One-step synthesis of CdS–TiO2–chemically reduced graphene oxide composites via microwave-assisted reaction for visible-light photocatalytic degradation of methyl orange†
Tian
Lv
a,
Likun
Pan
*a,
Xinjuan
Liu
a,
Ting
Lu
a,
Guang
Zhu
a,
Zhuo
Sun
a and
Chang Q.
Sun
b
aEngineering Research Center for Nanophotonics & Advanced Instrument, Ministry of Education, Department of Physics, East China Normal University, Shanghai, China. E-mail: lkpan@phy.ecnu.edu.cn; Fax: +86 21 62234321; Tel: +86 21 62234132
bSchool of Electrical and Electronic Engineering, Nanyang Technological University, Singapore 639798, Singapore
First published on 9th December 2011
Abstract
One-step synthesis of CdS–TiO2–chemically reduced graphene oxide (RGO) composites was carried out using microwave-assisted reduction of graphite oxide in CdS precursor solution with TiO2 suspension. The photocatalytic performance of CdS–TiO2–RGO composites in degradation of methyl orange was examined. Results show that the RGO addition could enhance the photocatalytic performance of CdS–TiO2 composites with maximum degradation efficiency of 99.5% under visible light irradiation as compared with the pure TiO2 (43%) and CdS–TiO2 (79.9%) composites due to the increase of specific surface area for more adsorbed MO and the reduction of electron–hole pair recombination with the introduction of RGO.
1. Introduction
Currently considerable attention has been paid to semiconductor oxide photocatalysis for water treatment due to its good ability to destroy pollutants and broad compound applicability.1–4 Among the various semiconductor oxides, TiO2 has been proven to be the promising photocatalyst for widespread environmental applications due to its intriguing optical and electrical properties, low cost, stability, nontoxicity, and abundance.5,6 However, the wide band gap of TiO2 (3.2 eV) limits its photocatalytic property in the UV region which only occupies 3–5% of the total solar spectrum. To improve the sunlight photocatalytic efficiency, many promising studies are reported to couple TiO2 with a narrow band gap semiconductor material, typically CdS (2.4 eV), to extend the absorption to the visible light range.7–10 In the meantime, the photoexcited electrons in CdS can be efficiently injected into TiO2 because the conduction band position of CdS is higher than that of TiO2.11,12Unfortunately, the quick recombination of photo-generated charge carriers still exists in the CdS–TiO2 composite and the recombination significantly decreases its photocatalytic performance. Therefore, conductive nanomaterials with a work function lower than the conduction band of TiO2, such as carbon nanotubes (CNTs)13,14 and Pt,15–17 have been incorporated into the CdS–TiO2 composite to form hybrid materials to solve this problem. In the photocatalysis process, these nanomaterials can act as an excellent electron-acceptor/transport material to effectively facilitate the migration of photo-induced electrons and hinder the charge recombination in electron-transfer processes due to the electronic interaction between the CdS–TiO2 composite and them, which enhances the photocatalytic performance.
Recently, graphene, a rising star in the carbon family, has generated increasing interest both in fundamental science and potential applications due to its good conductivity, high electron mobility, superior chemical stability and large surface-to-volume ratio.18–24 Similar to CNTs, graphene can also act as an excellent electron-acceptor/transport material.25 Efforts have been made to combine TiO2 and graphene to obtain hybrid materials with superior photocatalytic performance.26–31 Li et al.32 reported the incorporation of chemically reduced graphene oxide (RGO) in TiO2 to form TiO2–graphene composites by visible-light photocatalytic reduction of graphite oxide (GO). However, the study on the incorporation of RGO into CdS–TiO2 composites for photocatalysis has not yet been reported. Thus, the exploration of the CdS–TiO2–RGO hybrid is necessary to gain the understanding of the individual contribution from RGO, CdS and TiO2 to form the basis for photocatalysis design and application.
In this work, we report the one-step synthesis of CdS–TiO2–RGO composites using microwave-assisted reduction of GO in CdS precursor solution with TiO2 suspension. Microwave irradiation heats the reactant to a high temperature in a short time by transferring energy selectively to microwave absorbing polar solvents. Thus it can facilitate mass production in a short time span with little energy consumption33,34 and form an intimate contact between the components.35–37 Such a microwave-assisted reaction is hardly employed to synthesize CdS–TiO2–RGO composites though it has been used successfully to fabricate RGO,36–40 CdS41–43 or CdS–TiO2,44 respectively. The photocatalytic efficiency of the as-synthesized CdS–TiO2–RGO composite in degradation of methyl orange (MO) under visible light irradiation is much improved compared with the pure TiO2 and CdS–TiO2.
2. Experimental
Commercial graphite powder (Sianpharm Chemical Reagent Co, Ltd.) was used as the starting reagent for the synthesis of GO via a modified Hummers method.21,22,31,45 1.8 mg ml−1 GO suspension, 7 ml 0.45 mM CdCl2 solution and 3 ml 0.64 mM Na2S solution were added into 10 ml 0.6 mM TiO2 (Degussa P25) suspension and then the solution was sonicated for 30 min to produce uniform dispersion. The pH value of solution was adjusted to 10–11 by adding NaOH. It was observed that the color of suspension had changed into yellow, indicating the possibility of spontaneous CdS formation. The solution was then put into an automated focused microwave system (Explorer-48, CEM Co.) and irradiated at 150 °C under the power of 100 W for 10 min. It was observed that the color of suspension had changed into grayish-black, indicating the chemical reduction of the GO sheets.46 The obtained RGO consists of four to five atom layers.36 The as-synthesized CdS–TiO2–RGO samples with 0, 0.72, 1.43, 2.13, 2.82, and 3.50 wt.% RGO, named as CTG-0, CTG-1, CTG-2, CTG-3, CTG-4 and CTG-5, were isolated by filtration. They were washed several times using distilled water, and finally dried in a vacuum oven at 60 °C for 24 h. TiO2–RGO samples with 0.72, 1.43 and 2.13 wt% RGO, named as TG-1, TG-2 and TG-3, and CdS–RGO samples with 0.72, 1.43 and 2.13 wt% RGO, named as CG-1, CG-2 and CG-3, were also synthesized by direct microwave assisted reaction in aqueous solution for comparison.Microwave-assisted synthesis of CdS–TiO2–RGO composites is a one-step process. RGO is synthesized by direct microwave assisted reduction of GO suspension. Due to the distribution of carboxylic acid groups on GO, TiO2 nanoparticles dispersed on the carbon support are found to accumulate along the wrinkles and edge of RGO.26 Meanwhile, positively charged Cd2+ ions form complexes with TiO2 nanoparticles and S2− from Na2S can move quickly to react with Cd2+ producing uniform CdS nuclei under microwave irradiation. The CdS nuclei grow, crystallize, and stabilize on TiO2 under the microwave irradiation, leading to a good contact between CdS and TiO2.37,47
The surface morphology, structure and composition of the samples were characterized by field-emission scanning electron microscopy (FESEM, Hitachi S-4800), high-resolution transmission electron microscopy (HRTEM, JEOL-2010), energy dispersive X-ray spectroscopy (EDS, JEM-2100) and X-ray diffraction spectroscopy (XRD, Holland PANalytical PRO PW3040/60) with Cu Kα radiation (V = 30 kV, I = 25 mA), respectively. The Brunauer–Emmett–Teller (BET) specific surface areas of the samples were evaluated on the basis of nitrogen adsorption isotherms measured at 77 K using a BELSORP-max nitrogen adsorption apparatus (Japan Inc.). All the samples were degassed at 150 °C before nitrogen adsorption measurements. The BET surface area was determined using the adsorption data in the relative pressure (P/P0) range of 0.05–0.35. The UV-vis absorption spectra and diffuse reflectance spectra with BaSO4 as a standard were recorded using a Hitachi U-3900 UV-vis spectrophotometer.
The photocatalytic performance of the as-prepared samples was evaluated through the experiment of photocatalytic degradation of MO under visible light irradiation. 150 mg CdS–TiO2–RGO powders were dispersed into 100 ml MO aqueous solution (10 mg l−1). The mixed suspensions were continuously stirred for 0.5 h in the dark to reach an adsorption–desorption equilibrium. Under the ambient conditions and stirring, the mixed suspensions were exposed to visible light irradiation produced by a 400 W metal halogen lamp with a cut off filter (λ > 400 nm). The irradiation intensity was approximately 8.7 mW cm−2 measured using a chromameter (CS-100A). At certain time intervals, 2 ml of the mixed suspensions were extracted and centrifugated to remove the photocatalyst. The degradation process was monitored by measuring the absorption of MO in the filtrate at 463 nm using a UV-vis absorption spectrometer.
3. Results and discussion
Fig. 1(a) shows the FESEM images of as-synthesized RGO. The RGO nanosheets are curled and corrugated. Fig. 1(b) and (c) display the FESEM images of CTG-0 and CTG-4. As compared with CTG-0, it is observed that RGO sheets are decorated densely by CdS–TiO2 in CTG-4, which displays a good combination between them. From the low-magnification HRTEM image of CTG-4 in Fig. 1(d), it is seen that some nanoparticles are attached onto the surface of the RGO sheet. The RGO sheets act as bridges for the connection between different nanoparticles. Fig. 1(e) shows a high-magnification HRTEM image of CTG-4. The larger crystallite is identified as TiO2 nanoparticles. The lattice spacing measured for this crystalline plane is 0.352 nm, corresponding to the (101) plane of anatase TiO2 (JCPDS#21–1272). Around the edge of TiO2 crystallite, fine crystallites are observed. The crystallites connecting to the TiO2 have lattice fringes of 0.335 nm which are ascribed to the (111) plane of CdS (JCPDS#80-0019). The existence of CdS and TiO2 in the composite is confirmed by the presence of Cd, S, Ti and O peaks in the EDS profile (Fig. 1(f)). Furthermore, the BET specific surface areas of CTG-0, CTG-2, and CTG-4 are found to be 1.3, 4.6 and 8.3 m2 g−1, respectively, indicating that the increase of RGO can supply more surface sites to accommodate more adsorbed MO, which is beneficial to the photocatalytic degradation of MO.48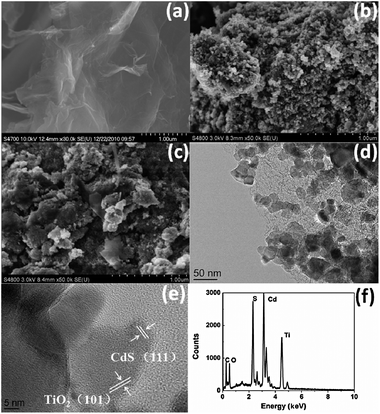 | ||
| Fig. 1 FESEM images of (a) RGO, (b) CTG-0 and (c) CTG-4; (d) low-magnification and (e) high-magnification HRTEM images of CTG-4; (f) EDS spectrum of CTG-4. | ||
The XRD patterns of CdS, RGO, TiO2, CTG-0 and CTG-4 are shown in Fig. 2. The main peaks located at 26.6°, 43.8°, and 51.8° correspond to the (111), (220) and (311) planes of CdS (JCPDS#80-0019). RGO sheets exhibit a (002) diffraction peak at 26° and a (100) peak at 44.5°.49 Compared with the XRD pattern of pure TiO2 in which the peaks correspond to anatase and rutile phases of TiO2 (JCPDS#21-1272), three main peaks of CdS appear in the XRD profiles of CTG-0 and CTG-4. This finding further confirms the existence of CdS on TiO2. The signals corresponding to CdS indicate that CdS crystallites are mainly present as a cubic structure. No typical diffraction peaks of carbon species are observed, which may be due to the low amount and relatively low diffraction intensity of RGO.50
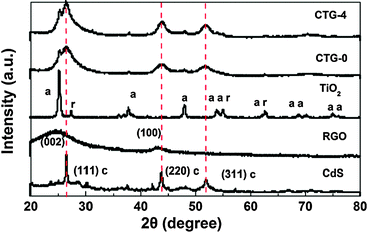 | ||
| Fig. 2 XRD spectra of CdS, RGO, TiO2, CTG-0 and CTG-4 with notations of a—anatase TiO2, r—rutile TiO2, c—cubic CdS. | ||
Fig. 3 shows the UV-vis absorption spectra of TiO2, CTG-0, CTG-1, CTG-2, CTG-3, CTG-4 and CTG-5. It can be seen that the RGO shows an absorption peak at ∼240 nm, which is generally regarded as the excitation of π-plasmon of graphitic structure.51,52 The absorption peak near 350 nm is dominated by TiO2. The addition of CdS results in the appearance of an absorption peak near 500 nm. The increase of the absorbance with the increase of RGO content in the composite, also observed by others,26–28 is suggested to be due to the absorption contribution from RGO, the increase of surface electric charge of the oxides and the modification of the fundamental process of electron–hole pair formation during irradiation.53,54 Furthermore, the diffuse reflectance spectra were also used to approximate the optical absorbance precisely through the Kubelka–Munk function:55K/S = F(R) = (1 − R)2/2R and the similar result as UV-vis absorption spectra was revealed (see Fig. S1, ESI†).
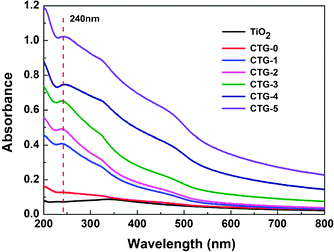 | ||
| Fig. 3 UV-vis absorption spectra of TiO2, CTG-0, CTG-1, CTG-2, CTG-3, CTG-4 and CTG-5. | ||
The photocatalytic degradation of MO under visible light irradiation was conducted to evaluate the photocatalytic performance of TiO2, CTG-0, CTG-1, CTG-2, CTG-3, CTG-4 and CTG-5, as shown in Fig. 4a. It is observed that the concentration of MO is hardly reduced under visible light irradiation in the absence of the photocatalyst. The CdS–TiO2 composite (CTG-0) exhibits much higher photocatalytic degradation efficiency (79.9%) than the pure TiO2 (43%) because the incorporation of CdS extends the absorption to the visible light range. When the RGO is introduced into the composite, the degradation efficiency is increased to 82.1%, 93.4%, and 98.5% for CTG-1, CTG-2 and CTG-3 and reaches the maximum value of 99.5% for CTG-4. The enhancement of the photocatalytic performance should be ascribed to the increase of the amount of adsorbed MO in the presence of RGO with giant two-dimensional planar structure in the composite,26 as indicated by BET measurement, and the stepwise structure of energy levels constructed in the dye–CdS–TiO2–RGO composite,31 as shown in Fig. 5. Two photoinduced degradation paths, one initiated by CdS excitation and another from the self-sensitized destruction of the dyes,56,57 are at work simultaneously under visible light: (i) considering the potential of the conduction band of TiO2 (−4.2 eV), CdS (−3.98 eV) vs. vacuum and the work function of RGO (−4.4 eV),58–61 photo-induced electrons easily transfer from photoexcited dye (MO*) to RGO via the conduction bands of CdS and TiO2, which react with adsorbed oxidants, usually O2, to produce reactive oxygen radicals (O2−). The increase of the amount of adsorbed MO can cause its more efficient self-sensitized degradation; (ii) the photoexcited electrons at the CdS conduction band also easily transfer to RGO via the TiO2 conduction band, which could efficiently separate the photo-induced electrons and hinder the charge recombination in the processes of electron-transfer, thus enhance the photocatalytic performance. However, when the RGO content is further increased above its optimum value, the photocatalytic performance deteriorates. This deterioration should be due to the following possible reasons: (i) the excessive RGO can act as a kind of recombination center instead of providing an electron pathway;31,61,62 (ii) RGO simply filters light and decreases the number of charge carriers in the composite to be photogenerated.
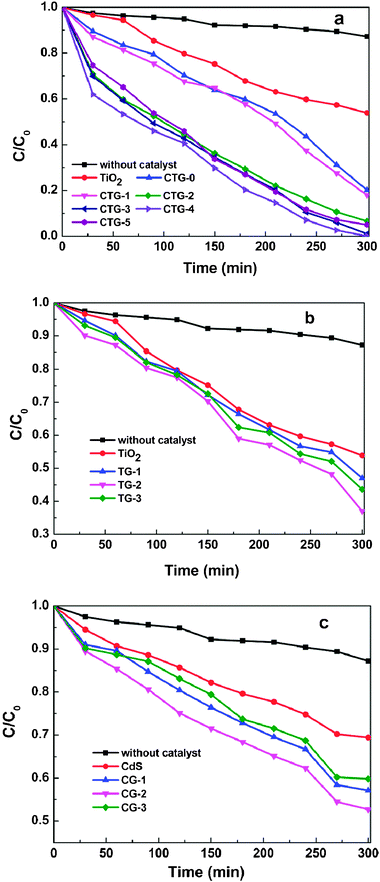 | ||
| Fig. 4 Photocatalytic degradation of MO by (a) TiO2, CTG-0, CTG-1, CTG-2, CTG-3, CTG-4 and CTG-5, (b) TiO2, TG-1, TG-2 and TG-3, (c) CdS, CG-1, CG-2 and CG-3 under visible light irradiation. | ||
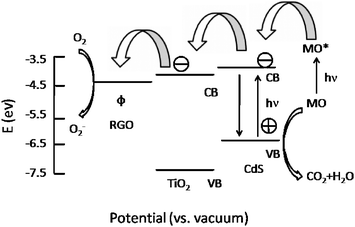 | ||
| Fig. 5 Schematic diagram of energy levels of MO, CdS, TiO2 and RGO. CB, VB, and Φ are conduction band, valence band and work function. | ||
Because both CdS excitation and self-sensitized degradation of dyes contribute to the photocatalytic performance in this work, and self-sensitized destruction of the dyes still exists if only using wide-band gap TiO2 under visible light irradiation,63 the photocatalytic activities of TiO2–RGO in degradation of MO under visible light irradiation are investigated in order to discriminate the contribution of CdS from self-sensitized degradation of dyes. The degradation efficiencies, as shown in Fig. 4b, for TG-1, TG-2 and TG-3 are 53%, 63% and 57%, respectively. Therefore, the large contribution of CdS can be obviously observed from the enhanced degradation efficiencies of CdS–TiO2–RGO composites as compared with TiO2–RGO composites.
Fig. 4c shows the degradation results of MO under visible light irradiation in the presence of pure CdS and CdS–RGO composites. The degradation efficiencies for pure CdS, CG-1, CG-2 and CG-3 are 30%, 43%, 48% and 40%, respectively. Although CdS can absorb the visible-light radiant energy,64 CdS nanoparticles are easily aggregated in aqueous solution during the preparation process, which accordingly results in their rather lower efficiency.65 As is expected, an enhanced photocatalysis of the CdS–RGO composite is observed, which is attributed to migration of photo-induced electrons from the CdS conduction band to RGO. However, compared to CdS–TiO2–RGO composites, affinity between CdS–RGO and dye may decrease, thus affecting the photooxidation reaction rate.66
4. Conclusions
CdS–TiO2–RGO composites synthesized via microwave-assisted reduction of GO in CdS precursor solution with TiO2 suspension show much improved efficiency for photocatalytic degradation of MO. In the composites, CdS extends the absorption of TiO2 from the UV region to visible light range while RGO increases the specific surface areas of the composites and facilitates the charge transportation and separation. The results of photocatalytic experiments indicate that (i) CdS–TiO2–RGO composites exhibit a better photocatalytic performance than the pure TiO2, CdS–TiO2 composites and TiO2–RGO composites; (ii) the photocatalytic performance of CdS–TiO2–RGO is dependent on the proportion of RGO in the composites and the composite with 2.82% RGO achieves a highest MO degradation efficiency of 99.5%; (iii) the enhanced photocatalytic performance is ascribed to the increase of specific surface area for more adsorbed MO and the reduction of photoelectron–hole pair recombination with the introduction of RGO.Acknowledgements
Financial support from Special Project for Nanotechnology of Shanghai (No. 1052nm02700) and the Key laboratory of new ceramics and fine processes at Tsinghua University is gratefully acknowledged.Notes and references
- C. T. Chen, J. Q. Kao, C. Y. Liu and L. Y. Jiang, Catal. Sci. Technol., 2011, 1, 54 CAS.
- R. Guttel, M. Paul and F. Schuth, Catal. Sci. Technol., 2011, 1, 65 Search PubMed.
- C. C. Ou, C. S. Yang and S. H. Lin, Catal. Sci. Technol., 2011, 1, 295 CAS.
- M. Zarezade, S. Ghasemi and M. R. Gholami, Catal. Sci. Technol., 2011, 1, 279 CAS.
- J. Shang, M. Chai and Y. F. Zhu, J. Solid State Chem., 2003, 174, 104 CrossRef CAS.
- J. Zhang, Y. P. Zhang, Y. K. Lei and C. X. Pan, Catal. Sci. Technol., 2011, 1, 273 CAS.
- Y. Xie, G. Ali, S. H. Yoo and S. O. Cho, ACS Appl. Mater. Interfaces, 2010, 2, 2910 CAS.
- J. Bai, J. Li, Y. Liu, B. Zhou and W. Cai, Appl. Catal., B, 2010, 95, 408 CrossRef CAS.
- S. C. Hayden, N. K. Allam and M. A. El-Sayed, J. Am. Chem. Soc., 2010, 132, 14406 CrossRef CAS.
- A. H. Zyoud, N. Zaatar, I. Saadeddin, C. Ali, D. Park, G. Campet and H. S. Hilal, J. Hazard. Mater., 2010, 173, 318 CrossRef CAS.
- G. Zhu, F. F. Su, T. Lv, L. K. Pan and Z. Sun, Nanoscale Res. Lett., 2010, 5, 1749 CrossRef CAS.
- W. Zhao, Z. Bai, A. Ren, B. Guo and C. Wu, Appl. Surf. Sci., 2010, 256, 3493 CrossRef CAS.
- B. Huang, Y. Yang, X. Chen and D. Ye, Catal. Commun., 2010, 11, 844 CrossRef CAS.
- W. Lee, J. Lee, S. Lee, W. Yi, S. H. Han and B. W. Cho, Appl. Phys. Lett., 2008, 92, 153510 CrossRef.
- V. Daskalaki, M. Antoniadou, G. L. Puma, D. I. Kondarides and P. Lianos, Environ. Sci. Technol., 2010, 44, 7200 CrossRef CAS.
- H. Park, W. Choi and M. R. Hoffmann, J. Mater. Chem., 2008, 18, 2379 RSC.
- Q. Kang, Q. Z. Lu, S. H. Liu, L. X. Yang, L. F. Wen, S. L. Luo and Q. Y. Cai, Biomaterials, 2010, 31, 3317 CrossRef CAS.
- T. N. Lambert, C. A. Chavez, B. Hernandez-Sanchez, P. Lu, N. S. Bell, A. Ambrosini, T. Friedman, T. J. Boyle, D. R. Wheeler and D. L. Huber, J. Phys. Chem. C, 2009, 113, 19812 CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- M. Pumera, Energy Environ. Sci., 2011, 4, 668 CAS.
- T. Lu, Y. P. Zhang, H. B. Li, L. K. Pan, Y. L. Li and Z. Sun, Electrochim. Acta, 2010, 55, 4170 CrossRef CAS.
- H. B. Li, T. Lu, L. K. Pan, Y. P. Zhang and Z. Sun, J. Mater. Chem., 2009, 19, 6773 RSC.
- W. T. Zheng and C. Q. Sun, Energy Environ. Sci., 2011, 4, 627 CAS.
- W. T. Zheng, Y. M. Ho, H. W. Tian, M. Wen, J. L. Qi and Y. A. Li, J. Phys. Chem. C, 2009, 113, 9164 CAS.
- P. Wang, Y. Zhai, D. Wang and S. Dong, Nanoscale, 2011, 3, 1640 RSC.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380 CrossRef CAS.
- Y. J. Wang, R. Shi, J. Lin and Y. F. Zhu, Appl. Catal., B, 2010, 100, 179 CrossRef CAS.
- K. Zhou, Y. Zhu, X. Yang, X. Jiang and C. Li, New J. Chem., 2011, 35, 353 RSC.
- Y. Zhang and C. Pan, J. Mater. Sci., 2011, 46, 2622 CrossRef CAS.
- J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175 CrossRef CAS.
- G. Zhu, T. Xu, T. Lv, L. K. Pan, Q. Zhao and Z. Sun, J. Electroanal. Chem., 2011, 650, 248 CrossRef CAS.
- H. B. Li, W. Zhang, L. Zou, L. K. Pan and Z. Sun, J. Mater. Res., 2011, 26, 970 CrossRef CAS.
- S. Horikoshi, H. Abe, K. Torigoe, M. Abe and N. Serpone, Nanoscale, 2010, 2, 1441 RSC.
- X. J. Liu, L. K. Pan, T. Lv, T. Lu, G. Zhu, Z. Sun and C. Q. Sun, Catal. Sci. Technol., 2011, 1, 1189 CAS.
- G. Zhu, L. K. Pan, T. Xu, Q. Zhao, B. Lu and Z. Sun, Nanoscale, 2011, 3, 2188 RSC.
- T. Lu, L. K. Pan, H. B. Li, G. Zhu, T. Lv, X. J. Liu, Z. Sun, T. Chen and D. H. C. Chua, J. Alloys Compd., 2011, 509, 5488 CrossRef CAS.
- G. Zhu, L. K. Pan, T. Xu and Z. Sun, ACS Appl. Mater. Interfaces, 2011, 3, 1472 CAS.
- W. Chen, L. Yan and P. R. Bangal, Carbon, 2010, 48, 1146 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Chem. Mater., 2009, 21, 5004 CrossRef CAS.
- B. G. Choi, H. Park, M. H. Yang, Y. M. Jung, S. Y. Lee, W. H. Hong and T. J. Park, Nanoscale, 2010, 2, 2692 RSC.
- R. M. Nirmal, K. Pandian and K. Sivakumar, Appl. Surf. Sci., 2011, 257, 2745 CrossRef.
- Y. Liu, T. Tan, B. Wang, R. Zhai, X. Song, E. Li, H. Wang and H. Yan, J. Colloid Interface Sci., 2008, 320, 540 CrossRef CAS.
- A. L. Wangshington and G. F. Strouse, Chem. Mater., 2009, 21, 3586 CrossRef.
- S. M. Wang, L. Qing, B. Xie, J. Wu and Y. T. Qian, Mater. Chem. Phys., 2002, 78, 288 Search PubMed.
- H. B. Li, L. K. Pan, T. Lu, Y. K. Zhan, C. Y. Nie and Z. Sun, J. Electroanal. Chem., 2011, 653, 40 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463 CrossRef CAS.
- S. Kundu, H. Lee and H. Liang, Inorg. Chem., 2009, 48, 121 CrossRef CAS.
- Y. P. Zhang and C. X. Pan, J. Mater. Sci., 2011, 46, 2622 CrossRef CAS.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782 CrossRef CAS.
- Y. L. Chen, Z. A. Hu, Y. Q. Chang, H. W. Wang, Z. Y. Zhang, Y. Y. Yang and H. Y. Wu, J. Phys. Chem. C, 2011, 115, 2563 CAS.
- J. L. Wu, X. P. Shen, L. Jiang, K. Wang and K. M. Chen, Appl. Surf. Sci., 2010, 256, 2826 CrossRef CAS.
- X. Wang, L. J. Zhi, N. Tsao, J. L. Tomovic and K. Mullen, Angew. Chem., Int. Ed., 2008, 47, 2990 CrossRef CAS.
- T. G. Xu, L. W. Zhang, H. Y. Cheng and Y. F. Zhu, Chem. Rev., 2010, 101, 382 Search PubMed.
- Y. Miyake and H. Tada, J. Chem. Eng. Jpn., 2004, 37, 630 CrossRef CAS.
- Y. P. Zhang and C. X. Pan, J. Mater. Sci., 2011, 46, 2622 CrossRef CAS.
- D. Zhao, C. Chen, Y. Wang, W. Ma, J. Zhao, T. Rajh and L. Zang, Environ. Sci. Technol., 2008, 42, 308 CrossRef CAS.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef.
- Y. L. Lee and Y. S. Lo, Adv. Funct. Mater., 2009, 19, 604 CrossRef.
- R. Czerw, B. Foley, D. Tekleab, A. Rubio, P. M. Ajayan and D. L. Carroll, Phys. Rev. B: Condens. Matter, 2002, 66, 033408 CrossRef.
- N. Yang, J. Zhai, D. Wang, Y. S. Chen and L. Jiang, ACS Nano, 2010, 4, 887 CrossRef CAS.
- X. Y. Zhang, H. P. Li, X. L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801 RSC.
- B. J. Li and H. Q. Cao, J. Mater. Chem., 2011, 21, 3346 RSC.
- J. Y. Wang, Z. H. Liu, Q. Zheng, Z. K. He and R. X. Cai, Nanotechnology, 2006, 17, 4561 CrossRef CAS.
- H. Y. Zhu, R. Jianga, L. Xiao, Y. H. Chang, Y. J. Guan, X. D. Li and G. M. Zeng, J. Hazard. Mater., 2009, 169, 933 CrossRef CAS.
- J. C. Tristao, F. Magalhaes, P. Corio and M. C. Sansiviero, J. Photochem. Photobiol., A, 2006, 181, 152 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00452f |
| This journal is © The Royal Society of Chemistry 2012 |