The average Pd oxidation state in Pd/SiO2 quantified by L3-edge XANES analysis and its effects on catalytic activity for CO oxidation†
Received
14th October 2011
, Accepted 16th December 2011
First published on 19th December 2011
Abstract
The palladium oxidation state of an SiO2-supported palladium catalyst was quantitatively determined by Pd L3-edge XANES (X-ray absorption near-edge structure) analysis. By changing the time of CO-reduction pre-treatment at 673 K, a series of 5 wt% Pd-loaded SiO2 catalysts (PdOx/2/SiO2) containing different amounts of the Pd metal and PdO phases were prepared, and the average oxidation number (x) was estimated from the number of CO2 molecules formed during the CO-reduction treatment. L3-edge XANES spectra of these samples and a reference sample (Pd powder) were recorded, and the white line area of the spectra was evaluated. A good linear relationship was confirmed between the white line area intensity and the average oxidation number (x), indicating that the oxidation state of Pd in structurally unknown Pd samples could be quantitatively determined by a simple XANES analysis. To demonstrate the utility of this method in a catalytic study, the effect of the oxidation number (x) on the CO oxidation activity of PdOx/2/SiO2 was also examined, and metallic Pd0 sites in PdOx/2/SiO2 were shown to be active species.
Introduction
Pt, Pd, and Rh have been indispensable elements in industrial heterogeneous catalysts1–18 for electrocatalysis, hydrogenation, oxidation, and automotive catalysis. Toward a knowledge-based development of metal catalysts, fundamental studies of the correlation between structure (size, shape, and oxidation state) and reactivity have been an important topic in catalysis. For the oxidation of CO and hydrocarbons, the oxidation state of metal species strongly affects the activity of supported Pt,3–10 Pd,11–16 and Rh17,18 catalysts. The effect of Pt oxidation state on the activity has been most extensively studied. Generally, Pt in the metallic state shows higher catalytic performance than in the oxidized state. However, a few reports showed that partially oxidized Pt exhibited higher activity than the metallic one.7 For supported Pd catalysts, Yazawa et al.11 studied the relation between the Pd oxidation state and their catalytic activity for propane combustion and concluded that partially oxidized Pd was effective. Newton and co-workers14 recently demonstrated that the Pd(I) and Pd(0) sites are active species of Al2O3-supported Pd with Pd particle size 2.5 nm and 3.5 nm, respectively, for CO elimination under CO/(NO + O2) cycling conditions. These examples demonstrate that the oxidation state of the active metal species depends on the structural factors (type of metals and support materials and particle size) and the reaction conditions, and hence, continuous efforts should be devoted to elucidate the effect of the metal oxidation state on the catalytic activity. For this purpose, the oxidation states of Pt, Pd, and Rh should be conveniently and accurately estimated. Although XPS (X-ray photoelectron spectroscopy) has been the most commonly used method for this purpose, the XPS spectra of metal catalysts containing the metal species of various oxidation states generally give a poorly resolved broad feature.11,15,18
X-Ray absorption spectroscopy (XAS) is a powerful method to characterize the electronic state of metal catalysts.19 The white line feature at L3-edge X-ray absorption near-edge structures (XANES) of transition metal compounds is typically assigned to electron transition from 2p3/2 to 5d3/2 and 5d5/2, and hence the area of the white line can be an index for the oxidation state of the transition metals. Most of the L3-edge XANES studies of heterogeneous catalysts have been focused on 5d transition metals.1–10,20–22 In contrast, few attempts have been focused on L3-edge XAS studies of 4d (Pd,23–27 Ag,28 Rh29) and 3d (Cu,30 Co31) transition metals-based catalysts. Previously, Chen et al.32 reported a XANES study of the electronic structure of 4d transition metals (Mo, Tc, Ru, Pd, Ag) and showed that the white line area intensity of them increased linearly with increasing atomic number. The result illustrates the proportionality of the 4d-hole count to the white line area. Yoshida et al.4 demonstrated that the white line area at Pt L3-edge XANES increased linearly with increase in the O/Pt ratio in various Pt catalysts, indicating a quantitative correlation between the Pt oxidation state and the white line area intensity. Although Pd is an important element in oxidation catalysis, to the best of our knowledge, there are quite a few Pd L3-edge XANES studies of Pd catalysts for catalytic oxidation. Recently, we have reported a simple method of Rh L3-edge XANES analysis to quantify the oxidation states of Rh species in a series of supported Rh catalysts containing different amounts of Rh metal and Rh2O3 phases.29 A linear relationship between the average oxidation numbers determined by a chemical method (the number of CO2 molecules formed during pre-reduction by CO) and the white line intensities at the L3-edge Rh verifies that this method is effective for quantification of the average Rh oxidation number in structurally unknown Rh samples. In the present study, the same research strategy was applied to establish a Pd L3 edge XANES analysis for the quantification of the average Pd oxidation state (x) of Pd species in PdOx/2/SiO2 samples. A linear relationship between these values will verify the quantitative correlation between the Pd oxidation state and the white line area intensity. These samples are characterized by conventional methods (Pd K-edge XANES/EXAFS and XRD), and the advantages of Pd L3-edge XANES are discussed. To demonstrate the utility of this method in a catalytic study, we also study the effect of the Pd oxidation state on the catalytic activity of the PdOx/2/SiO2 samples for CO oxidation.
2. Experimental
2.1. Catalyst preparation
Pd(5 wt%)-loaded SiO2 catalysts were prepared by impregnating SiO2 (Q-10, Fuji Silysia Chemical Ltd., 300 m2 g−1) with aqueous HNO3 solution of Pd(NO3)2, followed by evaporation to dryness at 353 K, drying at 393 K for 12 h, calcination in air at 973 K for 1 h. As shown below, Pd species existed as PdO in the as-calcined catalyst, referred to as PdO/SiO2. This oxidized sample was finally reduced at 673 K in a flow of 0.4% CO/He (flow rate = 100 cm3 min−1), and the gas formed during the CO-reduction treatment was continuously analyzed by nondispersive infrared CO/CO2 analyzers (Horiba VIA510). By changing the time (0, 1, 2, 7, 10 min) of this CO-reduction treatment, a series of PdOx/2/SiO2 catalysts with different average oxidation numbers of Pd (x) were prepared as summarized in Table 1.
Table 1 Preparation conditions and structural information of the catalysts
| Catalysts |
t
CO/mina |
x
b
|
D/nmc |
L3 WL area (a.u.)d |
|
Time of CO-reduction treatment at 673 K.
Average oxidation number of supported Pd species estimated from CO2 formation during the CO-reduction at 400 °C.
Average particle size of Pd or PdO from XRD analysis.
White line area intensity at L3-edge after subtraction of the continuum absorption.
|
| PdO/SiO2 |
0 |
2.0 |
14.8 |
12.4 |
| PdO0.8/SiO2 |
1 |
1.6 |
— |
10.8 |
| PdO0.6/SiO2 |
2 |
1.3 |
— |
10.1 |
| PdO0.1/SiO2 |
7 |
0.24 |
— |
7.1 |
| Pd/SiO2 |
10 |
0 |
21.1 |
6.9 |
| Pd powder |
— |
0 |
— |
6.9 |
2.2. Characterization
X-Ray diffraction (XRD) patterns of the powdered catalysts were recorded with a Rigaku MiniFlex II/AP diffractometer with Cu Kα radiation. By using Si powder as an internal standard, the XRD intensity of different PdOx/2/SiO2 samples was normalized. The number of surface metal atoms in the fully reduced sample (Pd/SiO2) was estimated with the CO uptake of the samples at 195 K,33 after reducing the catalyst surface in H2 at 473 K, by the pulse-adsorption of CO in a flow of He. The average particle size was calculated from the CO uptake assuming that CO was adsorbed on the surface of spherical Pd particles at a CO/(surface Pd atom) ratio of 1/1.
Pd K-edge XAS measurement was carried out in a transmission mode with a Si(111) monochromator at BL14B2 of SPring-8 (Hyogo, Japan) operated at 8 GeV. The analyses of the extended X-ray absorption fine structure (EXAFS) and XANES were performed using the REX version 2.5 program (RIGAKU). The Fourier transformation of the k3-weighted EXAFS from k space to R space was carried out over the k range 2.5–15 Å−1. A part of the Fourier-transformed EXAFS in the R range of 1.0–3.6 Å were inversely Fourier-transformed, followed by the analysis with a usual curve fitting method in the k range of 2.5–14.0 Å−1. The parameters for the Pd–Pd and Pd–O shells were provided by the FEFF6. The number of free parameters34 for curve fitting can be estimated as Pfree = 2ΔkΔR/π ≈ 20, indicating that we can model the EXAFS data with five shells.
Pd L3-edge XANES spectra were obtained at the BL-9A station of the KEK-PF (Tsukuba, Japan) with a ring energy of 2.5 GeV and a stored current of 250–350 mA. The spectra were recorded in a transmission mode at room temperature with a Si(111) double-crystal monochromator. The background subtraction and normalization were performed by REX version 2.5. The white line area of normalized Pd L3-edge XANES spectrum was evaluated by subtracting the normalized XANES spectrum by an arctangent function, eqn (1):4,30
| | | a(E) = h[0.5 + π−1tan−1{(E − E0)/ω}], | (1) |
where
E: X-ray energy (eV),
a(
E): arctangent function,
h: height,
ω: width,
E0: inflection point (eV). In the present simulation,
h1 was 1 and
ω1, which should be related to the natural width of the 2p electron and the experimental conditions, was fixed as 0.33.
4,30 Note that changes in the
ω1 value in a range of 0.2–0.5 resulted in negligible changes in the white line area intensity (below ±0.6%). The value of
E0 was assumed to be the same as the peak position of the white line.
Catalytic CO oxidation was performed in a fixed-bed flow reactor (inner diameter = 4 mm) at 413 K. A gas mixture of CO/O2/He (0.46%/10%/balance) was fed to the catalyst (5 mg) at a flow rate of 100 cm3 min−1. The effluent gas was analyzed by nondispersive infrared CO/CO2 analyzers (Horiba VIA510). Reaction rates per gram of the PdOx/2/SiO2 catalysts for the CO oxidation were estimated under the conditions where CO conversion was below 15%.
3. Results and discussion
3.1. Conventional characterizations
Structural information derived from various characterization experiments is summarized in Table 1. The crystal phase was confirmed by XRD (Fig. 1). In the XRD pattern of the as-calcined sample, PdO/SiO2, lines due to the Pd metal were absent, and broad lines assignable to PdO were observed. By changing the time of CO-reduction pre-treatment at 673 K, we prepared a series of PdOx/2/SiO2 samples with various oxidation states. The average oxidation number (x) was estimated from the amount of CO2 molecules formed during the CO reduction treatment, according to the following stoichiometry: x/2CO + PdOx/2 = x/2 CO2 + Pd. As expected, the oxidation number (x) decreased with the time of CO-reduction, accompanying a decrease in the diffraction intensity of the PdO phase and an increase in that of the Pd metal. The number of CO2 molecules formed during CO-reduction at 673 K for 10 min corresponds to a completely reduced PdOx/2/SiO2 (x = 0) sample, thus the sample is referred to as Pd/SiO2. The result is consistent with the XRD result of this sample; there is a diffraction line due to the Pd metal but no diffraction lines due to PdO. Normalized XRD intensity due to the Pd metal or PdO is plotted as a function of the oxidation number (x) in Fig. 1b. It is qualitatively shown that the PdO/Pd ratio increases with x, but the result in Fig. 1b does not give a quantitative conclusion. The average crystallite size of the Pd metal in the Pd/SiO2 sample and PdO in the PdO/SiO2 sample was calculated from the half-width of the diffraction lines at 40.12° and 33.88°, respectively, in the XRD patterns using the Scherrer equation. The average size of the Pd metal in Pd/SiO2 (21.1 nm) was larger than that of PdO in PdO/SiO2 (14.8 nm). In summary, five kinds of the PdOx/2/SiO2 samples with different average oxidation numbers but with the same Pd loading (5 wt%) were prepared as listed in Table 1.
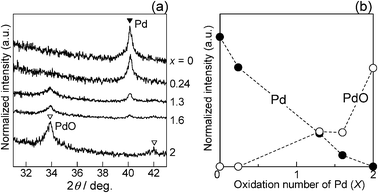 |
| | Fig. 1 (a) X-ray diffraction patterns of the PdOx/2/SiO2 samples with different average oxidation numbers (x). (b) Normalized XRD intensity due to Pd metal or PdO vs. x. | |
Fig. 2 shows Fourier transforms of k3-weighted Pd K-edge EXAFS, and Table 2 shows curve-fitting results. Peaks appearing at 1–2 Å are due to the adjacent oxygen atoms, and peaks at 2–3.5 Å are due to Pd atoms in Pd or PdO. The values of the coordination numbers for Pd–O and Pd–Pd species as well as the distances (EXAFS analysis) are listed in Table 2. The EXAFS of the PdO/SiO2 sample consists of a Pd–O contribution with a coordination number (CN) of 3.0 at a bond distance (R) of 2.02 Å and two Pd–Pd contributions: CN = 3.0 at R = 3.04 Å and CN = 4.4 at R = 3.42 Å. These Pd–O and Pd–Pd distances are consistent with the crystallographic data for PdO. The EXAFS of the Pd/SiO2 sample consisted of a Pd–Pd contribution of CN = 7.7 at R = 2.74 Å, which is nearly close to the crystallographic data of the Pd–Pd distance in the Pd metal (2.74 Å). These results are consistent with the results from the XRD analysis. Considering the crystallographic data of Pd and PdO (Table 2) and the XRD results, the EXAFS of the PdO0.8/SiO2, PdO0.6/SiO2 and PdO0.1/SiO2 samples could be fitted with four shells: a Pd–O shell at 2.02 Å and three Pd–Pd shells at the second (2.74 Å), the third (3.04 Å) and the fourth (3.42 Å) coordination spheres. The curve-fitting with the four shells was successful for the PdO0.8/SiO2 and PdO0.6/SiO2 samples. However, for PdO0.1/SiO2, the curve-fitting with two shells gave a good fitting value (Rf in Table 2), and increase of the number of Pd–Pd shells did not improve the fitting value. There are general tendencies that a decrease in the oxidation number (x) in the PdOx/2/SiO2 samples results in decreases in the CN of the Pd–O shell and the Pd–Pd shells at 3.04 and 3.42 Å and an increase in the CN of the Pd–Pd shell at 2.73–2.74 Å. This supports that the PdO/Pd ratio decreases with increase in x. For the PdOx/2/SiO2 (x = 2, 1.6, 1.3) samples, the ratio of CN values for the Pd–Pd shells at 3.04 Å and 3.42 Å is not consistent with the crystallographic value. Considering the above problems in the curve-fitting analyses, we can conclude that EXAFS analysis is useful to estimate a trend in the PdO/Pd ratio qualitatively but is not useful for a quantification of the PdO/Pd ratio as an index of the average oxidation state of Pd species.
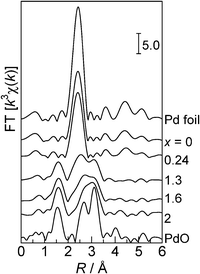 |
| | Fig. 2 Fourier transforms of Pd K-edge EXAFS for the reference compounds and the PdOx/2/SiO2 samples with different average oxidation numbers (x). | |
Table 2 Curve-fitting analysis of Pd L3-edge EXAFS
| Sample |
Shell |
CNa |
R/Åb |
σ/Åc |
R
f (%)d |
|
Coordination number.
Bond distance.
Debye–Waller factor.
Residual factor.
Crystallographic values (ICSD #29281 for PdO, ICSD #64916 for the Pd metal).
|
| PdO/SiO2 |
O |
3.0 |
2.02 |
0.047 |
1.2 |
| |
Pd |
3.0 |
3.04 |
0.064 |
|
| |
Pd |
4.4 |
3.42 |
0.059 |
|
| PdO0.8/SiO2 |
O |
2.7 |
2.03 |
0.046 |
1.0 |
| |
Pd |
1.4 |
2.74 |
0.089 |
|
| |
Pd |
3.1 |
3.04 |
0.071 |
|
| |
Pd |
4.1 |
3.42 |
0.067 |
|
| PdO0.6/SiO2 |
O |
2.1 |
2.03 |
0.038 |
1.1 |
| |
Pd |
2.1 |
2.74 |
0.078 |
|
| |
Pd |
3.1 |
3.04 |
0.077 |
|
| |
Pd |
3.1 |
3.42 |
0.060 |
|
| PdO0.1/SiO2 |
O |
0.5 |
2.02 |
0.059 |
0.8 |
| |
Pd |
7.2 |
2.73 |
0.079 |
|
| Pd/SiO2 |
Pd |
7.7 |
2.74 |
0.080 |
0.8 |
| PdO |
O |
(4)e |
(2.02)e |
— |
— |
| |
Pd |
(4)e |
(3.04)e |
— |
— |
| |
Pd |
(8)e |
(3.42)e |
— |
— |
| Pd metal |
Pd |
(12)e |
(2.74)e |
— |
— |
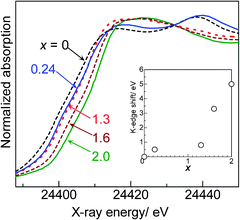
It is established that the K-edge threshold energy shifts to higher energy as the oxidation number of the X-ray absorbing atom is increased. Pd K-edge XANES spectra of the PdOx/2/SiO2 samples are shown in Fig. 3. In this study, the threshold was defined as the point at which intensity was 0.4 in the normalized XANES spectra. The threshold energy, which would be an indicator for the Pd oxidation state, is plotted in an inset of Fig. 3 as a function of x. Qualitatively, there is a tendency that an increase in the oxidation number (x) in the PdOx/2/SiO2 samples results in an increase in the K-edge energy. However, a poor correlation between x and the threshold energy indicates that the K-edge XANES shift is not useful in the quantitative determination of the average oxidation state of the PdOx/2/SiO2 samples.
3.2. Quantification of the Pd oxidation state by L3-edge XANES analysis
Pd L3-edge absorption spectra of the PdOx/2/SiO2 samples and Pd powder are shown in Fig. 4. As previously reported by Chen et al.,32 the XANES spectrum of Pd powder, a reference compound of metallic Pd, showed a small while line due to lack of the 4d-hole. In contrast, the XANES spectrum of the PdO/SiO2 sample exhibited a large white line peak at 3174.5 eV with a symmetric shape. This peak should be predominantly assignable to the electron transition from 2p3/2 to 5d3/2 and 5d5/2, and hence, it is expected that the area intensity of the white line can be an index for the oxidation state of Pd species. As the average oxidation number (x) of the PdOx/2/SiO2 samples increased, the area intensity of the white line increased, indicating an increase of vacancy in the d-states. Adopting the method in our previous study29 for the Rh L3-edge XANES analysis, the area intensity of the white line was determined as illustrated in Fig. 5. An arctangent function shown as a dashed line, which corresponds the continuum absorption, is subtracted from the raw spectrum (dots) to give a function corresponding to an absorption predominantly related to the electron transition from 2p3/2 to 5d3/2 and 5d5/2 (solid line). The white line area intensities thus estimated were listed in Table 1, and also plotted as a function of the average oxidation number of the PdOx/2/SiO2 samples and Pd powder as shown in Fig. 6. It is found that the white line area intensity increased linearly with the oxidation number. This clearly indicates that the white line area intensity at the Pd L3-edge XANES is a quantitative index of the Pd oxidation state. Therefore, it is concluded that the present method of Pd L3-edge XANES analysis can be used for the quantitative estimation of the average oxidation number of unknown Pd species. Taking into account the fact that conventional XPS spectra of Pd samples containing Pd species of different oxidation states give a poorly resolved broad feature,11 this method would be widely acceptable as a novel characterization method for a quantification of the Pd oxidation states. Since this method is based on the 4d-hole, this will become generally applicable to determination of the oxidation state of various Pd compounds.
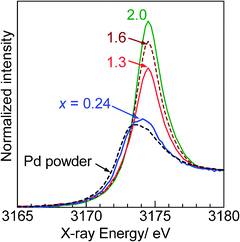 |
| | Fig. 4 Pd L3-edge XANES spectra of the representative PdOx/2/SiO2 samples with different average oxidation numbers (x) and Pd powder. | |
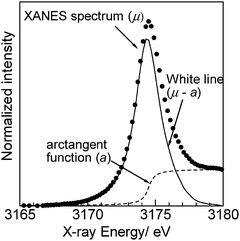 |
| | Fig. 5 Example for a method for Pd L3-edge XANES analysis. The spectrum was for the PdO/SiO2 sample. An arctangent function (dashed line) for the continuum absorption is subtracted from the raw spectrum (dots) to give a function corresponding to the white line peak (solid line). | |
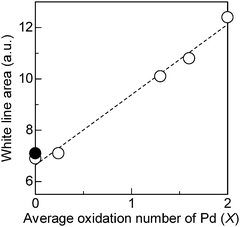 |
| | Fig. 6 Relation between the average oxidation number (x) and the white line area intensity of the Pd L3-edge XANES of (○) the PdOx/2/SiO2 samples and (●) Pd powder. | |
3.3. Structure–activity relationship
To demonstrate the utility of Pd L3-edge XANES analysis in a catalytic study, the effect of the Pd oxidation state on the catalytic activity of the PdOx/2/SiO2 samples for CO oxidation was examined. The rate of CO oxidation by O2 to CO2 was measured in the flowing mixture of CO and O2 at 413 K. As shown in Fig. 7, the rates per gram of the PdOx/2/SiO2 samples decreased with the average oxidation number. Taking into account the result that the size of the Pd metal in the Pd/SiO2 sample (21.1 nm) was larger than that of PdO in the PdO/SiO2 sample (14.8 nm), the variation of activity was not originated from the variation of the Pd dispersion. The result in Fig. 7 indicates that the Pd0 site is active species of the PdOx/2/SiO2 samples for CO oxidation under the present reaction conditions.
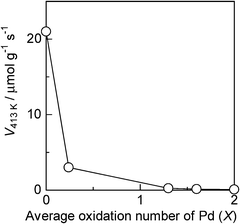 |
| | Fig. 7 Effect of the average oxidation number (x) of Pd in the PdOx/2/SiO2 catalyst on its reaction rate for CO oxidation at 413 K. Reaction conditions: 0.46% CO, 10% O2. | |
It should be noted that Pd L3-edge XANES analysis is useful to quantify the average oxidation states of bulk and surface Pd species. The oxidation state of surface Pd species may change during the reaction at 413 K. To discuss the oxidation state of surface active Pd species, in situ IR with CO as a probe is the best characterization method.12 Among various studies, a recent study by Zorn et al.12b is of importance, because it gives comprehensive results on the relationship between the Pd oxidation state and activity of PdOy/Al2O3 (y = 0–1) catalysts for CO oxidation, whose conditions are similar to those in this study. They carried out in situ CO adsorption IR experiments at low temperature (100 K) after the pretreatment of PdOy/Al2O3 catalysts under realistic CO oxidation conditions. Combining with various characterization results and density functional theory calculations, they concluded that the metallic Pd is responsible for the catalytic activity of Pd/Al2O3 for CO oxidation under realistic conditions at temperatures up to 553 K. Although our conclusion is not based on in situ characterization results, the results by Zorn et al.12b could support our conclusion that the Pd0 site is active species of PdOx/2/SiO2 for CO oxidation under the reaction conditions.
The above discussion was based on the accurate oxidation number of the Pd species, which was determined by both the chemical method recorded during the catalyst preparation and the spectroscopic method recorded after the catalyst preparation. However, for the samples that are not applicable with the chemical method, such as Pd catalysts supported by highly reducible supports and Pd catalysts with transition metal additives, the spectroscopic method will be the only way to determine the accurate oxidation number. In this study, we presented a simple L3-edge XANES analysis for the quantitative estimation of the average oxidation number of Pd species. It is claimed that, hereafter, this method will be available to discuss the effect of the Pd oxidation state on the activity of Pd catalysts, even when the determination of the Pd oxidation state is difficult by other characterization methods.
4. Conclusion
A simple method of Pd L3-edge XANES analysis was examined to quantify the oxidation states of Pd species in a series of Pd-loaded SiO2 catalysts containing different amounts of the Pd metal and PdO phases. The linear relationship between the average oxidation numbers determined by a chemical method (the number of CO2 molecules formed during pre-reduction by CO) and the white line area intensities at L3-edge Pd verifies that this method is effective for quantification of the average Pd oxidation number of unknown Pd species. This method reflecting the 4d-hole of Pd will become generally applicable to determination of the oxidation state of various Pd compounds.
Additionally, for the CO oxidation reaction by the PdOx/2/SiO2 (x = 0–2) samples, metallic Pd0 sites are shown to be active species under the present reaction conditions.
Acknowledgements
The XAS measurements at Spring-8 were conducted with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2008B2066). The XAS measurements at KEK-PF were performed under the approval of the Photon Factory Program Advisory Committee (No. 2008G024). This work was supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology via Grant-in-Aids for Scientific Research B (20360361) and for Young Scientists A (22686075).
References
- E. Principi, A. Witkowska, S. Dsoke, R. Marassic and A. D. Cicco, Phys. Chem. Chem. Phys., 2009, 11, 9987–9995 RSC.
- H. Yoshitake and Y. Iwasawa, J. Phys. Chem., 1992, 96, 1329–1334 CrossRef CAS.
- S. R. Bare, S. D. Kelly, B. Ravel, N. Greenlay, L. King and G. E. Mickelson, Phys. Chem. Chem. Phys., 2010, 12, 7702–7711 RSC.
- H. Yoshida, S. Nonoyama, Y. Yazawa and T. Hattori, Phys. Scr., 2005, T115, 813–815 CrossRef CAS.
- Y. Yazawa, N. Takagi, H. Yoshida, S. Komai, A. Satsuma and T. Hattori, Appl. Catal., A, 2002, 233, 103–112 CrossRef CAS.
- T. Tanabe, Y. Nagai, K. Dohmae, H. Sobukawa and H. Shinjoh, J. Catal., 2008, 257, 117–124 CrossRef CAS.
- J. Singh, E. M. C. Alayon, M. Tromp, O. V. Sofanova, P. Glatzel, M. Nachtegaal, R. Frahm and J. A. van Bokhoven, Angew. Chem., Int. Ed., 2008, 47, 9260–9263 CrossRef CAS.
- E. Becker, P.-A. Carlsson, H. Grönbeck and M. Skoglundh, J. Catal., 2007, 252, 11–17 CrossRef CAS.
- J.-D. Grunwaldt, M. Beier, B. Kimmerle, A. Baiker, M. Nachtegaal, B. Griesebock, D. Lützenkirchen-Hecht, J. Stötzel and R. Frahm, Phys. Chem. Chem. Phys., 2009, 11, 8779–8789 RSC.
- J. A. Anderson, B. Bachiller-Baeza and M. Fernández-García, Phys. Chem. Chem. Phys., 2003, 5, 4418–4427 RSC.
-
(a) Y. Yazawa, H. Yoshida, S. Komai, A. Satsuma and T. Hattori, Appl. Catal., B, 1998, 19, 261–266 CrossRef CAS;
(b) Y. Yazawa, H. Yoshida, N. Takagi, S. Komai, A. Satsuma and T. Hattori, J. Catal., 1999, 187, 15–23 CrossRef CAS.
-
(a) Y. Shen, G. Lu, Y. G. Yanqin, Y. Guo and X. Gong, Catal. Today, 2011, 175, 558–567 CrossRef CAS;
(b) K. Zorn, S. Giorgio, E. Halwax, C. R. Henry, H. Grönbeck and G. Rupprechter, J. Phys. Chem. C, 2011, 115, 1103–1111 CrossRef CAS.
- S. Hinokuma, H. Fujii, M. Okamoto, K. Ikeue and M. Machida, Chem. Mater., 2010, 22, 6183–6190 CrossRef CAS.
- A. Iglesias-Juez, A. Kubacka, M. Fernández-García, M. D. Michiel and M. A. Newton, J. Am. Chem. Soc., 2011, 133, 4484–4489 CrossRef CAS.
- S.-H. Oh and G. B. Hoflund, J. Phys. Chem. A, 2006, 110, 7609–7613 CrossRef CAS.
- A. Iglesias-Juez, A. Martínez-Arias, M. A. Newton, S. G. Fiddy and M. Fernández-García, Chem. Commun., 2005, 4092–4094 RSC.
- J.-D. Grunwaldt, L. Basini and B. S. Clausen, J. Catal., 2001, 200, 321–329 CrossRef CAS.
- K. Dohmae, Y. Nagai, T. Tanabe, A. Suzuki, Y. Inada and M. Nomura, SIA Surf. Interface Anal., 2008, 40, 1751–1754 CrossRef CAS.
- S. R. Bare and T. Ressler, Adv. Catal., 2009, 52, 339–465 CrossRef CAS.
- M. Fernández-García, F. K. Chong, J. A. Anderson, C. H. Rochester and G. L. Haller, J. Catal., 1999, 182, 199–207 CrossRef.
- S. R. Bare, S. D. Kelly, F. D. Vila, E. Boldingh, E. Karapetrova, J. Kas, G. E. Mickelson, F. S. Modica, N. Yang and J. J. Rehr, J. Phys. Chem. C, 2011, 115, 5740–5755 CAS.
- E. Bus, R. Prins and J. A. van Bokhoven, Phys. Chem. Chem. Phys., 2004, 6, 3903–3906 RSC.
- I. Davoli, S. Stizza, A. Bianconi, M. Benfatto, C. Furlani and V. Sessa, Solid State Commun., 1983, 48, 475–478 CrossRef CAS.
- T. Kubota, Y. Kitajima, K. Asakura and Y. Iwasawa, Bull. Chem. Soc. Jpn., 1999, 72, 673–681 CrossRef CAS.
- M. W. Tew, J. T. Miller and J. A. van Bokhoven, J. Phys. Chem. C, 2009, 113, 15140–15147 CAS.
- K. Okumura, T. Tomiyama, S. Okuda, H. Yoshida and M. Niwa, J. Catal., 2010, 273, 156–166 CrossRef CAS.
- L. C. Witjens, J. H. Bitter, A. J. van Dillen, K. P. de Jong and F. M. F. de Groot, Phys. Chem. Chem. Phys., 2007, 9, 3312–3320 RSC.
- T. Miyamoto, H. Niimi, Y. Kitajima, T. Naito and K. Asakura, J. Phys. Chem. A, 2010, 114, 4093–4098 CrossRef CAS.
- K. Shimizu, T. Oda, Y. Sakamoto, Y. Kamiya, H. Yoshida and A. Satsuma, Appl. Catal. B, 2012, 111–112, 509–514 CrossRef CAS.
- K. Shimizu, H. Maeshima, H. Yoshida, A. Satsuma and T. Hattori, Phys. Chem. Chem. Phys., 2000, 2, 2435–2439 RSC.
- D. Bazin and L. Guczi, Appl. Catal., A, 2001, 213, 147–162 CrossRef CAS.
- J. Chen, E. Kemly, M. Croft, Y. Jcon, X. Xu, S. A. Shaheen and P. H. Ansari, Solid State Commun., 1993, 85, 291–296 CrossRef CAS.
- T. Tanabe, Y. Nagai, T. Hirabayashi, N. Takagi, K. Dohmae, N. Takahashi, S. Matsumoto, H. Shinjoh, J. N. Kondo, J. C. Schouten and H. H. Brongersma, Appl. Catal., A, 2009, 370, 108–113 CrossRef CAS.
-
B. K. Teo, EXAFS: Basic Principles and Data Analysis, Springer-Verlag, Berlin, 1986 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00422d |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.