, 242–251 CrossRef CAS.
- T. Tsoncheva, L. Ivanova, J. Rosenholm and M. Linden, Appl. Catal., B, 2009, 89, 365–374 CrossRef CAS.
- J. A. Z. Pieterse, R. W. van den Brink, S. Booneveld and F. A. de Bruijn, Appl. Catal., B, 2002, 39, 167–179 CrossRef CAS.
- X. Wang, H.-Y. Chen and W. M. H. Sachtler, Appl. Catal., B, 2000, 26, L227–L239 CrossRef CAS.
- C. Chupin, A. C. van Veen, M. Konduru, J. Despres and C. Mirodatos, J. Catal., 2006, 241, 103–114 CrossRef CAS.
- K. I. Hadjiivanov, Catal. Rev. Sci. Eng., 2000, 42, 71–144 CAS.
- E. Ivanova, K. Hadjiivanov, D. Klissurski, M. Bevilacqua, T. Armaroli and G. Busca, Microporous Mesoporous Mater., 2001, 46, 299–309 CrossRef CAS.
- A. Mihaylova, K. Hadjiivanov, S. Dzwigaj and M. Che, J. Phys. Chem. B, 2006, 110, 19530–19536 CrossRef CAS.
- K. Góra-Marek, B. Gil, M. Śliwa and J. Datka, Appl. Catal., A, 2007, 330, 33–42 CrossRef.
- J. Dědeček, D. Kaucký and B. Wichterlová, Microporous Mesoporous Mater., 2000, 35–36, 483–494 Search PubMed.
- V. Indovina, M. C. Campa and D. Pietrogiacomi, J. Phys. Chem. B, 2008, 112, 5093–5101 CAS.
- J. Dědeček, L. Čapek, D. Kaucký, Z. Sobalík and B. Wichterlová, J. Catal., 2002, 211, 198–207 Search PubMed.
- L. Drozdová, R. Prins, J. Dědeček, D. Kaucký, Z. Sobalík and B. Wichterlová, J. Phys. Chem. B, 2002, 106, 2240–2248 CrossRef.
- L. F. Cordoba, G. A. Fuentes and C. M. de Correa, Microporous Mesoporous Mater., 2005, 77, 193–201 CrossRef CAS.
- C. W. Lee, P. J. Chong, Y. C. Lee, C. S. Chin and L. Kevan, Catal. Lett., 1997, 48, 129–133 CrossRef CAS.
- F. Seyedeyn-Azad and D.-K. Zhang, Catal. Today, 2001, 68, 161–171 CrossRef CAS.
- B. M. Abu-Zied, W. Schwieger and A. Unger, Appl. Catal., B, 2008, 84, 277–288 CrossRef CAS.
- K. Jíša, J. Nováková, M. Schwarze, A. Vondrová, S. Sklenák and Z. Sobalik, J. Catal., 2009, 262, 27–34 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00465h |
| This journal is © The Royal Society of Chemistry 2012 |