Periodic mesoporous organosilica functionalized sulfonic acids as highly efficient and recyclable catalysts in biodiesel production†
Babak
Karimi
*,
Hamid M.
Mirzaei
and
Akbar
Mobaraki
Institute for Advanced Studies in Basic Sciences (IASBS)—Department of Chemistry, Zanjan 45137-66731, Islamic Republic of Iran
First published on 20th January 2012
Abstract
Novel water tolerant sulfonic acid based Periodic Mesoporous Organosilicas (PMOs) having either phenylene (1a) or ethyl (1b) as bridge and methylpropyl sulfonic acid as functionalized group have been developed. The materials have been then employed in efficient biodiesel production via direct transesterification of sunflower oil, canola oil, corn oil, refined olive oil, and extracted oil from olive sludge with methanol. By comparing the catalytic performance of these acids with well-known SBA-15-PrSO3H, it was revealed that catalyst 1b bearing an ethyl bridging group is a more reactive catalytic system in biodiesel production. On the other hand, while the water adsorption experiment shows that among the described PMO functionalized acids, 1a shows more hydrophobic character, in contrast, we interestingly found that 1b consistently exhibited higher catalytic performance in biodiesel formation, which relies on a balance between hydrophobic-hydrophilic- and pore size of the mesochannels in this catalyst.
1 Introduction
Today's most important source of energy and chemicals is crude oil and consumption of this resource enhances everyday as countries develop. The International Energy Agency (IEA) foresees that fossil fuels will continue to dominate energy supplies in 2030 with the transportation sector expected to account for two-thirds of the growth, and global daily oil demand will possibly reach 115 million barrels per day in 2030.1 However, this would strongly diminish the reserved crude oil resources, and result in the increase in the price of petroleum. Additionally, as this energy resource is not renewable, emission of CO2 and other particulates would increase greenhouse-gas effects.2–4 Thus, finding new alternative energy resources would be of prime importance. As a consequence, owing to ever increasing environmental standards, and economic pressures, there is encouragingly interest toward the use of more renewable energy sources. It seems that a possible approach to address these problems is to gradually replace the traditional fossil oil sources with biofuels having renewable capabilities.The idea of using vegetable oil as fuel for diesel engines was demonstrated by Radulf Diesel, the inventor of the diesel engine.5 In contrast to petroleum diesel which contains hydrocarbons, biodiesel consists of monoalkyl esters of long chain fatty acids. This property provides several advantages over petroleum diesel: it is safe, renewable, nontoxic, and biodegradable; it contains no sulfur and is a better lubricant.6 Finally, biodiesel production deals with a positive impact, by enhancing rural revitalization.6 The traditional method used for biodiesel production is transesterification catalyzed by sodium hydroxide as a base catalyst.7 This catalyst is corrosive for equipment and also reacts with free fatty acids to form unwanted soap by-products, requiring expensive separation.8 Other methods include: (i) direct use and blending of vegetable oil, (ii) use of microemulsions with short chain alcohols,7 and (iii) thermal cracking (pyrolysis) of vegetable oils.9
To prevent soap formation, the use of homogeneous sulfuric acid was shown to be beneficial for biodiesel production using either esterification or transesterification.10 The problem with strong liquid acid catalysts is that they are slow and require higher reaction temperature in comparison with homogeneous base catalysts. While acid catalyzed processes could produce biodiesel from the low-cost feedstock thus lowering the production cost, in contrast the separation processes involving base catalyst systems produce a large volume of hazardous wastes. Such drawbacks actually make the base-catalyzed protocols impractical from both commercial and environmental points of view.11
Recently, tight environmental legislation created new opportunities for catalysis and catalytic processes toward clean technology.12 Along this line, study on economical assessment of different biodiesel production processes using homogeneous alkali and acid catalysts, as well as heterogeneous acid catalysts and supercritical methods, showed that heterogeneous acid-catalyzed processes have met with promising success, as they have the highest rate-of-return, lowest capital investment, and need technically simple apparatus.13
Nevertheless, in order to commercialize the biodiesel production using solid acids, as a matter of fact, there are still major challenges to overcome some important limitations concerning widespread applications of solid acids such as catalyst poisoning, coking, sintering, and leaching.14 Therefore, it seems that one strategy to circumvent these problems could be based on the development of novel efficient catalysts by tuning their synthetic parameters in a designated manner. In this context, several approaches based on sulfated metal oxides,15–19 zeolites,20 resins,21 heteropolyacids,22–25 and sulfonated carbon26–28 have been used to develop the efficient heterogeneous solid catalysts. However, despite promising improvements, the leaching of the active sites, thermal stability, and low surface area are still important issues that remain unresolved. Moreover, in spite of the plethora of methods for esterification of fatty acids using solid acids, only a few such studies have been described for a direct transesterification reaction with vegetable oils.29–31
In recent years, organic–inorganic nanocomposites based on ordered mesoporous silicas32–34 with tunable structure and tailored composition have been extensively investigated in broad applications ranging from adsorbent,35–41 gas separation,42,43 catalysis,44–48 to biological uses.49 One of the attractive features of these materials is that they combine in a single solid, the chemical functionalities arising mainly from an anchored organic moiety with high surface area, ordered narrow pore size distribution and highly mechanical stable structure through an inorganic–organic cooperative mechanism. Along the line of this property, quite recently Melero et al. have elegantly shown that SBA-15-PrSO3H is a promising and recyclable catalyst for transesterification of various vegetable oils, by emphasizing the possibility to control both acidity and hydrophobicity of the catalysts.50,51
Hybrid periodic mesoporous organosilica was first introduced in 1999, built from bridge organosilica precursor [(R′O)3Si–R–Si(OR′)3], through the surfactant self-assembly approach analogous to that used in preparation of ordered mesoporous silicates.52–57 In these materials, the bridging organic moieties which are well distributed inside the inorganic pore wall not only improve hydrothermal and mechanical stabilities, but also higher organic loading and greater avoidance of channel blockage can be achieved in comparison with mesoporous materials functionalized with terminal organic groups. These unique properties cause PMOs to be excellent candidates for application in catalysis,55,58 especially where high water tolerance and hydrothermal stability of the catalysts are needed. Although much is known about the preparation of PMOs, the number of applications that exploit their fascinating hybrid nature are quite limited.55,58–60 In particular, to the best of our knowledge, there is currently no example of employing PMO functionalized materials in biodiesel production. Considering the high loading of organic groups inside the pore wall of PMOs, we reasoned that the anchoring of sulfonic acid functionalities on a suitable PMO having an appropriate concentration of organic functional groups may result in a highly active and hydrophobic acid catalyst that could be used in biodiesel production from vegetable oils. Along the line of this hypothesis and our continuous interest in developing novel nano-structured catalysts in recent years,61–66 herein, we wish to disclose our finding regarding the use of novel sulfonic acid based PMOs having either phenylene 1a or ethyl 1b as bridge and methylpropylsulfonic acid as functionalized group in biodiesel production via direct transesterification of a variety of vegetable oils, and extracted oil from olive sludge around our region in Zanjan province, Iran (Scheme 1).
 | ||
| Scheme 1 Sulfonic acid based PMOs having either phenylene 1a or ethyl 1b. | ||
In order to optimize the hydrophobic nature of the catalyst in the vicinity of the sulfonic acid groups, we have chosen to employ MPMDS, since it has been shown that the presence of a methyl group bound to the same silicon atom bearing the sulfonic acid group has a remarkable shielding effect against polar molecules.67,68 Therefore, the close methyl group may prevent polar starting material (methanol) and by-products (glycerin and water) from poisoning the active sulfonic acid sites, thus increasing the durability of the catalyst and guaranteeing fast and efficient mass transfer phenomena.69
2 Experimental
2.1 Materials and methods
BTEB (1,4-bis(triethoxysilyl)benzene), BTEE (1,2-bis(triethoxysilyl)ethane), MPMDS (3-mercaptopropylmethyl dimethoxysilane), tetraethoxysilane (TEOS), 3-mercaptopropyl trimethoxysilane (MPTMS) and triblock co-polymer P123 (Eo70 Po20 Eo70) were obtained from Aldrich and used as received. Organosulfonic acid-functionalized mesoporous organosilicas Ph–PMO–Me–PrSO3H 1a and Et–PMO–Me–PrSO3H 1b were synthesized by a modification according to the methods of Kim et al. and Hamoudi etal. reported elsewhere respectively.70,71 SBA-15-PrSO3H and SBA-15-Ph–PrSO3H were synthesized according to the methods we previously presented in the literature (see ESI†).64,652.2 Synthesis of Ph–PMO–Me–PrSO3H
In a typical one-step synthesis, 0.66 g of pluronic P123 was dissolved in 23.6 g of deionized water, 0.57 g of H2O2 (30 wt%), and 0.13 g of HCl (37 wt%). Then 0.47 g of BTEB and 0.0902 g (30 mol% in total silica precursors) of MPMDS were added to the solution. The resulting mixture was agitated for 2 h at 40 °C and thereafter aged for 24 h at 100 °C. The resulting solid material was filtered and air-dried. To extract the residual block co-polymer, the solid material (0.5 g) was stirred in acetone (60 ml) for 10 h at 56 °C, followed by washing with deionized water. The final products were obtained after drying the samples in an oven for 1 day at 100 °C.2.3 Synthesis of Et–PMO–Me–PrSO3H
In this synthesis procedure, P123 (1.95 g) was added under vigorous stirring to 70 ml of HCl solution (2 N). After complete dissolution of the surfactant at 35 °C, BTEE (2.77 g) was added and the agitation was continued for 3 h before the dropwise addition of MPMDS (0.478 g). This was followed by stirring for 24 h at 35 °C. Then the suspension was aged for 24 h at 87 °C. The solid material was separated by filtration, washed with deionized water and dried at room temperature. The surfactant was removed by solvent extraction with anhydrous ethanol in a Soxhlet apparatus for 24 h (entry 1, Table 1). Typically, 0.2 g of extracted material was contacted with 8 g of H2O2 (30 wt%) and the suspension was stirred at room temperature for 24 h. After filtration and washing with deionized water and warm ethanol separately, the oxidized samples were acidified in 100 ml 0.1 M H2SO4 solution during 2 h. Next, the samples were washed thoroughly with deionized water until neutral pH, filtered and vacuum dried at 60 °C overnight.| Entry | PMO | S BET a | V p b | Pore sizec | Sulfur contentd | Acid capacitye |
|---|---|---|---|---|---|---|
| a BET surface area (m2 g−1). b Total pore volume (cm3 g−1). c BJH pore size diameter (nm). d Sulfur content measured by elemental analysis (mmol g−1). e Determined by titration after ion-exchange (mmol H+ g−1). f Determined by titration after ion-exchange after the 4th reaction cycle. | ||||||
| 1 | 1a | 404 | 0.27 | 2.4 | 0.6 | 0.40 |
| 2 | Et–PMO–Me–PrSH | 213 | 0.24 | 3.5 | — | — |
| 3 | 1b | 318 | 0.27 | 3.4 | 0.8 | 0.50 |
| 4 | SBA-15-PrSO3H | 682 | 0.92 | 6.2 | — | 1.20 |
| 5 | SBA-15-Ph–PrSO3H | 349 | 0.62 | 5.4 | — | 0.80 |
| 6 | 1b (recycled) | 642 | 0.5 | 3.2 | — | 0.32f |
2.4 Characterization
The textural properties of the functionalized mesoporous organosilicas were determined by nitrogen adsorption–desorption isotherms at 77 K with a BELSORB-max system (SIP). The surface area and pore size distribution were calculated with the BET and BJH methods, respectively. The N2-sorption isotherms indicate that the pore sizes of all samples are in the mesoporous range (Table 1). The materials exhibited a type IV isotherm pattern with indistinct H2 hysteresis loops, which are characteristics of the mesoporous structure with relatively broad pore size distributions. The calculated Brunauer–Emmet–Teller (BET) surface areas are 404 and 318 m2 g−1, for 1a and 1b, respectively. The total pore volumes for 1a and 1b were 0.27 cm3 g−1 (Table 1). Organic material present in the solids was determined by elemental analysis. The organic composition of the modified mesoporous materials was also determined by thermogravimetric analysis (TGA) and differential thermoanalysis (DTA), with heating from room temperature to 800 °C under an argon flow. The ion exchange capacities of the sulfonic acid functionalized mesoporous organosilicas were determined by acid–base titration and pH analysis (Table 1).2.5 Reactor design
Biodiesel production was carried out in a home-designed reactor. It was made of stainless steel which has a low magnetic property with a pressure gauge and two valves (Fig. 1). The internal surface of the reactor was protected by PTFE Teflon. | ||
| Fig. 1 The home-designed reactor for biodiesel production. | ||
2.6 Extraction of oil from olive sludge
Olive sludge was an industrial sludge which was derived from a huge olive oil industry in Tarom, Zanjan, Iran (Fig. 2). The extraction method was simple and was carried out using n-hexane and by a Soxhlet apparatus in 2 hours. Next, the solution was evaporated in a rotary evaporator. The final residue which consisted of 2–3 wt% of sludge was used as an extracted oil sample for biodiesel production. | ||
| Fig. 2 A typical image of the residue of the olive oil industry (Tarom, Zanjan, Iran). | ||
3 Results and discussions
3.1 Transesterification reactions
The activity of 1a and 1b solid catalysts was tested for the biodiesel formation from various types of vegetable oils. In a typical reaction, canola oil (2.5 g), methanol and the catalyst were placed together inside the reactor and allowed to react under conditions reported in Table 2. After the completion of the reaction (Table 2), the reactor was cooled down in a water bath. The mixture was simply filtered and washed with 20 ml of dichloromethane to recover the catalyst. The residual methanol and solvent were removed under vaccum. In the final two-phase product, the lower phase which is the by-products glycerol and water and the upper phase which is biodiesel and unreacted oil were separated by simple decantation. Biodiesel yield was assessed by 1H-NMR.72 Finally, the reaction catalyst was vacuum dried at 60 °C in an oven, to employ in another reaction run.| Entry | Catalyst | Molar ratio (oil![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol) :methanol) |
Catalysta (wt%) | Temperature/°C | Pressure/bar | Time/h | Yieldb (%) |
|---|---|---|---|---|---|---|---|
| a Weight of catalyst referred to canola oil weight. b 1H-NMR yield. c P 0 = 10 bar argon was applied prior to the reaction. | |||||||
| 1 | 1b | 1:50 |
6 | 180 | 5 | 2.5 | 70 |
| 2 | 1b | 1:70 |
6 | 180 | 7 | 2.5 | 79 |
| 3 | 1b | 1:100 |
6 | 180 | 12 | 2.5 | 88 |
| 4 | 1b | 1:150 |
6 | 180 | 18 | 2.5 | 98 |
| 5 | 1b | 1:100 |
8 | 180 | 12 | 2.5 | 89.5 |
| 6 | 1b | 1:50 |
6 | 150 | 10 | 4 | 93 |
| 7 | 1b |
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :100 :100
|
6 | 150 | 12 | 4 | 98 |
| 8 | 1b | 1:100 |
6 | 150 | 15c | 4 | 70 |
| 9 | 1b | 1:50 |
6 | 120 | 5 | 12 | 40 |
| 10 | 1a | 1:100 |
8.7 | 150 | 12 | 4 | 53 |
| 11 | 1a | 1:150 |
8.7 | 180 | 18 | 2.5 | 62 |
| 12 | SBA-15-PrSO3H | 1:100 |
3 | 150 | 12 | 4 | 78 |
| 13 | SBA-15-Ph–PrSO3H | 1:100 |
4 | 150 | 12 | 4 | 72 |
| 14 | H2SO4 | 1:150 |
0.32 | 180 | 18 | 2.5 | 99.5 |
The preliminary optimizations have been achieved by emphasizing four important parameters: (i) the molar ratio of oil to methanol, (ii) the reaction temperature, (iii) catalyst loading, and (iv) the reactor pressure (Table 2). First, we preferred to use spherical glasses with 5 mm diameter since our reactor was not equipped with mechanical stirring. In the next step, several experiments were carried out at different temperatures to find the effectiveness of this parameter (entries 1, 6, and 9). As can be clearly seen, while temperatures higher than 180 °C were found to significantly result in extensive catalyst decomposition, and thus not to be beneficial in catalyst efficiency, the use of temperatures lower than 150 °C gave remarkably lower biodiesel yields even after prolonged reaction times under otherwise identical reaction conditions (Table 2, entries 1 and 9). The impact of the methanol ratio and the optimum reaction temperature was also surveyed together, and it was found that the best results were obtained using an oil/methanol ratio of 1/100 at 150 °C (Table 2, entry 7). It is also worth mentioning that further increasing either catalyst loading from 6 wt% up to 8 wt% or the temperature to 180 °C resulted in a negative effect on the total efficiency of the process (Table 2, entries 3 and 5). Next, we turned our attention to pressure. Owing to the fact that the system we examined is a three-phase system, this reaction would inherently tend to prohibit easy diffusion. Therefore, it seems reasonable to speculate that by increasing the reactor pressure, the diffusion problem might be diminished to a suitable level, thus improving the biodiesel production. In contrast, we found that increasing the reactor pressure to 10 bar using argon was shown to give much worse result, leading to moderate yields of 70% within 4 hours (Table 2, entry 8). This unexpected result is possibly due to the fact that extra primitive pressure leads to an increase in the boiling point of methanol and consequently the effective pressure of methanol drops down. A direct comparison between 1b comprising ethyl bridge in the PMO framework, SBA-15-PrSO3H, and SBA-15-Ph–PrSO3H was also accomplished under the same reaction conditions and demonstrated that these catalysts showed significantly lower performances than that observed using 1b toward biodiesel production. These results clearly imply that the large differences between the various sulfonic acid based catalysts caused greatly by textural properties of the support probably originated from pore size distribution and the nature of the functionalized organic groups inside the PMO walls.
Under the optimized reaction conditions using 1b (Table 2, entry 7), we managed to investigate the generality of our catalyst system with respect to different vegetable oils in biodiesel production (Table 3). As summarized in Table 3, our catalyst system is also quite effective for efficient reaction of refined olive oil, sunflower oil, corn oil as well as the extracted olive oil from olive sludge, giving the corresponding biodiesel in excellent isolated yields.
3.2 Catalyst water tolerance
Waste cooking oils, non-edible oils, and alcohols often contain small quantities of water. For homogeneous catalysts, the water content has to be reduced significantly (<0.03%). Also, most heterogeneous catalysts reported so far are strongly affected by the presence of water.30 Li et al. reported that by adding an excess amount of water to the reaction mixture, their catalyst would retain its catalytic activity until 4 wt% of water is present in the reaction mixture. However, despite the excellent water tolerance property of this catalyst in the presence of 4 wt% of water, its reactivity significantly diminished in the presence of just 10 wt% of water.27 This observation encouraged us to investigate the effect of water concentration on the performance of our catalyst system using the same protocol with slight modification (Fig. 3).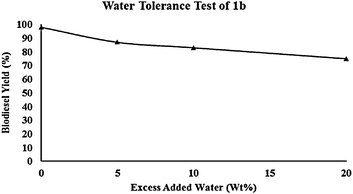 | ||
| Fig. 3 Effect of excess water addition on catalyst efficiency (conditions: molar ratio canola oil/methanol = 1/100, T = 150 °C, 6 wt% 1b catalyst, 4 h). | ||
Gratifyingly, we found that our catalyst activity reached 75% yield by addition of 20 wt% water to the reaction mixture while the same reaction without water gave 98% yield. In another test we investigated SBA-15-PrSO3H water tolerance and it was shown that by addition of 20 wt% water, biodiesel yield decreased by about 50% from its authentic value (Fig. 4).
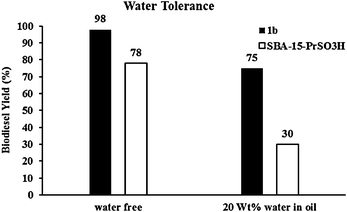 | ||
| Fig. 4 Comparing water tolerance test of 1b with SBA-15-PrSO3H (conditions: molar ratio oil/methanol = 1/100, T = 150 °C, 6 and 3 wt% 1b and SBA-15-PrSO3H catalyst, respectively). | ||
These results show that our catalyst is highly water tolerant. To gain more insight into the hydrophobicity of the catalyst, water adsorption isotherms of 1a, 1b, and SBA-15-PrSO3H were measured (Fig. 5). As can be seen SBA-15-PrSO3H showed a large water uptake at saturated vapor pressure owing to capillary condensation of water, confirming that the nanospaces of SBA-15 in this catalyst are more hydrophilic and thus were readily filled with liquid water. In contrast, our sulfonic acid based PMOs 1a and 1b showed much lower (still not negligible) water condensation, indicating that nanopores are highly hydrophobic in nature.
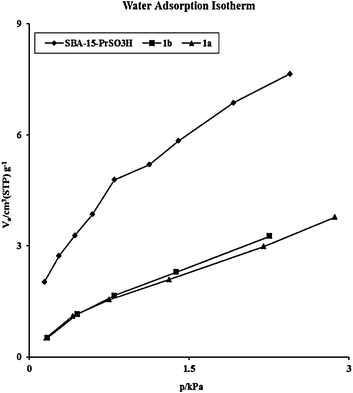 | ||
| Fig. 5 Water adsorption isotherm of solid acid catalysts. | ||
It is also interesting to note that in the cases of both 1a and 1b the water adsorption isotherms are of type IV, which is indicative of weak adsorbent–adsorbate interaction.73 These results also show that 1a has somewhat a more-hydrophobic nature than 1b. However, our studies demonstrated that higher isolated yields and activities were consistently observed within shorter reaction times when using 1b rather than more hydrophobic 1a under the described reaction conditions (Table 2). Therefore, it seems that one of the reasons for the somewhat higher activity of 1b than 1a is most likely due to the differences in the hydrophobic–hydrophilic nature of these two materials. It is clear that during the biodiesel production, the starting materials (triglyceride and methanol) should be first diffused into the nanopores of the catalysts, where the catalytic sites are located, followed by the departure of the reaction products (fatty methylester) from the pores which leaves the reaction sites for the next available reactants. Based on this model, while the higher hydrophobicity of 1a increases the relative concentration of the hydrophobic reactant (triglyceride) in the pores, it may prevent the diffusion of the hydrophilic reaction partner (methanol) into the pores and block the active sites. Notably, inspection of the data in Table 2 also confirmed that 1a showed significantly lower performance than SBA-15-PrSO3H having a higher hydrophilic backbone than 1a towards biodiesel production (Table 2, entries 10 and 12). These observations imply that high surface hydrophobization does not always improve the catalytic activity of a typical catalyst for the described reaction. Therefore, the reason that biodiesel formations are more prominent in the case of 1b than SBA-15-PrSO3H and 1a is presumably due to a combination of moderate hydrophobic nature of the nanospaces (mesochannels) and the suitable pore opening of 1b which results in a fast mass transfer and an increased concentration of both hydrophobic and hydrophilic starting materials inside the channels where sulfonic acid moieties are located.
3.3 Reusability of Et–PMO–Me–PrSO3H catalyst
The highly active acid-functionalized mesoporous organosilica 1b catalyst was easily recovered from the reaction mixture, washed with dichloromethane and subsequently vacuum dried at 60 °C before another reaction was performed. The recovered material could be added to a fresh substrate and it was shown that this catalyst can be effectively used for 3 successive cycles (Fig. 6).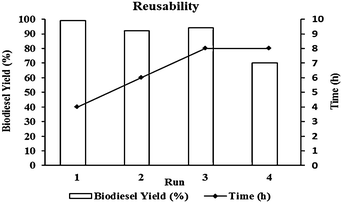 | ||
| Fig. 6 Reusability of 1b catalyst (conditions: molar ratio canola oil/methanol = 1/100, T = 150 °C, 6 wt% 1b catalyst). | ||
In order to show the thermal stability of our 1b catalyst system during the described biodiesel production protocol and the recycling stages, the loading of sulfonic acid groups into the recycled catalyst was measured in each reaction cycle. In each run, titration of ion-exchanged catalysts shows that there is indeed a little decrease in the proton capacity of the recycled catalyst. (Second run 0.44 mmol H+ g−1, third run 0.39 mmol H+ g−1 and forth run 0.32 mmol H+ g−1.) This might originate from two possibilities: decomposition of sulfonic acid groups arising either from high reaction temperature or solvolysis by methanol. However, even under such harsh reaction conditions, the majority of the acidic sites (∼64%) survived after the 4th reaction run (Table 1, entry 6).
4 Conclusions
In this paper, two novel water tolerant sulfonic acid based periodic mesoporous organosilicas (PMOs) having either phenylene (1a) or ethyl (1b) as bridge and methylpropyl sulfonic acid as functionalized group were synthesized and biodiesel production from some vegetable oils, refined olive oil and extracted oil from olive sludge was studied over these catalysts in a home designed reactor. It has been shown that 1b catalyst is more active than 1a and well-known SBA-15-PrSO3H in biodiesel formation. On the other hand, while the water adsorption experiment shows that among the described PMO functionalized acids, 1a shows more hydrophobic character, in contrast, we interestingly found that 1b consistently exhibited higher catalytic performance in biodiesel formation, which relies on a balance between hydrophobic-hydrophilic- and pore size of the mesochannels in this catalyst.Acknowledgements
The authors acknowledge the IASBS Research Council and Iran National Science Foundation (INSF) for support of this work. We also appreciate Mr. Ebrahim Toumari from IASBS for manufacturing of the reactor.References
- D. Leckel, Energy Fuels, 2009, 23, 2342–2358 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- Y.-H. P. Zhang, J. Ind. Microbiol. Biotechnol., 2008, 35, 367–375 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- R. Diesel, Engineering (London), 1912, 93, 395–406 CAS.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Energy Fuels, 2008, 22, 598–604 CrossRef CAS.
- F. Maa and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Adv. Synth. Catal., 2006, 348, 75–81 CrossRef CAS.
- W. Charusiri and T. Vitidsant, Energy Fuels, 2005, 19, 1783–1789 CrossRef CAS.
- E. Lotero, Y. Liu, D. E. López, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Jr, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- R. Luque, L. Herrero-davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 CAS.
- J. H. Clark and D. J. Macquarrie, Chem. Commun., 1998, 853–860 RSC.
- J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 11, 1285–1308 RSC.
- A. Sivasamy, K. Y. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278–300 CrossRef CAS.
- X.-R. Chen, Y.-H. Ju and C.-Y. Mou, J. Phys. Chem. C, 2007, 111, 18731–18737 CAS.
- K. Suwannakarn, E. Lotero, J. G. Goodwin, Jr and C. Lu, J. Catal., 2008, 255, 279–286 CrossRef CAS.
- C. Martins Garcia, S. Teixeira, L. Ledo Marciniuk and U. Schuchardt, Bioresour. Technol., 2008, 99, 6608–6613 CrossRef.
- Y.-M. Park, D.-W. Lee, D.-K. Kim, J.-S. Lee and K.-Y. Lee, Catal. Today, 2008, 131, 238–243 CrossRef CAS.
- D. E. López, J. G. Goodwin, Jr, D. A. Bruce and S. Furuta, Appl. Catal., A, 2008, 339, 76–83 CrossRef.
- A. Brito, M. E. Borges and N. Otero, Energy Fuels, 2007, 21, 3280–3283 CrossRef CAS.
- D. E. López, J. G. Goodwin, Jr and D. A. Bruce, J. Catal., 2007, 245, 381–391 CrossRef.
- M. G. Kulkarni, R. Gopinath, L. C. Meher and A. K. Dalai, Green Chem., 2006, 8, 1056–1062 RSC.
- M. Di Serio, M. Cozzolino, M. Giordano, R. Tesser, P. Patrono and E. Santacesaria, Ind. Eng. Chem. Res., 2007, 46, 6379–6384 CrossRef CAS.
- F. Chai, F. Cao, F. Zhai, Y. Chen, X. Wang and Z. Su, Adv. Synth. Catal., 2007, 349, 1057–1065 CrossRef CAS.
- J. Li, X. Wang, W. Zhu and F. Cao, ChemSusChem, 2009, 2, 177–183 CrossRef CAS.
- M.-H. Zong, D.-Q. Duan, W.-Y. Lou, T. J. Smith and H. Wu, Green Chem., 2007, 9, 434–437 RSC.
- R. Luque and J. H. Clark, ChemCatChem, 2011, 3, 594–597 CrossRef CAS.
- R. A. Arancon, H. R. BarrosJr, A. M. Balu, C. Varga and R. Luque, Green Chem., 2011, 13, 3162–3167 RSC.
- M. Hara, ChemSusChem, 2009, 2, 129–135 CrossRef CAS.
- R. Jothiramalingam and M. K. Wang, Ind. Eng. Chem. Res., 2009, 48, 6162–6172 CrossRef CAS.
- M. D. Serio, M. Cozzolino, M. Giordano, R. Tesser, P. Patrono, E. Santacesaria, N. Federico, C. Universitario and M. S. Angelo, Ind. Eng. Chem. Res., 2007, 46, 6379–6384 CrossRef.
- A. Stein, Adv. Mater., 2003, 15, 763–775 CrossRef CAS.
- H. Lee, S. I. Zones and M. E. Davis, Nature, 2003, 425, 385–388 CrossRef CAS.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS.
- X. Feng, G. E. Fryxell, L. Q. Wang, A. Y. Kim, J. Liu and K. M. Kemner, Science, 1997, 276, 923–926 CrossRef CAS.
- Y. Mori and T. J. Pinnavaia, Chem. Mater., 2001, 13, 2173–2178 CrossRef CAS.
- L. Mercier and T. J. Pinnavaia, Adv. Mater., 1997, 9, 500–503 CrossRef CAS.
- H. Yoshitake, New J. Chem., 2005, 29, 1107–1117 RSC.
- G. Rodríguez-López, M. D. Marcos, R. Martínez-Máñez, F. Sancenón, J. Soto, L. A. Villaescusa, D. Beltrán and P. Amorós, Chem. Commun., 2004, 2198–2199 RSC.
- M. Yasui, M. Ikeda, K. Takimiya, J. Ohshita, S. Yamanaka and K. Inumaru, Chem. Lett., 2004, 33, 1582–1583 CrossRef CAS.
- A. Sayari, S. Hamoudi and Y. Yang, Chem. Mater., 2005, 17, 212–216 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 46, 446–458 CrossRef CAS.
- P. J. E. Harlick and A. Sayari, Ind. Eng. Chem. Res., 2007, 45, 3248–3255 CrossRef.
- A. Corma and H. Garcia, Chem. Rev., 2002, 102, 3837–3892 CrossRef CAS.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS.
- Z. L. Lu, E. Lindner and H. A. Mayer, Chem. Rev., 2002, 102, 3543–3578 CrossRef CAS.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589–3614 CrossRef CAS.
- E. Lindner, M. Kemmler, F. Auer and H. A. Mayer, Angew. Chem., Int. Ed., 1999, 38, 2154–2174 CrossRef.
- M. Hartmann, Chem. Mater., 2005, 17, 4577–4593 CrossRef CAS.
- J. A. Melero, L. F. Bautista, G. Morales, J. Iglesias and D. Briones, Energy Fuels, 2009, 23, 539–547 CrossRef CAS.
- J. A. Melero, L. F. Bautista, G. Morales, J. Iglesias and R. Sánchez-Vázquez, Chem. Eng. J. (Amsterdam, Neth.), 2010, 161, 323–331 CAS.
- S. Inagaki, S. Gouan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- C. Ishii, T. Asefa, N. Coombs, M. J. MacLachlan and G. A. Ozin, Chem. Commun., 1999, 2539–2540 RSC.
- F. Hoffmann, M. Cornelius, J. Morell and M. Fröba, Angew. Chem., Int. Ed., 2006, 45, 3216–3251 CrossRef CAS.
- B. Hatton, K. Landskron, W. Whitnall, D. Perovic and G. A. Ozin, Acc. Chem. Res., 2005, 38, 305–312 CrossRef CAS.
- J. A. Melero, R. van Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS.
- S. Fujita and S. Inagaki, Chem. Mater., 2008, 20, 891–908 CrossRef CAS.
- Hoffmann, M. Cornelius, J. Morell and M. Fröba, J. Nanosci. Nanotechnol., 2006, 6, 265–288 CAS.
- W. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634–2642 CrossRef CAS.
- B. Karimi and D. Enders, Org. Lett., 2006, 8, 1237–1240 CrossRef CAS.
- B. Karimi, S. Abedi, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2006, 45, 4776–4779 CrossRef CAS.
- B. Karimi, A. Biglari, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2007, 46, 7210–7213 CrossRef CAS.
- B. Karimi and D. Zareyee, Org. Lett., 2008, 10, 3989–3992 CrossRef CAS.
- B. Karimi and D. Zareyee, J. Mater. Chem., 2009, 19, 8665–8670 RSC.
- B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Chem.–Eur. J., 2010, 16, 8047–8053 CAS.
- I. K. Mbaraka, D. R. Radu, V. S.-Y. Lin and B. H. Shanks, J. Catal., 2003, 219, 329–336 CrossRef CAS.
- I. K. Mbaraka and B. H. Shanks, J. Catal., 2005, 229, 365–373 CrossRef CAS.
- J. Dacquin, H. E. Cross, D. R. Brown, T. Düren, J. J. Williams, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 1383–1391 RSC.
- E. Cho and D. Kim, J. Phys. Chem. Solids, 2008, 69, 1142–1146 CrossRef CAS.
- S. Hamoudi, S. Royer and S. Kaliaguine, Microporous Mesoporous Mater., 2004, 71, 17–25 CrossRef CAS.
- G. Gelbard, O. Bres, R. M. Vargas, F. Vielfaure and U. F. Schuchardt, J. Am. Oil Chem. Soc., 1995, 72, 1239–1241 CrossRef CAS.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by powders and porous solids, Academic Press, London, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00444e |
| This journal is © The Royal Society of Chemistry 2012 |