“On-water” rhodium-catalysed hydroformylation for the production of linear alcohols†
Olivier
Diebolt
,
Christian
Müller
and
Dieter
Vogt
*
Eindhoven University of Technology, Den Dolech 2, 5600MB Eindhoven, The Netherlands. E-mail: d.vogt@tue.nl; Fax: +31 40 245 5054; Tel: +31 40 247 2730
First published on 21st December 2011
Abstract
Optimisation of the reaction conditions for the rhodium-catalysed aldehyde hydrogenation under hydroformylation conditions showed that water used as co-solvent enhances both rate and selectivity towards primary alcohols. One-pot hydroformylation–hydrogenation using rhodium as the only transition metal yielded alcohols in excellent selectivities and good linearities.
Introduction
The hydroformylation reaction was discovered in 1938 by Otto Roelen for cobalt-based systems at Ruhrchemie in Germany.1 In the early days research and development focused mainly on those cobalt-based catalysts.2 Later other transition metals were shown to exhibit interesting activities for this transformation as well.3 In particular Rh-based systems in combination with phosphorus ligands showed excellent activities4 and led to many industrial applications. Nowadays ligand-modified Rh-systems are preferred due to their superior activity and linear selectivity allowing processes to be operated at much lower pressure and temperature and giving less undesired by-products. However, Co-based systems remain an exception due to their hydrogenation activity. Due to the protic nature of the cobalt hydride, these systems show high isomerisation activity and due to the harsh conditions at which they are applied, aldehydes can be directly reduced to the corresponding alcohols. This is interesting for lower value internal alkene streams as obtained e.g. in the Shell Higher Olefin Process, SHOP. Here detergent range internal alkenes (C12–C18) can be directly converted to surfactant alcohols with decent, yet not really satisfying linearity.5Thus it is of high interest to apply the highly active and linear selective Rh-systems tandem transformation. However, Rh-catalysts usually show only very low hydrogenation activity (Scheme 1).
 | ||
| Scheme 1 Tandem hydroformylation–hydrogenation. | ||
Amongst the many ligands studied in the literature for the Rh-catalysed tandem hydroformylation–hydrogenation, few of them allow the formation of alcohols with satisfying linearity. A major breakthrough was made in the 1990s by Cole-Hamilton and co-workers when a new catalytic system based on Rh in combination with a tri-n-alkylphosphine ligand was reported.6 High activities and selectivities towards alcohols were obtained under mild conditions of pressure and temperature. Nevertheless, the linearities obtained were rather poor (2 to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). This problem was later circumvented by using cooperative ligand effects.7 The combination of PEt3 and Xantphos allowed the formation of alcohols in excellent yields and selectivities.7 Importantly, it was shown that aldehydes are not intermediates in this reaction and that the high activity of highly σ-donating tris-alkyl-phosphine was due to the favored protonation of the acylrhodium intermediate to a hydroxycarbene, leading to the direct production of alcohols. Breit and co-workers reported in 2009 on a supramolecular catalyst for this reaction showing very good activity under mild conditions: For example, 80% of 1-nonanol could be obtained at temperatures as low as 40 °C with a l/b ratio of 11.5.8 Recently, Nozaki and co-workers reported on a new catalytic system using a combination of rhodium and ruthenium.9 The linearities obtained were very satisfying (l/b of up to 22) and up to 90.1% yield of linear alcohol could be obtained from 1-octene. The major drawback of this catalytic system is its price, since 1 mol% of rhodium and 2.5 mol% of the expensive Shvo's catalyst were necessary for this transformation.
:1). This problem was later circumvented by using cooperative ligand effects.7 The combination of PEt3 and Xantphos allowed the formation of alcohols in excellent yields and selectivities.7 Importantly, it was shown that aldehydes are not intermediates in this reaction and that the high activity of highly σ-donating tris-alkyl-phosphine was due to the favored protonation of the acylrhodium intermediate to a hydroxycarbene, leading to the direct production of alcohols. Breit and co-workers reported in 2009 on a supramolecular catalyst for this reaction showing very good activity under mild conditions: For example, 80% of 1-nonanol could be obtained at temperatures as low as 40 °C with a l/b ratio of 11.5.8 Recently, Nozaki and co-workers reported on a new catalytic system using a combination of rhodium and ruthenium.9 The linearities obtained were very satisfying (l/b of up to 22) and up to 90.1% yield of linear alcohol could be obtained from 1-octene. The major drawback of this catalytic system is its price, since 1 mol% of rhodium and 2.5 mol% of the expensive Shvo's catalyst were necessary for this transformation.
Optimisation of the nonanal reduction reaction
Aiming at a monometallic catalyst for this tandem reaction, we decided to first optimise the reduction of linear aldehydes by syngas using a rhodium precursor. Xantphos, giving excellent linearities in Rh-catalysed hydroformylation,10 was used as ligand (see the results in Table 1). Cole-Hamilton and co-workers used ethanol as solvent for their catalytic system.6 Hence, we started our study on the reduction of n-nonanal using this solvent. The reaction being very slow under milder conditions, the temperature was set to 160 °C with a syngas pressure of 36 bar (ratio of CO:H2 1:2). Under these conditions, only 16.5% of n-nonanol were produced within half an hour (Table 1, entry 1). Running the reaction in the presence of H2 instead of syngas afforded a nearly complete reaction producing mainly n-nonanol (Table 1, entry 2). This reaction demonstrates the expected catalyst poisoning effect by CO. Previous studies on the hydrogenation of linear aldehydes by a ruthenium catalyst showed that the use of water as co-solvent improves both activity and selectivity, especially limiting the formation of acetal in alcohol solvents.11,12 Moreover, Cole-Hamilton and co-workers also mentioned the beneficial effect of additional water in the tandem hydroformylation–hydrogenation.6b We reasoned that mixtures of ethanol:water could improve the hydrogenation activity of the catalyst. The reaction under syngas was carried out in a mixture of ethanol:water 1:1. No significant effect was noticed; the reaction being even slower and producing acetals (ca. 10%) despite the presence of water as co-solvent (Table 1, entry 3). This proves again the high acetalisation activity of the present catalytic system in alcohol solvents.11 Increasing the ratio of ethanol:water to 1:9 increased both selectivity and activity towards n-nonanol, obtaining 76.3% alcohol with a selectivity higher than 99% (Table 1, entry 4). Diethoxynonane was only produced in traces in this case (below 0.2%). Interestingly, not only protic and polar co-solvents (ethanol, methanol, isopropanol and tert-amyl alcohol, entries 4 to 7) are suitable for this reaction, but also toluene, dimethylacetamide and tetrahydrofuran gave good results (entries 8 to 10). Even the basic triethylamine allowed for 47.7% conversion under the same conditions (entry 11). The reaction could also be performed in neat water (entry 12). Isopropanol was selected as co-solvent of choice for this reaction since 90.0% of 1-nonanol was produced in a very short time (entry 7).

| Entry | Solvent | Nonanol (%) | Selectivity (%) |
|---|---|---|---|
| a Reaction conditions: nonanal (2 mmol), [Rh(CO)2(acac)] (10 μmol, S/Rh = 200), Xantphos (30 μmol, L/Rh = 3), solvent (2 mL). b H2 (20 bar) used instead of syngas. c Significant amounts of acetal (ca. 10–15%) were also produced. d Conversion is >99.5%; 5% acetal was formed together with 4.8% of aldol condensation product. | |||
| 1 | EtOH | 16.5c | 57 |
| 2 | EtOHb | 90.1d | 90.1 |
| 3 | EtOH:H2O (1:1) |
9.8c | 34.2 |
| 4 | EtOH:H2O (1:9) |
76.3 | >99 |
| 5 | MeOH:H2O (1:9) |
57.9 | >99 |
| 6 |
t
AmOH:H2O (1:9) |
34.3 | >99 |
| 7 |
i
PrOH:H2O (1:9) |
90.0 | >99 |
| 8 | toluene:H2O (1:9) |
40.4 | >99 |
| 9 | DMA:H2O (1:9) |
84.7 | >99 |
| 10 | THF:H2O (1:9) |
72.9 | >99 |
| 11 | Et3N:H2O (1:9) |
47.7 | >99 |
| 12 | H2O | 46.2 | >99 |
The beneficial effect of water can be explained by the fact that this very polar reaction medium pushes the long hydrophobic aliphatic chains together, increasing the local substrate concentration. This was described as “on water” effect by Sharpless et al. in 2005.13 Also, this very protic and polar medium may favour the formation of cationic rhodium species, known for their good activity in polar multiple bond reduction.14–16 The same study was thus conducted using the readily available PnBu3 and PPh3. PnBu3 showed excellent activity for the reduction of nonanal to nonanol under syngas at only 80 °C. The optimum solvent mixture in this case was methanol:water 1:3.17 When using PPh3 as ligand, large amounts of aldol condensation products were obtained and no selective system based on Rh/PPh3 was found for this reduction.17
Tandem hydroformylation–hydrogenation
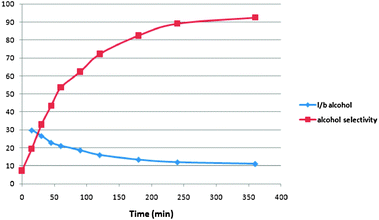
The tandem hydroformylation–hydrogenation reaction was then performed under the optimised conditions using 1-octene as substrate [Rh(CO)2(acac) (S/Rh = 1000) and Xantphos (P/Rh = 6)]. The reaction profile is depicted in Fig. 1. As expected, the hydroformylation reaction is very fast and nearly complete conversion is obtained before the reaction temperature is reached (ca. 10 min.). The aldehyde hydrogenation is slower and the alcohol selectivity reached 92.5% after 6 h of reaction. Fig. 2 shows the evolution of the l/b ratio of alcohol and the alcohol selectivity in time. Not surprisingly, the l/b ratio decreases in time and is relatively low due to the high reaction temperature (l/b = 11). The overall yield in linear alcohol is 85.8% (Table 2, entry 1). It is important to note that in this case the aldehyde is clearly a reaction intermediate.18 | ||
| Fig. 1 Reaction profile of one-pot hydroformylation–hydrogenation of 1-octene. | ||
 | ||
| Fig. 2 Evolution of l/b ratio and alcohol selectivity in time. | ||
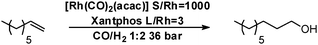
| Entry | Temperature profile | l/b alcohol | Alcohol selectivityc | Yield in nonanol |
|---|---|---|---|---|
| a In all runs conversion was >99.5%. b Reaction profile of this reaction: see Fig. 3 and 4. c Amongst hydroformylation products, aldehyde and alcohols. | ||||
| 1 | 160 °C for 6 h | 11.0 | 92.5% | 85.8% |
| 2 | 100 °C for 3 h, then 160 °C for 6 hb | 15.2 | 93.9% | 88.1% |
| 3 | 110 °C for 4 h, then 160 °C for 20 h | 14.3 | 99.7% | 93.2% |
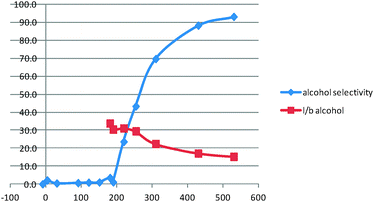
In order to improve the l/b ratio of the final product, we decided to use different reaction temperatures for the hydroformylation and for the hydrogenation reaction. Two temperature profiles were investigated (see Table 2). Setting up the starting temperature to 100 °C for 3 h and then heating to 160 °C for 6 h afforded a very satisfying l/b ratio of 15.2 with good alcohol selectivity (Table 2, entry 2 and Fig. 3). However, after the 3 h treatment at 100 °C, the l/b ratio of the intermediate aldehyde was 33–34 (Fig. 4). Moreover, significant amounts of internal octenes were formed (3.5%). We reasoned that the drop in linearity was due to the hydroformylation–hydrogenation sequence of remaining internal octenes at 160 °C, producing branched alcohols (5.8% at the end of the reaction). Setting up the initial reaction temperature to 110 °C for 4 h led to a similar l/b ratio of 32 for the intermediate aldehyde and only 1.4% of internal octenes. Letting the reaction stir at 160 °C overnight provided excellent alcohol selectivity with a l/b ratio of 14.3 (Table 2, entry 3). These reaction conditions could produce an unprecedented 93.2% yield of 1-nonanol. Of important note, compared to precedent reports in the literature,9 the reaction is very clean and only octane is formed in traces as a secondary product (see ESI†).
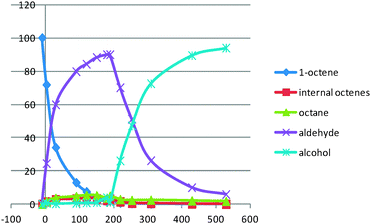 | ||
| Fig. 3 Reaction profile of the reaction (Table 2, entry 2). | ||
Next, the tandem hydroformylation–hydrogenation of 1-octene using Rh/PnBu3 in methanol:water (1:3) was performed at 80 °C (see Scheme 2). Despite the significant formation of octane (11%), the reaction proceeded very fast and reached 94.0% alcohol selectivity in only 2 h. An alcohol selectivity of 99.8% was obtained by setting up the reaction for 18 h.
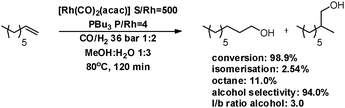 | ||
| Scheme 2 Tandem hydroformylation–hydrogenation using an Rh/PnBu3 system. | ||
Scope of the reaction
With these optimised reaction conditions in hand, we decided to investigate the scope of this transformation (see Table 3). Reaction profiles are depicted in the ESI.† Usually, aqueous catalytic systems are more suitable for short chain alkenes due to their solubility in water and hence better interaction with the hydrophilic catalysts. However in the present case, the catalyst is soluble in the organic phase. 1-Decene and 1-dodecene showed indeed good conversions of 93.4% and 99.2%, respectively (Table 3, entries 5 and 6). Nevertheless these two examples emphasize the importance of substrate dependence. First, 1-decene was selectively hydroformylated but the surprisingly low aldehyde hydrogenation activity produced alcohols with a selectivity of only 69.2% (Table 3, entry 5). In contrast, the aldehyde reduction activity was very high for 1-dodecene as substrate (95.0% alcohol selectivity) but the l/b ratio of the produced alcohol was poor (Table 3, entry 6).
| Entry | Substrate | Reaction conditionsb | Conv.c (%) | Hydrod (%) | Alcohol selectivitye (%) | l/b ratio |
|---|---|---|---|---|---|---|
|
a Reaction profiles of these reactions: see ESI.
b Reaction conditions: A: L is Xantphos (L/Rh = 3), iPrOH:H2O 1:9, 160 °C, 6 h. B: L is Xantphos (L/Rh = 3), iPrOH:H2O 1:9, temperature profile: 110 °C for 4 h, then 160 °C for 20 h. C: L is PBu3 (L/Rh = 4), MeOH:H2O 1:3, 80 °C, 6 h. D: L is PBu3 (L/Rh = 4), MeOH:H2O 1:3, 80 °C, 24 h.
c Conversion amongst octenes.
d Hydrogenation product.
e Amongst hydroformylation products, aldehyde and alcohols.
|
||||||
| 1 | 1-Octene | A | 100 | 2.8 | 92.5 | 11.0 |
| 2 | B | 100 | 3.1 | 99.7 | 14.3 | |
| 3 | C | 99.1 | 9.9 | 96.8 | 2.7 | |
| 4 | D | 99.9 | 11.4 | 99.8 | 2.4 | |
| 5 | 1-Decene | B | 93.4 | 6.0 | 69.2 | 14.6 |
| 6 | 1-Dodecene | B | 99.2 | 8.9 | 95.0 | 4.6 |
| 7 | trans-2-Octene | A | 98.4 | 5.4 | 67.0 | 1.15 |
| 8 | C | 17.0 | 0.1 | 24.6 | 0.2 | |
| 9 | D | 51.6 | 0.2 | 90.3 | 0.01 | |
| 10 | Cyclooctene | C | 83.3 | 0.0 | 88.5 | / |
| 11 | D | 92.5 | 0.0 | 97.2 | / | |
A catalytic system able to produce linear hydroformylation products and especially alcohols from internal alkenes would be of very high interest.19–21 Our attention then turned to the tandem hydroformylation–hydrogenation reaction of internal alkenes. Although Rh/Xantphos systems are well-known for their low hydroformylation and alkene isomerization rates, the high temperatures used here may allow for some activity. Gratifyingly, the tandem reaction using trans-2-octene as substrate gave 98.4% conversion and 67.0% alcohol selectivity. However, the linearity was expectedly poor (Table 3, entry 7). To this end ligands that tolerate the protic conditions and at the same time combine high isomerization and hydroformylation activity with good linear selectivity will be required. Using PBu3 as ligand at 80 °C gave poor activity in this case (Table 3, entry 8), with only 51.6% conversion observed after 24 h (Table 3, entry 9). Moreover, the linear alcohol was only formed as traces. Under the same conditions (PBu3, 80 °C), cis-cyclooctene reacted smoothly to give cycloctylmethanol in good conversion and selectivity (Table 3, entries 10 and 11). These reactions proceeded very cleanly and no side-products were formed.
The reaction was also performed using water-soluble allyl alcohol as substrate, but the standard conditions described here did not allow the formation of 1,4-butanediol or subsequent cyclic products. Changing the co-solvent for better catalyst solubility also proved unsuccessful. This emphasizes again the high substrate dependence of the present catalytic system.
Conclusions
The Rh-catalysed reduction of linear aldehydes under syngas pressure was investigated. Three common hydroformylation ligands, Xantphos, triphenylphosphine and tri-n-butylphosphine, were tested. The use of water as main solvent significantly improved the activity and the selectivity of the overall reaction. Interestingly, all the co-solvents tested showed good activity in aldehyde hydrogenation and the reaction was also efficient in neat water. The formation of cationic rhodium species, known for their high activity in hydrogenation, is likely to be favoured by highly polar and protic solvents. Optimum reaction conditions could be found using either Xantphos or PnBu3 as ligand.The feasibility of tandem hydroformylation–hydrogenation reaction was investigated. 1-Octene could be converted to 1-nonanol by an Rh/Xantphos system in excellent conversion, selectivity and with satisfying linearity. Up to 93.2% of 1-nonanol could be obtained from 1-octene which is—to the best of our knowledge—the highest reported yield for this transformation.
The substrate scope showed significant substrate dependence. Higher alkenes showed either slower hydrogenation activity or lower linearity. In aqueous media, the length of the alkyl chains influences the catalyst solubility and the interaction between the water and the organic phase. The solvent mixture, the pressure and the temperature may be adapted for each substrate. Internal alkenes are also suitable for this transformation and trans-2-octene and cis-cyclooctene could be converted in high yield to their corresponding alcohols. A future challenge here will be the development of ligands that are stable under protic conditions and enable high isomerisation activity combined with high linear selectivity.
Acknowledgements
This work has been funded by the ACTS/ASPECT programme (O.D.). C.M. gratefully acknowledges support from NWO (Vidi grant). We thank Ton Staring for technical assistance.Notes and references
- B. Cornils, W. A. Herrmann and M. Rasch, Angew. Chem., Int. Ed. Engl., 1994, 33, 2144–2163 CrossRef.
- F. Hebrard and P. Kalck, Chem. Rev., 2009, 109, 4272–4282 CrossRef CAS and references therein. Also see ESI† for parameter screening in cobalt-catalysed hydroformylation of 1-octene.
- R. Jennerjahn, I. Piras, R. Jackstell, R. Franke, K.-D. Wiese and M. Beller, Chem.–Eur. J., 2009, 15, 6383–6388 CrossRef CAS; R. A. Sanchez-Delgado, J. S. Bardley and G. Wilkinson, J. Chem. Soc., 1976, 399–405 Search PubMed; I. Piras, R. Jennerjahn, R. Jackstell, A. Spannenberg, R. Franke and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 280–284 CrossRef; H. C. Clark and V. K. Jain, Coord. Chem. Rev., 1984, 55, 151–204 CrossRef.
- C. K. Brown and G. Wilkinson, Tetrahedron Lett., 1969, 10, 1725–1726 CrossRef.
- P. N. Bungu and S. Otto, Dalton Trans., 2007, 2876–2884 RSC; P. N. Bungu and S. Otto, Dalton Trans., 2011, 9238–9249 RSC; C. Crause, L. Bennie, L. Damoense, C. L. Dwyer, C. Grove, N. Grimmer, W. J. van Rensburg, M. M. Kirk, K. M. Mokheseng, S. Otto and P. J. Steynberg, Dalton Trans., 2003, 2036–2042 RSC; A. Polas, J. D. E. T. Wilton-Ely, A. M. Z. Slawin, D. F. Foster, P. J. Steynberg, M. J. Green and D. J. Cole-Hamilton, Dalton Trans., 2003, 4669–4677 RSC.
- J. K. McDougall, M. C. Simpson and D. J. Cole-Hamilton, Polyhedron, 1993, 12, 2877–2881 CrossRef CAS; J. K. McDougall, M. C. Simpson, M. J. Green and D. J. Cole-Hamilton, J. Chem. Soc., Dalton Trans., 1996, 1161–1172 RSC; P. Cheliatsidou, D. F. S. White and D. J. Cole-Hamilton, Dalton Trans., 2004, 3425–3427 RSC.
- I. I. F. Boogaerts, D. F. S. White and D. J. Cole-Hamilton, Chem. Commun., 2010, 46, 2194–2196 RSC.
- L. Diab, T. Šmejkal, J. Geier and B. Breit, Angew. Chem., Int. Ed., 2009, 48, 8022–8026 CrossRef CAS.
- K. Takahashi, M. Yamashita, T. Ichihara, K. Makano and K. Nozaki, Angew. Chem., Int. Ed., 2011, 49, 1–4 Search PubMed.
- M. Kranenburg, Y. E. M. van der Burgt, P. C. J. Kamer and P. W. N. M. van Leeuwen, Organometallics, 1995, 14, 3081–3089 CrossRef CAS.
- A catalytic system producing acetals in high yields and selectivities has been developed and will be reported elsewhere.
- Ruthenium–Xantphos catalysed reduction of nonanal was studied. See ESI†.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS.
- H. Fujitsu, E. Matsumura, K. Takeshita and I. Mochida, J. Chem. Soc., Perkin Trans., 1981, 2650–2655 RSC; T. V. Rajanbabu, Y.-Y. Yan and S. Shin, J. Am. Chem. Soc., 2001, 123, 10207–10213 CrossRef CAS; B. Hamers, E. Kosciusko-Morizet, C. Müller and D. Vogt, ChemCatChem, 2009, 1, 103–106 CrossRef; A. Behr, M. Becker and S. Reyer, Tetrahedron Lett., 2010, 51, 2438–2441 CrossRef.
- Cationic dihydridoiridium species have been isolated and showed to be the active species in the hydrogenation of polar double bonds: R. H. Crabtree, H. Felkin, T. Fillebeen-Khan and G. E. Morris, J. Organomet. Chem., 1979, 168, 183–195 CrossRef CAS.
- For an example of equilibrium between neutral and cationic rhodium species in polar media, see: A. J. Sandee, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2001, 123, 8468–8476 CrossRef CAS.
- See ESI† for more details on Rh–PBu3 and Rh–PPh3-catalysed reduction of nonanal.
- In the catalytic system described by Cole-Hamilton and co-workers, once formed, the aldehyde does not leave the coordination sphere of the catalyst and is directly hydrogenated. See ref. 6.
- For selected examples of production of linear aldehydes from internal alkenes, see: L. A. van der Veen, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 1999, 38, 336–338 CrossRef CAS; D. Selent, D. Hess, K.-D. Wiese, D. Röttger, C. Kunze and A. Börner, Angew. Chem., Int. Ed., 2001, 40, 1696–1698 CrossRef; H. Klein, R. Jackstell, K.-D. Wiese, C. Borgmann and M. Beller, Angew. Chem., Int. Ed., 2001, 40, 3408–3411 CrossRef; A. Behr, D. Obst, C. Schulte and T. Schosser, J. Mol. Catal. A: Chem., 2003, 206, 179–184 CrossRef; Y. Yan, X. Zhang and X. Zhang, J. Am. Chem. Soc., 2006, 128, 16058–16061 CrossRef; D. Sémeril, C. Jeunesse, D. Matt and L. Toupet, Angew. Chem., Int. Ed., 2006, 45, 5810–5814 CrossRef; M. Janssen, L. Bini, B. Hamers, C. Müller, D. Hess, A. Christiansen, R. Pranke and D. Vogt, Adv. Synth. Catal., 2010, 51, 1971–1975 Search PubMed.
- For an excellent example of palladium-catalysed methoxycarbonylation producing linear ester see: C. Jímenez Rodríguez, D. F. Foster, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2004, 1720–1721 RSC.
- To the best of our knowledge, no efficient rhodium-based catalytic system producing linear alcohols from internal alkenes has been reported so far.
Footnote |
| † Electronic supplementary information (ESI) available: Calibration curves, optimisation of aldehyde reduction reaction using PBu3 and PPh3, optimisation of aldehyde reduction using Ru3(CO)12/Xantphos, reaction profiles and parameter screening in cobalt-catalysed hydroformylation of 1-octene. See DOI: 10.1039/c2cy00450j |
| This journal is © The Royal Society of Chemistry 2012 |