Synthesis of N-heterocyclic compounds over zeolite molecular sieve catalysts: an approach towards green chemistry†
V. V. Krishna Mohan
Kandepi‡
* and
Nama
Narender
Catalysis Group, Indian Institute of Chemical Technology, Uppal Road, Tarnaka, Hyderabad-500607, India
First published on 23rd December 2011
Abstract
The need for environmentally benign reactions is very important in view of today's eco-friendly conscious attitude. “Benign by Design” represents the 12 principles of Green Chemistry as articulated by John Warner and Paul Anastas (Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30) These principles have given chemists a framework for the evaluation of the chemical procedures and help chemists to develop synthetic procedures which are more efficient, create less waste, use and produce less toxic substances. Many chemists already considered some of these principles without giving much attention. The difference between traditional chemistry and green chemistry is that the green chemistry demands issues of sustainability and environmental impact; in essence we design chemicals that are inherently more benign. In recent years, the use of solid acid catalysts such as zeolites and zeotype molecular sieve (D. W. Breck, Zeolite Molecular Sieves, Wiley, New York, 1974; A. Dyer, An Introduction to Zeolite Molecular Sieves, John Wiley & Sons, Chichester, 1988) catalysts in the manufacture of fine chemicals, chemical intermediates, drugs and drug intermediates has attracted increasing interest as green catalysts. Owing to the special features of zeolites such as shape selectivity, thermal stability, controlled variability, reusability and eco-friendly nature, these catalysts are most sought after in green chemistry. This perspective particularly illustrates the application of zeolites and zeotype catalysts for the synthesis of various N-heterocyclic compounds. For proposed reaction mechanisms see ESI.†
 V. V. Krishna Mohan Kandepi | Dr V. V. Krishna Mohan Kandepi received his BSc and MSc from Andhra University, Visakhapatnam, India, and received his PhD from Indian Institute of Chemical Technology (IICT), Hyderabad, India. His doctoral work focused on development of organic processes over zeolite-molecular sieves, in particular synthesis of N-heterocyclic compounds. Later he worked as a guest postdoctoral fellow at Leibniz Institute für Katalysis (University of Rostock, Germany) and also worked as a postdoctoral fellow (2007–2010) at Instituto de Tecnologica Química e Biológica (Universidade Nova de Lisboa, Lisbon, Portugal). He is presently employed as a Research Scientist at ecoLogic Technologies Pvt. Ltd. Hyderabad, India. |
 Nama Narender | Dr Nama Narender received his BSc and MSc from Osmania University, Hyderabad, India, and received PhD (1995) from Indian Institute of Chemical Technology (IICT), Hyderabad, India. His doctoral work focused on development of organic processes over heterogeneous catalysts such as hydrotalcites. Later he worked as a postdoctoral fellow at INETI, Lisbon, Portugal. Since 2001, he is working as a Research Scientist at Indian Institute of Chemical Technology (IICT), Hyderabad, India. Basically his research involves the development of organic processes via green chemical techniques. |
1. Introduction
Catalysis plays a crucial role in providing fuels, fertilizers, pharmaceuticals, many other commodities and fine chemicals. It also helps in strengthening of environmental protection.1 More than 60% of all chemical products and 90% of chemical processes are based on catalysis. Recently, in the manufacture of bulk chemicals, traditional and environmentally unacceptable processes have largely been replaced by many catalytic alternatives.Heterogeneous catalysis involves a reaction in which one or more of the constituents are in different phases. The advantages of using heterogeneous catalysts are: low cost, tolerance to a wide range of temperatures and pressures, easy and inexpensive removal from the reaction mixture by filtration or centrifugation, easy and safe disposal, safe storage, long lifetime, eco-friendly nature and regenerability. The catalyst may be pure, mixed with other catalysts or dispersed on an inert support e.g. metals, metal oxides, mixed metal oxides, zeolites and clays. Zeolites2 have the ability to act as catalysts for chemical reactions that take place within the internal cavities. An important class of reactions is that catalysed by hydrogen-exchanged zeolites, whose framework-bound protons give rise to very high acidity. This is exploited in many organic reactions, including crude oil cracking, isomerisation and fuel synthesis. Zeolites can also serve as oxidation or reduction catalysts, often after metals have been introduced into the framework. Examples are the use of titanium ZSM-5 in the production of caprolactam, and copper zeolites in NOx decomposition. Depending on metal ion incorporated/isomorphic substitution in a zeolite framework, quantitative and qualitative estimation of the framework and extra framework metal ions can be performed using XANES, MAS-NMR, FT-IR and XRD techniques besides the catalyst activity. Underpinning all these types of reactions is the unique microporous nature of zeolites, where the shape and size of a particular pore system exert a steric influence on the reaction, controlling the access of reactants and products. Thus zeolites are often said to act as shape-selective catalysts.3
An economic process is not necessarily green, especially if waste treatment is ignored or neglected. An industrial process may breach one or more of green chemistry principles and still economical, even if complex waste treatment adds to the cost and diminishes economic viability. Hence for a given product, the following guidelines should be the part of selection of a route to construct a green process: (i) selection of feedstock (cost, energy and waste generation, etc. for the given feedstock), (ii) selection of reaction paths (minimized energy requirements by the use of selective catalysts), (iii) selection of catalysts (efficiency, easy separation from products, recycling of catalysts, non-hazardous and safe handling), (iv) down-stream processes/unit operations (minimizing the number of stages necessary to obtain the product), (v) minimizing not only the amount of pollutants, but also the volume of waste (effluent gases and solid) and (vi) recycling of auxiliary, side and intermediate products into the process. Application of zeolites as catalysts provides many advantages as stated above to transform a hazardous process into a green chemical process.
Zeolites also have some disadvantages over conventional solid catalysts in a certain number of applications. Both are discussed in terms of the adjustment of the acidity and basicity, structural effects, concentration and adsorption effects, coke formation and deactivation. Actually, the main advantages of zeolites over conventional catalysts are due to the great acid strength they have and to their great adaptability to almost all types of reactions. The main limitations of zeolites are (i) the great sensitivity to deactivation by irreversible adsorption or steric blockage of heavy secondary products, (ii) the impossibility of using their microporosity for the synthesis of bulky molecules. Because of the greater polarity of functional compounds, it is more difficult to exploit the shape selectivity of zeolites for their reactions than in the case of hydrocarbons, (iii) coke formation at high temperatures and (iv) formation of side products under harsh conditions. Increasingly, attention has been focused on fine tuning the properties of zeolite catalysts in order to carry out very specific syntheses of high-value chemicals and to reduce the problems described above.
Heterocyclic compounds are widely distributed in nature and are essential to human life; they play a vital role in metabolism. Because of their extensive practical use, the literature of the subject is correspondingly vast. Due to wide-ranging biological activity of pyridine derivatives the preparation of these N-heterocyclic compounds remains a current topic of interest.4–7
Since several years we have been working in the area of synthesis of N-heterocyclic compounds over zeolite molecular sieve catalysts. In view of potential applications of zeolites in organic synthesis and importance of N-heterocyclic compounds, we have been working on the efficient synthesis of 2-methyl-5-ethyl pyridine, various highly substituted pyridines, various annelated pyridines, N,N′-dimethyl piperazine, etc. The outcome is discussed in the respective topics.
Since many decades, several methods have been developed to prepare pyridines and substituted pyridines.8–12 Pyridine and quinoline and their derivatives are generally synthesized by condensation of amines and carbonyl compounds.13–18
2. Synthesis of pyridines and picolines
2.1 Synthesis of pyridines
Pyridine bases are industrially important compounds. Pyridine, 2-picoline and 2-methyl-5-ethyl pyridine possess a strong demand, whereas 3-picoline and 4-picoline have a limited market. Pyridine and picolines are useful intermediates in the synthesis of herbicides, surfactants and pharmaceuticals. Industrially, pyridine and its derivatives are synthesized by condensation of aldehydes and ammonia over an amorphous aluminosilicate catalyst promoted by ThO2, ZnO or CdO with 40–60% yield.17 These catalysts suffer from the common drawbacks such as poor catalytic activity, selectivity, thermal stability and regeneration. Pyridine has been produced commercially from coal tar since 1920s. During 1950s, few synthetic processes were developed to provide alternatives to isolation from coal tar.19 There are few selective commercial processes for the synthesis of pyridine and its derivatives and almost all these manufacturing processes produce pyridine along with a series of alkylated pyridines. The reaction of aldehydes or ketones with ammonia is the most general synthetic reaction for the manufacture of pyridine bases and allows the preparation of various pyridine derivatives. Reaction of acetaldehyde and formaldehyde with ammonia is the most widely used method for pyridine production.19 Pyridine can also be prepared from cyclopentadiene by ammoxidation, or from 2-pentenenitrile by cyclization and dehydrogenation. Furfuryl alcohol or furfural reacts with ammonia in the gas phase to give pyridine.19Pyridine and 3-picoline occupied nearly 80% of world consumption of pyridines in 2010.20 Agricultural chemicals (mainly the nonselective contact herbicide paraquat) account for most consumption of pyridine. Consumption of 3-picoline, which is used to produce niacinamide/niacin (vitamin B3), is anticipated to grow at an average annual rate of nearly 2%, as a result of the demand for niacinamide/niacin in developing regions such as Asia for use in animal feed. 2-Methyl-5-ethylpyridine (MEP) is also used entirely to produce niacin. The largest market for 2-picoline is 2-vinylpyridine and 2-vinylpyridine is used as a component of styrene-butadiene-2-vinylpyridine terpolymer latexes (SBV latexes), which are used as tire cord adhesives and in other adhesives for binding textiles to elastomers. Consumption of pyridine for paraquat declined substantially in the United States in late 2006, as a result of the termination of paraquat production at Bayport, Texas, by Syngenta Crop Protection. Additionally, a ban on paraquat use in the European Union has dampened the prospects for future consumption of pyridine in Europe; future consumption of pyridine for paraquat in Europe will depend largely on the export demand. China will account for most growth in pyridine demand for paraquat and other agricultural chemicals such as chlorpyrifos, which is derived from 3-picoline. Global pyridine production and consumption growth will continue to be driven by Asia, in particular China and India. Other Asian countries (Republic of Korea, Taiwan, Indonesia, Malaysia, Singapore and Thailand) will have moderate growth of about 2% annually. No growth is expected in Japan in the next five years. Similarly, no market growth is expected for Canada or Central and South America. Central and Eastern Europe will experience growth, but from a very small base.
In 1991, Kulkarni and Subrahmanyam21 reported the synthesis of pyridines with propylene oxide, propylene glycol, ethylene glycol, acetaldehyde or acetone22 and ammonia over modified ZSM-5 and SiO2–Al2O3 promoted by Pb, Cu, Cr, etc. Van der Gaag et al.23 reported a method for the synthesis of pyridine from ethanol and ammonia in the presence of oxygen over ZSM-5. In this reaction, carbon dioxide was formed as a major product due to the presence of oxygen in the feed. Pyridines were also synthesized from acetaldehyde over various modified SiO2–A12O3 and pentasil type zeolite catalysts.24–29 Cobalt-modified HZSM-5 (Si/Al = 70) zeolite catalysts were reported for the synthesis of pyridine and picolines prepared by ion exchange with Co(CH3COO)2 solution, and incipient wetness impregnation with aqueous solutions of Co(NO3)2 and Co(CH3COO)2, respectively.30 The yield of pyridine was slightly increased from 42% to 50% by using Co-ZSM-5. The effects of Co on the catalytic activity are mainly attributed to the modification of surface acidity and the shape selectivity due to the narrowing of the pore opening. The reaction of acetaldehyde and ammonia over HZSM-5 affords 2-picoline and 4-picoline. Whereas the reaction of acetaldehyde, formaldehyde and ammonia over HZSM-5 leads to the formation of pyridine and 3-picoline.31 Reddy et al. studied the critical process parameters in order to achieve high selectivity with minimal coke formation.32
Synthesis of pyridine and picolines from ethanol, formaldehyde and ammonia over various ZSM-5 catalysts in the absence of oxygen was reported (Scheme 1).33 The reaction was carried out at 420 °C with 0.5 h−1 weight hourly space velocity (WHSV). The amount of formaldehyde present in the feed has a significant role in the formation of pyridines. The reaction of ethanol and ammonia without formaldehyde over Pb-ZSM-5 resulted in only 6% yield of pyridine and 55% of both (2-picoline and 3-picoline) picolines. This is a clear indication of the importance of the formaldehyde towards the formation of pyridine. The pyridine and picoline formation is explained by an imine mechanism. The imine formation was reported by Golunski and Jackson.17 Ethanol with ammonia over ZSM-5 at 420 °C leads predominantly to the formation of ethylimine. Thus it is believed that the imine resulted from the reaction of acetaldehyde with ammonia over ZSM-5 via dehydrogenation (Scheme I, ESI†).
 | ||
| Scheme 1 Synthesis of pyridine and picoline over the HZSM-5 zeolite catalyst. | ||
Reddy et al. reported a study on mass transfer and kinetics of vapor phase synthesis of pyridine through catalytic aminocyclization.34 Interrelationship of catalyst activity and process parameters and their effect on conversion and selectivity were also reported by Reddy et al.35–37 Process factors contributing to the catalyst deactivation have also been investigated to evolve a model to simulate its time-on-stream performance. In conclusion, the HZSM-5 catalyst with silica to alumina ratio of 240 is found to provide the best product distribution with least coke formation. The diffusion studies revealed that the optimum particle size of the catalyst particle is 600 μm for these conditions.
2.2 Synthesis of monomethyl pyridines (picolines)
The synthesis of 2-picoline was carried out with acetone, formaldehyde, methanol and ammonia over modified ZSM-5(Si/Al = 30), ZSM-5(Si/Al = 150) and ZSM-5(Si/Al = 280) catalysts. PbZSM-5 and WZSM-5 were found to be best catalysts for the selective synthesis of 2-picoline.20 2,6-Lutidine was found as the major side product. In order to get high activity in this dehydrocyclization reaction, an optimum number of active centres are required. With the increase in the Si/Al ratio, 2-picoline/4-picoline ratio was also increased. Pb-ZSM-5 and WZSM-5 have a promoting effect in increasing the yield of 2-picoline. The yield of 2-picoline was increased from 20% to 40% with increase in the formaldehyde mole equivalent from 0.25 to 1. Picolines are produced today by the condensation of acetaldehyde, formaldehyde and ammonia in the gas-phase, which simultaneously produces large quantities of pyridine. A selective and suitable alternative method for the syntheses of picolines has yet to be developed. Given the thermodynamic properties of these molecules and reactions involved, it does not seem likely to expect a selective process for picoline.3. Synthesis of lutidines and collidines
3.1 Synthesis dimethylpyridines (lutidines)
Yasuda and Abe38 reported the synthesis of 3-picoline and 3,5-lutidine from acrolein, propionaldehyde, formaldehyde and ammonia over CdO–SiO2–Al2O3. The yield of 3,5-lutidine was very low (13%). Selective synthesis of 3,5-lutidine was achieved from propionaldehyde, formaldehyde and ammonia over modified ZSM-5 catalysts.39 The substitution of transition metal cations in H-ZSM-5(30) increased the yield of collidines from 6% to 31%. The maximum yield (56.1% at 68.2% conversion) of 3,5-lutidine was obtained at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio of propionaldehyde and formaldehyde over PbZSM-5(30). Synthesis of 3,5-lutidine was also carried out using propanol, formaldehyde and ammonia with methanol over modified ZSM-5 catalysts.40 The maximum yield (63.9% at 88.6% conversion) of 3,5-lutidine was observed at 400 °C and the selectivity was decreased upon increasing the temperature. Due to coking the shape selectivity for 3,5-lutidine was improved, because collidines could not diffuse out. The coking reduced the intersection or pore diameter of the catalyst. If 2-propanol was used as reactant instead of n-propanol, 2,6-lutidine was observed as a major product. This is due to the presence of –OH group at the second carbon atom in 2-propanol. The active sites for the cyclization and dehydrogenation are Brønsted acidic centres and cations.41 In conclusion, selective synthesis of 3,5-lutidine was achieved by using n-propanol, formaldehyde and ammonia. Whereas, 2,6-lutidine was achieved by replacing n-propanol with 2-propanol in the above process.
:1 mole ratio of propionaldehyde and formaldehyde over PbZSM-5(30). Synthesis of 3,5-lutidine was also carried out using propanol, formaldehyde and ammonia with methanol over modified ZSM-5 catalysts.40 The maximum yield (63.9% at 88.6% conversion) of 3,5-lutidine was observed at 400 °C and the selectivity was decreased upon increasing the temperature. Due to coking the shape selectivity for 3,5-lutidine was improved, because collidines could not diffuse out. The coking reduced the intersection or pore diameter of the catalyst. If 2-propanol was used as reactant instead of n-propanol, 2,6-lutidine was observed as a major product. This is due to the presence of –OH group at the second carbon atom in 2-propanol. The active sites for the cyclization and dehydrogenation are Brønsted acidic centres and cations.41 In conclusion, selective synthesis of 3,5-lutidine was achieved by using n-propanol, formaldehyde and ammonia. Whereas, 2,6-lutidine was achieved by replacing n-propanol with 2-propanol in the above process.
3.2 Synthesis of trimethylpyridines (collidines)
The selective synthesis of 2,3,5-trimethylpyridine (2,3,5-collidine) is an important task, since this isomer of collidine is the starting material for many drugs, such as Omeprazole (antiulcer). Coal tar is the most important commercial source of collidines. Synthesis of collidines includes ammonolysis of 2-butanone over a two component metal oxide, cyclotrimerization of propyne with acetonitrile and refluxing N-2-butylacetamide in hexamethylphosphoramide.In the above process, the maximum yield of collidines was 15–30%.42,43 Srinivas et al.44 reported a process for the synthesis of 2,3,5-collidine using 2-butanone, formaldehyde and ammonia as reactants and ZSM-5 as a shape selective catalyst (Scheme 2).
 | ||
| Scheme 2 Synthesis of collidines over zeolite catalysts. | ||
In this work, various H-ZSM-5 zeolites, such as H-ZSM-5(30), H-ZSM-5(40), and H-ZSM-5(280), modified with 5 wt% Pb, Mn, La, Fe, Cu and Co by an impregnation method were used. In general, cyclization reactions need catalysts such as those having both metal ions and as well as protons. Because, both have the potential to act as bifunctional catalysts since all required steps for the cyclization can occur at a single residence time of molecules inside the pores of a zeolite framework. A Pb modified catalyst showed high conversion (∼91%) of 2-butanone with 43% selectivity to 2,3,5-collidine and 30% to 2,3,6-collidine. The maximum combined yield for both collidines was 66.43%.
4. Synthesis of 3,5-dipropylpyridine under vapour phase conditions
Srinivas et al.45 reported the synthesis of 3,5-dipropyl pyridine using valeraldehyde, formaldehyde and ammonia over modified ZSM-5 catalysts. The ZSM-5 zeolite was modified with various metal ions such as Co, Ni, Pb, La, and Zr. The reaction of valeraldehyde, formaldehyde and ammonia was carried out over above-mentioned modified ZSM-5 catalysts. The conversion of valeraldehyde was >90% over all the catalysts. Whereas, the yield was around 72% over PbZSM-5 and the yield was decreased in the case of Ni+2, Co+2, La+3 modified catalysts. The yield of 3,5-dipropyl pyridine over Zr+4 and Pb+2 was increased with increase in acidity, but the yield was decreased in the case of Ni+2, Co+2 and La+3 catalysts, indicating the formation of coke at higher acidity. This concludes that mild acidic conditions are favourable for the pyridine formation. Furthermore, the dehydrogenating nature of the metal ion should also be considered, which is an important step in the pyridine synthesis.46Furthermore the reaction was carried out with n-pentanol instead of valeraldehyde to understand the mechanism. The yields were comparatively low under the same reaction conditions. In this reaction, n-pentylamine and valeraldehyde were also observed as other products (Scheme 3).
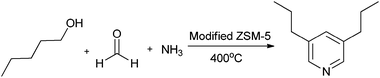 | ||
| Scheme 3 Possible reaction mechanism for the formation of 3,5-dipropyl pyridine over zeolite catalysts. | ||
5. Synthesis of 5-ethyl-2-methylpyridine under high pressure
Industrially, a large amount of 5-ethyl-2-methylpyridine (MEP) is required as starting material for the products such as nicotinic acid, its derivatives and 2-methyl-5-vinylpyridine (MVP). The condensation of acetaldehyde or paraldehyde with ammonia to form MEP was reported by a number of investigators47,48 but these processes suffer from low yields due to the formation of pyridine derivatives, high boiling compounds and large amounts of tar. Krishna Mohan et al. reported49 the synthesis of 5-ethyl-2-methylpyridine, using acetaldehyde and ammonia over zeolite catalysts under high pressure in the temperature range 150–225 °C (Scheme 4). | ||
| Scheme 4 Synthesis of 5-ethyl-2-methylpyridine (MEP) over zeolites. | ||
This reaction was studied over various zeolites and SiO2, SiO2–Al2O3, K10-montmorillonite. Among all these catalysts H-beta, HY, HZSM-5(40) and K10-montmorillonite showed better results compared to other catalysts. Best conversion of acetaldehyde and selectivity to MEP was obtained over H-beta catalysts. Moreover, with the increase in Si/Al ratio of HZSM-5, the conversion selectivity was decreased. This trend can be explained by the decrease in Brønsted acid sites while increasing the Si/Al ratio.50 Interestingly, if the reaction was carried out under vapour phase conditions (400–500 °C), the reaction afforded 2-methyl and 4-methylpyridines as major products and MEP in negligible amounts.51,52 However, the same reaction afforded very high selectivity and yield of MEP, when the reaction was carried out in an autoclave under high pressure. Thus it clearly indicates that the product formation is purely dependent on the reactor design such as an autoclave or a fixed-bed flow system in this case. An excess amount of ammonia more than the stoichiometric amount is necessary for this reaction. The excess ammonia available in the reaction mixture converts all acetaldehyde molecules into imine, which is the key intermediate for the formation of MEP. Moreover, the excess ammonia can avoid the polymerization of acetaldehyde. Excess ammonia after the optimum level is causing over-dilution of the reactants and reducing the active collisions between molecules.
6. Synthesis of highly substituted pyridines under high pressure
Narender et al.53 developed a process for the synthesis of trisubstituted pyridines with moderate selectivity and high conversion of aldehydes using zeolite catalysts under high pressure (Scheme 15). The process is a mild and efficient single-step procedure for the conversion of aliphatic aldehydes and aqueous ammonia to the corresponding 2,3,5-trisubstituted pyridines. The synthesis was carried out in an autoclave at high pressure (autogenous pressure) in a batch mode. The reactions of propionaldehyde, butyraldehyde, valeraldehyde and heptanaldehyde with aqueous ammonia (25%) were carried out over a H-beta catalyst and methanol as solvent (Scheme 5). | ||
| Scheme 5 Synthesis of 2,3,5-trisubstituted pyridines over zeolite catalysts. | ||
If the same reaction was carried out under vapour phase conditions, no significant selectivity to the corresponding product was observed. When acetaldehyde is used as reactant the product is 5-ethyl-2-methylpyridine (2,5-disubstituted pyridine). Whereas, the products were 2,3,5-trisubstituted pyridines when higher aldehydes (propionaldehyde, butyraldehyde, valeraldehyde and heptanaldehyde) were used as reactants. The difference in the formation of product can be attributed to the dynamic stability of the corresponding product. The higher aldehydes in this process afforded only long chain substituted pyridine derivatives and no pyridine ring enlargement was observed.
7. Synthesis of pyridine-2-carboxaldehyde from 2-picolyl alcohol
Pyridine-2-carboxaldehyde is used in the preparation of pralidoxime methiodide which is an antidote to poisoning by organophosphate acetyl-cholinesterase inhibitors.54 Pyridine-2-carboxaldehyde is also used in the production of bisacodyl, a laxative, and as an intermediate in pharmaceutical productions. Pyridine-2-carboxaldehyde was synthesized from the 2-picolylalcohol in the vapour phase reaction over modified silicoaluminophosphate catalysts,55 manganese dioxide56 and V–Mo oxide catalysts.57 Kamalakar et al.58 reported the synthesis of pyridine-2-carboxaldehyde from 2-picolylalcohol over modified ZSM-5(30) catalysts in the liquid phase at 70–80 °C with high yield (78%) (Scheme 6). | ||
| Scheme 6 Synthesis of pyridine-2-carboxaldehyde over modified ZSM-5 zeolites. | ||
The zeolite ZSM-5(30) was modified with 1 wt% Ti, V, Cr and Cu. These modified zeolites were applied for the liquid phase oxidation of 2-picolyl alcohol using tert-butylhydroperoxide and H2O2 as oxidants and methanol as solvent under reflux conditions. The yield of pyridine-2-carboxaldehyde was lower with H2O2 than tert-butylhydroperoxide. Liquid phase oxidative dehydrogenation of 3-picolyl alcohol was also carried out with tert-butylhydroperoxide over various TiZSM-5(30) pre-calcined at 800 °C, the maximum yield of pyridine-3-carboxaldehyde was ∼33%. In conclusion, the best yield and conversion of 2-picolyl alcohol and pyridine-2-carboxaldehyde were 98% and 82%, respectively, over the (1 wt%)Sb-(5 wt%)K-SAPO (DPA) catalyst.55
8. Synthesis of 2-phenyl pyridines using acetophenone, allyl alcohol and ammonia
Phenylpyridines are known as an integral backbone of several potent azapeptide HIV protease inhibitors, a moiety showing anti-HIV activity such as BMS-232632a.59 Strategies for the synthesis of the phenylpyridine nucleus were varied from the Chichibabin condensation reaction to Heck substitution reactions, but afforded low yields and cannot facilitate selective synthesis at the 2-position. In a synthetic route,60,61 homogeneous catalysts like Rh(I) were used for the regioselective alkylation of phenyl-pyridine with olefins. However, various substituted pyridines were often synthesized over microporous materials from cheaper chemicals via the Hantzsch route.62–67 Radha Rani et al. reported the synthesis of 2-phenyl pyridine using acetophenone, allyl alcohol and ammonia over zeolites and mesoporous materials.68 The reaction was studied over various zeolites (HY, H-beta, HX, HZSM-5), SiO2–Al2O3 and MCM-41 at 360 °C (Scheme 7). | ||
| Scheme 7 Synthesis of 2-phenyl pyridines from acetophenone. | ||
The conversion of acetophenone is dependent on the shape and pore size of the zeolite used. The selectivity of 2-phenyl pyridine was 67% and conversion of acetophenone was 99.7%. The large pore size of MCM-41 minimizes the diffusion constraints experienced by the reactants and as well as products, which can result in high conversion and selectivity. Whereas HY resulted in low selectivity compared to MCM-41 due to rapid coke formation. In the case of HZSM-5 the conversion of acetophenone and selectivity of 2-phenyl pyridine were low due to the small pore size. The diffusion of acetophenone was less than allyl alcohol through small pores of HZSM-5, because of this the formation 3-picoline was higher than the surface reaction leading to the formation of 2-phenyl pyridine. Although H-beta and HX were having a large pore size, the different crystal structures of these zeolites did not favour the formation of 2-phenyl pyridine in high yield.
9. Synthesis of N-methylpyrrolidine
Vapour phase synthesis of N-methyl pyrrolidine and tetrahydrofuran via intermolecular cyclization of diol and alkylamine over modified ZSM-5 catalysts was reported by Subba Rao et al.69 (Scheme 8). The reaction has been studied with 1,4-butanediol and methylamine over different modified ZSM-5 catalysts. The yield of N-methylpyrrolidine (NMP) was enhanced by using an excess methylamine. | ||
| Scheme 8 Synthesis of N-methyl pyrrolidine, N-methyl piperidine, N-ethyl pyrrolidine and N-ethyl piperidine; THF, Tetrahydropyran and 4-(N-methylamino)-1-butanol over zeolites. | ||
The optimum temperature to obtain more NMP was 300 °C. At lower temperatures i.e. <300 °C, tetrahydrofuran (THF) was obtained more than 90%. On the other hand, at >300 °C, side products were increased including aromatics. In the case of CrZSM-5, the NMP product selectivity was 64.2% at 300 °C. Nonetheless, the VZSM-5, MoZSM-5, and MnZSM-5 did not show much difference in the formation of NMP with respect to temperature, but the side products were increased up to 7%. Interestingly, VZSM-5 afforded NMP but the SAPO, VAPO and VSAPO did not produce NMP, even though the main constituent is vanadium. Either the channel structure of the ZSM-5 catalyst or the impregnated vanadium residing in the channels or pores was responsible for the above situation. Overall, the promoting effect of cations to form NMP is in the order of Cr > V > Mn > Mo > Pb = Cu > W. CrZSM-5 is a bifunctional catalyst with [Cr(OH)]2+ and H+ as active centres.
The bifunctionality and polarizability of the cation were responsible for the formation of NMP. Brønsted acidic centres were formed upon the dissociation of water. The polarizability of Cr3+ (or cations) is responsible for the dissociation of water resulting in the bifunctional nature of the catalyst. Because of the absence of amine in the feed, the product was only THF. The 1,4-butanediol interaction with Bronsted acidic center H+, followed by dehydration, resulted in the primary carbocation as shown in Scheme XIII (ESI†). The channel size (5.6 Å) of ZSM-5 may also restrict the formation of stable secondary cations, due to geometric constraints. Secondly, the reaction of methylamine with THF also took place. The remarkable feature of this system was the achievement of highest (∼98%) conversion of γ-butyrolactone to l-methyl-2-pyrrolidone under mild conditions. The reaction of aniline was found to be sluggish, because of the difficulty in diffusion of the product out of the channels of zeolite.
10. Selective synthesis of 3,4-dihydropyrimidin-2(1H)-ones
The first report on the Biginelli reaction involved a simple and straightforward procedure for the synthesis of DHPMs. A one-pot cyclocondensation of ethyl acetoacetate, an aromatic aldehyde and urea under strongly acidic conditions afforded the corresponding DHPMs.70 The dihydropyrimidinone moiety and its derivatives are an important class of compounds, as they are present in a large family of natural products and have a broad range of biological activities. They have fascinating therapeutic and pharmacological properties.71–74 Syntheses of these moieties by the Biginelli reaction over various homogeneous catalysts such as polyphosphate ester,75 LaCl3·7H2O76 and LiClO477 were reported. A heterogeneous catalyst such as KSF (montmorillonite)78–80 is used, but its reusability is limited for few runs only. Radha Rani et al.81 reported a single-step liquid phase cyclocondensation reaction with aromatic aldehydes, β-ketoesters and urea in toluene (Scheme 9) over zeolites (HY, HZSM-5 and MCM-41) with high yields and selectivity under reflux conditions. The HY catalyst furnished higher yields for aliphatic aldehydes, which normally shows extremely poor yields in the Biginelli reaction.70,79 | ||
| Scheme 9 Synthesis of dihydropyrimidine-2(1H)-ones via zeolite-catalyzed cyclocondensation. | ||
The yield of the Biginelli reaction is dependent on the type of substrates used. Highly functionalized or sterically hindered groups afforded reduced yields. A small amount of the catalyst is sufficient to obtain high yield, the amount of β-ketoesters used is lower than the stoichiometric quantity, in order to avoid the possibility of intra-condensation.
Kulkarni et al.82 reported the synthesis of dihydropyrimidin-2(1H)-ones over zeolite catalysts under solvent-free one-pot conditions to avoid limitations in using Lewis acid catalysts, elevated reaction temperatures, solvent mediated reactions and moderate yields of the products. Moreover, when aliphatic aldehydes and thiourea were used, low yields of DHPMs were obtained at room temperature. No substantial increase in the yield was observed even after prolonged time. The product was obtained in 98% yield at 50 °C. Excess ethyl acetoacetate did not produce Knoevenagel condensation side products under the similar reaction conditions.83
This methodology was applicable to aliphatic as well as aromatic aldehydes for the synthesis of dihydropyrimidinones with high yields. This catalyst (TS-1) is highly active with acid-sensitive aldehydes such as furfural without leading to the formation of any side products also. Appreciably, a variety of sensitive functional groups like the NO2, Cl, OH, OCH3 and conjugated double bonds did not affect the yield of the product. Further, the reaction has been studied successfully using thiourea to afford the corresponding dihydropyrimidin-2(1H)-thiones.
11. Intermolecular cyclization of ethanolamine to 1,4-diazabicyclo[2.2.2]octane
Zeolites as catalysts for the amination of ethanolamine to ethylenediamine, where 1,4-diazabicyclo[2.2.2]octane (DABCO), a minor product, was reported.84 Budnik and Sandner reported a process for the synthesis of DABCO using N-aminoethyl piperazine over zeolite catalysts.85 DABCO is a useful compound as a catalyst in the synthesis of industrially important polyurethane foams86 and also as a structure directing agent87 for the synthesis of zeolites.Budnik and Sander reported the synthesis of 1-azabicyclo[2.2.2]octanes or 1,4-diazabicyclo[2.2.2]octanes from acyclic or heterocyclic amines or mixtures using high silica zeolite catalysts at high temperatures between 250–550 °C.85 Srinivas et al. reported the selective synthesis of DABCO from the intermolecular cyclization of ethanolamine over modified ZSM-5 catalysts under vapour phase conditions at 300 °C (Scheme 10).88
![Zeolite catalyzed synthesis of 1,4-diazabicyclo[2.2.2]octane and formation of pyrazine.](/image/article/2012/CY/c2cy00162d/c2cy00162d-s10.gif) | ||
| Scheme 10 Zeolite catalyzed synthesis of 1,4-diazabicyclo[2.2.2]octane and formation of pyrazine. | ||
Surface area of the catalysts was decreased upon impregnation with various metal cations, compared to their protonated state (except Cu and La), due to the presence of metal ions and their oxide species in cavities of the modified zeolites. In order to increase the selectivity of DABCO, the HZSM-5(30) catalyst was modified with various metal ions such as V, Cr, Mn, Cu, Pb, Zr and La. Among these metal modified catalysts CrZSM-5(30), PbZSM-5(30), ZrZSM-5(30), WZSM-5(30) gave better selectivity. Decreased selectivity was observed over CuZSM-5(30), this may be due to the reductive nature of Cu ions, which leads to the formation of dehydrogenated products like pyrazine and its derivatives. It was also found that the catalysts having medium acidic sites gave better results. The medium strength acidic sites were found to be responsible for the selective synthesis of DABCO. At lower temperatures (<400 °C) the formation of pyrazine and its derivatives was low, therefore the selectivity of piperazine and DABCO was more.89 At elevated temperatures, an increase in Lewis acidic sites is known,90 this may be due to dehydration of Brønsted acidic sites which can lead to the formation of pyrazine derivatives. Additionally, at elevated temperatures the degradation of ethanolamine to aliphatic amines is also possible which leads to decrease in the selectivity of DABCO.
12. Intermolecular cyclization of diethanolamine and methylamine to N,N′- dimethylpiperazine under high pressure
Piperazines and substituted piperazines are very important intermediates in the synthesis of quinoline type antibacterial drugs.91 In particular the substituted piperazines are used in the treatment of intestinal worms, e.g., as anthelmintics and chemotherapeutics. N-Methylpiperazine was synthesized using diethanolamine and methylamine over zeolites at 30–80 atmosphere H2 pressure and at 250–350 °C reaction temperature.92 The yield of N-methylpiperazine was >90% at 300 °C and 30–80 atm H2 pressure using HZSM-5(30). The yield of N-methylpiperazine and N,N′-dimethylpiperazine was <10% at 300 °C under atmospheric pressure.Narender et al.50 reported a process for the selective synthesis of N,N′-dimethylpiperazine over zeolites under high pressure (Scheme 11). The reaction of diethanolamine and methylamine was carried out over HZSM-5(30) in the temperature range 200–300 °C in an autoclave. The yield of N,N′-dimethylpiperazine was maximum (68%) with 96% conversion of diethylamine at 300 °C reaction temperature and 100 atmosphere autogeneous pressure. In contrast, when the reaction was carried out using water as a solvent, the yield of N,N′-dimethylpiperazine was <10% at 66 atmospheric autogeneous pressure and at 285 °C.
 | ||
| Scheme 11 Synthesis of N,N′-dimethylpiperazine over zeolite catalysts. | ||
The Brønsted acidic centers present at the intersections of ZSM-5 catalysts are expected to be the active centres for this reaction. Thus, HZSM-5(30) showed higher activity than HZSM-5(280). The high yield under high pressure with respect to methylamine is mainly due to the drastic increase in the active collisions and miscibility. The selectivity of N,N′-dimethylpiperazine is also strongly influenced by the reactor design such as fixed bed continuous flow and autoclave batch mode.
13. Intermolecular cyclization of diethanolamine and methylamine to N-methylpiperazine
Piperazines and substituted piperazines are important intermediates in the synthesis of quinoline type antibacterial drugs.93 Piperazine is used in the synthesis of drugs such as norfloxacin, ciprofloxacin and enrofloxacin. In particular, N-methylpiperazine is used in the synthesis of the ofloxacin, amifloxacin, fleroxacin and difloxacin type of antibacterial drugs. Nagaiah et al.92 reported the synthesis of N-methylpiperazine using diethanolamine and methylamine over zeolites (Scheme 12). | ||
| Scheme 12 Synthesis of N-methylpiperazine over zeolite catalysts. | ||
The yield of N-methylpiperazine was more than 90% based on diethanolamine at about 100% conversion of diethanolamine over the HZSM-5(30) catalyst. The reaction between diethanolamine and methylamine (1:2 mole ratio) was carried out at 300 °C with 30 cm3 per minute of H2 gas flow or without hydrogen at 1 atmospheric pressure and 0.5 h−1 WHSV. The yield of N-methylpiperazine was <10% at about 15% conversion of diethanolamine over HZSM-5(30). The yield of N-methylpiperazine increases at 250 °C with increase in H2 pressure. Due to the presence of hydrogen as the reactant the formation of N-methylpyrazine was not obtained. The reaction was also studied over HZSM-5(280), HY and H-mordenite zeolites. The best yield of N-methylpiperazine was obtained over HZSM-5(280) at 80 atm H2 pressure and at 300 °C. HY and HZSM-5(280) showed fast deactivation than HZSM-5(30).
14. Amino cyclization of terminal (α,ω)-diols
Reports are available for the amino cyclization reaction over zeolites, using amino-alcohols,94 and showed that the selective synthesis of the desired products can be obtained in the presence of zeolite catalysts with strong acidic sites and narrow channels. Radha Rani et al.95 reported the synthesis of cyclic amines from terminal diols (α,ω) over modified ZSM-5 zeolites. Vapor-phase amino intramolecular cyclization of (α,ω)-diols with ammonia to N- and O-containing cyclic compounds over the modified ZSM-5 catalyst is shown in Scheme 13. | ||
| Scheme 13 Synthesis of cyclic amines over zeolite molecular sieves. | ||
14.1. Reaction of 1,4-butane diol with ammonia
The reaction of 1,4-butanediol with ammonia over various ZSM-5(30) catalysts at 250 °C with 0.5 h−1 weight hourly space velocity (WHSV) afforded tetrahydrofuran (THF) and pyrrole. The reaction over the HZSM-5(30) catalyst showed that although there is high conversion of the reactant (99%), the yield of pyrrolidine is low (41%). Among all the catalysts studied, CuZSM-596 and FeZSM-5 catalysts have afforded selectively pyrrolidine (95–99% product at 99% conversion) with high activity. LaZSM-5 afforded the pyrrole (dehydrogenated) product with high selectivity (93%).14.2. Reaction of 1,5-pentanediol with ammonia
In the reaction of 1,5-pentanediol with ammonia over various ZSM-5(30) catalysts the products were piperidine, pyridine, 5-amino-1-pentanol and pyran at 280 °C with 0.5 h−1 WHSV. When the reaction was carried out over the HZSM-5 catalyst with different Si/Al ratios cyclized products, mainly piperidine and pyran, were obtained. The maximum yield (piperidine, 81%) was obtained over HZSM-5(40). Additionally, the ZSM-5 was modified with different metal ions which are having different dehydrogenation activity. The completely or partially dehydrogenated piperidine product (pyridine) or dehydro piperidine was found in negligible amounts. However, among all the metal ion impregnated catalysts studied, the nickel ion showed high activity and selectivity (90%) towards the formation of saturated cyclic amines.9714.3. Reaction of 1,6-hexanediol with ammonia
The reaction of 1,6-hexanediol with ammonia was carried out over different ZSM-5(30) catalysts at 300 °C with 0.5 h−1 WHSV, which afforded hexamethyleneimine selectively (49%) and 50–80% conversion of 1,6-hexanediol. The formation of hexamethyleneimine by way of both dehydrocyclization of aminol and amination of the oxepane was dominant (88 wt% yield) over CeZSM-5. Moreover, hexamethyleneimine acts as the intermediate for the synthesis of ε-caprolactam. The direct synthesis of ε-caprolactam was attempted via the cyclization of 6-amino caproic acid and water (1:5 volume ratio) at 350 °C over various ZSM-5 catalysts. The high yield (75%) of caprolactam was obtained over PbZSM-5(30). A comparison for the reaction temperatures required to obtain the desired product reveals a distinct influence of the length of hydrocarbon backbone on the reactants (diols) OH–(CH2)n–OH. The optimized reaction temperature was directly proportional to increase in the chain length.
15. Synthesis of piperazine and pyrazine
Piperazine and pyrazine are mainly used in synthesis of perfumes and as starting materials for several medicines and agrochemicals. Methods of preparation of piperazines and pyrazines include vapor phase dehydration, dehydrogenation and deamination by using suitable catalysts with different reagents at elevated temperatures98–100 were reported. The cyclization of a terminal amino alcohol was reported by Subrahmanyam et al.101N-(2-Hydroxyethyl) ethylenediamine undergoes only dehydrocyclization, yielding piperazine as a major product over zeolite catalysts, whereas dehydrocyclization occurs together with dehydrogenation resulting in pyrazine as a major product over chromite catalysts. Certainly, over zeolites, the piperazine was formed only at 260 °C and above. Among all the catalysts studied HZSM-5(280) is producing 80% piperazine at 260 °C (Scheme 14). | ||
| Scheme 14 Zeolite catalysed synthesis of piperazine and pyrazine. | ||
16. Synthesis of annelated pyridines
Phenanthridines and substituted phenanthridines are useful as trypanocidal, antibacterial agents, antivirals and antidepressants, antispasmodics, pesticides and as corrosion inhibitors in oil and gas wells. Annelated pyridines are generally prepared by the condensation of cyclic ketones with an appropriate nitrogen containing species to build up the pyridine ring.Krishna Mohan et al. reported102 the synthesis of a set of diannelated pyridines using cyclic ketones such as cyclopentanone, cyclohexanone, cycloheptanone and cyclooctanone, aliphatic aldehydes such as formaldehyde, acetaldehyde, propionaldehyde, butyraldehyde and valeraldehyde and ammonia (25% aqueous) as reactants and zeolites as catalysts (Scheme 15).
 | ||
| Scheme 15 Synthesis of annelated pyridines using zeolite catalysts under high pressure. | ||
In the reaction of cyclohexanone, formaldehyde and ammonia, the products are 1,2,3,4,7,8,9,10-octahydrophenanthridine (OHP) (selectivity 32–61%) and 1,2,3,4,5,6,7,8-octahydroacridine (OHA) (selectivity 8–42%) using the H-beta zeolite as catalyst. The effect of temperature has a strong influence on selectivity to OHA and OHP, high temperature favoured the formation of OHA, whereas low temperature favoured OHP. The same reaction was carried out under atmospheric pressure and the selectivity to OHP and OHA is <10% at 150 °C. In the similar way (i) 1,2,3,4,5,8,9,10,11,12-decahydrodicyclohepta[b,d] pyridine (55%) and 1,2,3,4,5,7,8,9,10,11-decahydrodicyclohepta[b,e] pyridine (25%) using cycloheptanone, formaldehyde and ammonia (ii) 1,2,3,4,5,6,9,10,11,12,13,14-dodecahydrodicycloocta[b,d] pyridine (29%) and 1,2,3,4,5,6,8,9,10,11,12,13-dodecahydrodicycloocta[b,e] pyridine (60%) using cyclooctanone, formaldehyde and ammonia (iii) 1,2,3,6,7,8-hexahydrodicyclopenta[b,d] pyridine (51%) and 1,2,3,5,6,7-hexahydrodicyclopenta[b,e] pyridine (48%) using cyclopentanone, formaldehyde and ammonia were synthesized.
The same strategy has been extended to different aldehydes. Interestingly, from acetaldehyde to valeraldehyde the products were only 6-substituted OHP and did not yield their corresponding 9-substituted octahydroacridines. This is due to the restriction of the transition state by the zeolite channels. This means the transition state for the reactions which can yield 9-substituted octahydroacridines was too large to be accommodated within the pores of the zeolites used in this reaction. Thus, only 6-substituted octahydrophenanthridines were selectively formed inside the zeolite.
17. Synthesis of calixpyrroles and porphyrins
Calix[4]pyrroles and calix[4]arenes have attracted much attention because of their properties as binders of anionic species, transition metals and neutral substrates. Selective synthesis of these calix[4]pyrroles in high yields by an environmentally benign process is an important topic of interest. Radha Kishan et al. studied the synthesis of calix[4]pyrroles and calix[4]arenes over zeolites as catalysts.103 Nevertheless, due to their small pore size, they have limited use in the synthesis of macrocycles. However, these molecules were synthesized by using one dimensional molecular sieves like Al-MCM-41. In the reaction of cyclohexanone with pyrrole, tetraspirocyclohexyl calix[4]pyrrole (TCP) was obtained with high yield and selectivity. Along with TCP, the acyclic oligomers, linear dimer, trimer and tetramer (4a, 5 and 6, Fig. 1) were also formed. The high reactivity and selectivity of TCP (ca. 14.20 Å) in Al-MCM-41 are attributed to the large pore size (ca. 30 Å) and high surface area (980 m2 g−1). While very low yield and selectivity were found with HY (7.5 Å), HZSM-5(30) (5.6 Å) and SAPO-5 (7.3 Å) zeolites, due to their small pore size than the product TCP (Scheme 16).![Calix[4]pyrroles and linear dimers and monomers.](/image/article/2012/CY/c2cy00162d/c2cy00162d-f1.gif) | ||
| Fig. 1 Calix[4]pyrroles and linear dimers and monomers. | ||
![Synthesis of tetraspirocyclohexyl calix[4]pyrrole over MCM-41 and HY catalysts.](/image/article/2012/CY/c2cy00162d/c2cy00162d-s16.gif) | ||
| Scheme 16 Synthesis of tetraspirocyclohexyl calix[4]pyrrole over MCM-41 and HY catalysts. | ||
The synthesis of substituted calix[4]pyrroles was also carried out by condensation of different cyclic ketones with pyrrole over Al-MCM-41, the conversion of pyrrole was in between 60 and 95% and cyclic tetramer was in between 3 and 70% (Fig. 1). With cyclopentanone, the reaction afforded tetraspirocyclopentyl calix[4]pyrrole 1b. While the ring size is increased, the reactivity and selectivity are decreased. This might be because the increased ring size may have some electronic or steric effects on the reactivity. However, when cycloheptanone and cyclooctanone were used, initially, 4b and 4c dimers were only formed after 10 h. But after continuation of the reaction for 3 to 5 days the respective cyclic tetramers tetraspirocycloheptyl calix[4]pyrrole 1c and tetraspirocyclooctyl calix[4]pyrrole 1d were formed. Whereas, in the case of 2-methyl cyclohexanone tetraspiro(2-methylcyclohexyl) calix[4]pyrrole 2 was obtained in very low yield and in the case of acetone, octamethyl calix[4]pyrrole 8 was obtained in high yields. By increasing the chain length of the ketones, i.e. by using methyl ethyl ketone and pentan-3-one, tetramethyltetraethyl calix[4]pyrrole 10 and octaethyl calix[4]pyrrole 11 were obtained after 3 and 5 days respectively. The reason for long reaction time could also be due to the same factors as observed in cyclic ketones of higher ring size. Radha Kishan et al.104 also reported the synthesis of calix[4]pyrroles and porphyrins over zeolites under microwave irradiation.
18. Synthesis of isoquinoline, Beckmann rearrangement of E,E-cinnamaldoxime on rare earth exchanged (Ce3+, La3+, and Re3+) HFAU-Y zeolites
Isoquinoline derivatives, particularly 5-substituted isoquinoline-1-ones and 3,4-dihydro-5-[4-(1-piperidinyl)-butoxy]-1(2H)-isoquinolinone (DPQ), are potent inhibitors of poly(ADP-ribose) polymerase.105,106 The previous one has potential therapeutic applications in several diseases, like myocardial infarction, cancer, rheumatoid arthritis, diabetes, stroke, hemorrhagic shock, and retro-viral infections.107–112 These alkaloids are potential anti-tumor agents. Dihydroquinolinium salts are extensively used as potential catalysts for the synthesis of optically active epoxides in asymmetric synthesis.113 Bischler–Napieralski cyclization is one of the best routes for the synthesis of isoquinoline. In this reaction, the amide derived from a substituted phenyl ethylamine was cyclized under dehydrative acidic conditions. Industrially used dehydrating agents include P2O5, PCl5 and POCl3 in boiling xylene or decaline.114 Thomas et al. reported115 a vapour phase Beckmann rearrangement of cinnamaldoxime to isoquinoline over rare earth metal modified HY zeolites and also reported the same reaction over various zeolite catalysts such as ZSM-5, H-beta, H-mordenite and K10 montmorillonite clay, etc.116E,E-Cinnamaldoxime (I) under ambient reaction conditions on zeolites and alumina undergoes E–Z isomerisation followed by Beckmann rearrangement leading to the formation of isoquinoline (II) as a major product and dehydration of cinnamaldoxime affording cinnamonitrile as a minor product (Scheme 17). | ||
| Scheme 17 Synthesis of isoquinoline over zeolite catalysts. | ||
The reaction mechanism is depicted in Scheme XVI (ESI†). All the rare earth exchanged catalysts consistently showed a high value of acidity in the weak and medium acidic regions. Rare earth metal exchanged zeolites with Brønsted acidic centers are prepared by the hydrolysis of rare earth metal cations at the sodalite and supercages.117 Low acidic site strength of LaHY could be due to the presence of a large number of La3+ ions in the sodalite cages than Ce3+ in the case of the CeHY zeolite.118,119 The formation of inaccessible Brønsted acid sites in the sodalite cages is due to the migration of La3+ from supercages in the electrostatic repulsive field of residual cations upon heat treatment.119,120 Brønsted acidic sites can be formed in zeolites containing multivalent cations by thermal removal of water initially present in the pores. A water molecule dissociates and the proton can be formed, with a negatively charged oxygen framework giving the bridging hydroxyl groups, which is a catalytically active Brønsted acidic site. These sites that are formed in the sodalite cages are non-accessible acid sites. According to the Hirschler–Plank mechanism, cation migration influences the number of cations that are available for the formation of accessible Brønsted acidic sites during the thermal treatment.121 All catalysts studied showed very high activity and selectivity for the formation of desired product isoquinoline. Cinnamonitrile and cinnamaldehyde were the main by-products.
However, zeolites showed better results compared to SiO2. The HY zeolite and its rare earth metal modified counterparts are highly selective for the production of isoquinoline with conversion >97.5% and yield of isoquinoline >86%. The lanthanum modified HY zeolite afforded a maximum amount of the desired product (yield 95.6%). An optimum number of acidic sites on HY and other zeolite catalysts were expected to work well in effecting the intramolecular cyclization of the intermediate compound ultimately converting to isoquinoline. The mordenite zeolite was active than any other zeolite (yield 92.3%) for this reaction. However, SiO2 afforded only 73.9% of the desired product with a conversion of 55%. Silica, with its weak acidic nature, is inefficient in effecting the E–Z isomerization of E,E-cinnamaldoxime, which is the most crucial step in the formation of desired product.
19. Synthesis of 2-substituted indoles by heteroannulation with Pd–NaY zeolite catalysts
The indole nucleus is found in many natural products and useful in pharmaceutical agents and material polymers.122 Due to its interesting chemical properties a lot of chemists were inspired to design and synthesize a variety of indole derivatives.123 However, many synthetic methods have been developed for indoles, in particular palladium-mediated indole synthesis is one of the most interesting research fields.124–126 Specifically, the palladium-catalyzed annulation of o-haloanilines with alkynes has received much attention, as it is very a convenient method to synthesize 2- or 2,3-substituted indoles.127–136 Syntheses of 2-substituted indoles by palladium-catalyzed heteroannulation over Pd–NaY,137 Pd-SBA-15,138,139 and SO3H-MCM-41140 zeolite catalysts were reported by Hong et al., Batail et al. and Sheng et al. respectively (Scheme 18). | ||
| Scheme 18 Synthesis of substituted indoles over modified zeolites. | ||
Pd modified zeolites were prepared according to reported methods141–143 by an ion exchanged NaY zeolite. These Pd containing zeolites have SBET of 765 cm2 g−1 and an average pore diameter of 14.6 Å. The reaction was studied over various halide sources, bases, temperatures and over Pd-zeolite catalysts. The reaction afforded 2-phenylindole as the major product instead of N-acetyl-2-phenylindole. The deprotection of the acetyl group in the indole is expected due to the acidic nature of the zeolite. Indole was obtained in 80% yield when Cs2CO3 was used as base. Whereas, the mixture of desired indole and the Sonogashira coupling products were obtained using other carbonate or acetate bases. Indole was obtained using LiCl, 1.0 wt% Pd(II)–NaY and Cs2CO3 at 140 °C. But at the same time at 120 °C, the same reaction did not yield any of the desired products. The catalytic activity of the Pd modified zeolites depends on the palladium concentration and palladium species. The heteroannulation reaction using the 1 wt% Pd(II)–NaY catalyst showed good catalytic activity.
The reactions using N-acetyl o-iodoanilide with alkyl, aryl and hetero aryl substituted terminal alkynes afforded 2-substituted indoles in good to moderate yields along with deprotection of the N-acetyl group. The reaction using acetylene also gave indole after purging the acetylene into the reaction mixture. The reactions using N-Bn or N-Ts 2-iodoaniline with a terminal alkyne afforded N-protected 2-substituted indoles in moderate yields.
Tryptophols are a kind of indoles bearing a C-3 hydroxyethyl side chain and are pharmaceutically important compounds. Several methods were available to synthesize the tryptophols, such as Fischer indole methodology,144 the reduction of indole acetic acid derivatives,145 or of 3-substituted-dioxindoles,146 the ring opening of epoxides by indole compounds under the catalysis of Bi(OTf)3147 or by the reaction with epoxides under pressure in the presence of Yb(OTf)3148 and the domino reaction of aryl hydrazines and silyl-protected ω-(hydroxyoalkyl) alkynes.149
Sheng et al. reported the synthesis of tryptophols over sulfonic acid functionalized MCM-41 catalysts.150 6H-Indolo[2,3-b]quinolines were synthesized over RuY zeolite catalysts151 and these catalysts afforded 65% product.
20. Synthesis of bis(indolyl)methanes via an electrophilic substitution reaction of indoles over zeolite catalysts
Diindolylmethane (DIM) (or bis(indolyl)methane) is the most active substance for promoting favorable estrogen metabolism in women and men.152 DIM increases the body's natural metabolism of hormones and promotes good estrogen (2-hydroxyestrogen).153 This indole antioxidant has improved symptoms of fibromyalgia.154 DIM can prevent cancer due to its ability to modulate certain cancer causing estrogen metabolites.155 Moreover DIM induces apoptosis in human cancer cells.156 DIM is demonstrated to have significant physiological activity157and also useful applications as breast cancer preventative.158 Karthik et al. reported159 that zeolite catalyzed synthesis of bis(indolyl)methanes from indole with aromatic aldehydes at room temperature afforded good to excellent yields. When indole was reacted with benzaldehyde or substituted benzaldehydes over zeolite catalysts in dichloromethane as a solvent, bis(indolyl)methanes were formed in high yields. The electrophilic substitution reaction of indole with aromatic aldehydes proceeded easily at room temperature to produce the corresponding bis(indolyl)methanes in excellent yields. The reaction is depicted in Scheme 19. | ||
| Scheme 19 Synthesis of bis(indolyl)phenylmethane over zeolite catalysts. | ||
The yield of bis(indolyl)phenylmethane increases in the order of HZSM-5 < Hb < HY, due to the increasing acid site density of the catalysts in the same order. The Zn2+ ion exchanged Y zeolite showed better activity than the unmodified HY zeolite due to its higher Lewis acidity. The yield of the product increases with decrease in particle size of the zeolites, H-ZSM-5 (3.1 μm) > Hb (1 μm) > HY (0.5 μm). The number of external surface acidic sites was reduced with increase in the crystal size of the zeolite.160–162 The yield of bis(indolyl)phenylmethane was high over zeolites with a lower Si/Al ratio and smaller particle size, since these zeolites have more density of external acidic sites. The product might be formed on the external surface of the zeolite due to product's bulky nature (dimensions: 1.0622 × 1.0875 × 1.1167 nm). In order to support this concept, the reaction was carried out on passivated HY (SPHY) and H-beta (SPH-beta) zeolites. The yield of bis(indolyl)phenylmethane was drastically suppressed on the surface passivated zeolites. It is assumed that all acidic sites on the external surface were passivated by the amorphous silica layer.
Karthik et al.162 and Ma et al.163 reported the zeolite catalyzed electrophilic substitutions of indole and substituted indole with various aldehydes and ketones, affording excellent yields of bis(indolyl)methanes at room temperature. This process offers an easy access to the naturally occurring and bioactive bis(indolyl)methanes. The first successful synthesis of Vibrindole A (Scheme 20)164–167 over environmentally benign zeolite catalysts was reported by these authors.
 | ||
| Scheme 20 Synthesis of Vibrindole A over zeolite catalysts. | ||
21. Conversion of γ-butyrolactone into N-methyl-2-pyrrolidone over modified zeolites
N-Methyl-2-pyrrolidone (NMP) is one of the important chemicals in the industry due to its use in many chemical reactions, thermal stability, nontoxicity, and low viscosity. NMP is also used for the preparation of semiconductors. Moreover, NMP is used as a solvent in membrane processing and organic synthesis, like polymerization of functional resins such as polyphenylene sulfide, polyimide, polyetherketone and aramide. In general, NMP has been synthesized by a noncatalytic reaction of γ-butyrolactone (GBL) and methylamine under high pressure between 50 and 120 bar.168–170 NMP could also be produced from the catalytic hydrogenation of N-(α-hydroxyalkyl)-2-pyrrolidone171 and from the catalytic hydrogenation of maleic anhydride, maleic acid and/or fumaric acid in the presence of ammonia.172 Hatada et al.173 reported the synthesis 1-propyl-2-pyrrolidone with GBL and polyamine over different metal ion exchanged Y-zeolites at 260 °C and atmospheric pressure. The yields of pyrrolidone obtained were 62% and 34% over the Ca and Cu exchanged Y zeolites respectively. In another example, the yield of NMP is 56% with the reaction of GBL and 40% aqueous methylamine solution, over a Cu ion exchanged Y zeolite at 280 °C. They also reported the results of the ring transformation of GBL with ammonia to 2-pyrrolidinone over various metal ion exchanged Y zeolites.Akzo Nobel published US patent on the two step synthesis of NMP from maleic anhydride.174 The first step was the catalytic reduction of maleic anhydride with Cu chromite as a catalyst, whereas in the second step methylamine and water were introduced into the product of the first step over the modified zeolite X or Y as a catalyst at 275 °C and near atmospheric pressure. They also mentioned that NMP yield was about 96% and could be recovered in high purity by distillation. Yoon et al.175 reported the possibility of using zeolites in the one step ring conversion of GBL with methylamine to NMP in high yield (Scheme 21).
 | ||
| Scheme 21 Synthesis of N-methyl-2-pyrrolidone over zeolites. | ||
The major side product besides NMP was N-methyl-2-hydroxy butyric acid amide, which was considered as an intermediate in the transformation of GBL with methylamine into NMP.176 Surprisingly, in the feed container itself GBL reacted rapidly with 40% aqueous methylamine to afford N-methyl-2-hydroxy butyric acid amide with conversion higher than 80% at room temperature and atmospheric pressure. Nonetheless, the formation of NMP was not observed under similar conditions. When the reaction temperature was increased, a small amount of NMP was observed. Whereas, when a catalyst was used, the selectivity to NMP increased significantly except in the case of Na-mordenite at 260 °C. In particular the H-beta catalyst was very active and selective for the formation of NMP at 215 °C. In contrast, NaX and NaY zeolites were very active and selective only when the reaction was carried out as in the literature.177,178
22. Synthesis of 2-methylquinoxaline from 1,2-phenylenediamine and 1,2-propanediol over modified HY zeolites
2-Methylquinoxaline (2-MQ) and its derivatives have very significant importance as intermediates in the synthesis of biologically active antimicrobial compounds and germicides. Many substituted 2-methylquinoxalines have also shown fungicidal and antiviral activity.179 2-MQ N,N-dioxides substituted in the 3rd position (e.g. with amidino, amide, hydrozino, carbonyl and ester group) are effective bactericides. Antibiotics of the triosin and quinomycin series have been shown to contain a quinoxaline-2-carboxylic acid residue.180 Recently, a number of catalysts have been reported for the synthesis of quinoxalines. These include the bi-catalyzed oxidative coupling,181 RuCl2-(PPh3)3-TEMPO,182 MnO2,183 POCl3,184 CuSO4·5H2O,185 Zn[(L)proline] in HOAc,186 task-specific ionic liquid,187 montmorillonite K10188 and Ni-nanoparticles.189 Synthesis of quinoxaline derivatives by the condensation of 1,2-diamine with 1,2-dicarbonyl compounds using binary metal oxides supported on Si-MCM-41 mesoporous molecular sieves was reported by Ajaikumar and Pandurangan.190 Best yield (97%) of the product was obtained with the use of 17% ZrO2/4% Ga2O3/MCM-41 catalyst within 2 h, whereas under these conditions, the catalyst support does not show any significant catalytic activity (25% after 12 h) in the quinoxaline synthesis. However, its catalytic activity is increased drastically after impregnation with an appropriate wt% of ZrO2, Al2O3, Ga2O3, In2O3, and La2O3. Among these catalysts, ZrO2/MCM-41 showed lower activity compared with those of ZrO2/MxOy/MCM-41 (where M = Al, Ga, In and La) catalysts. The activity of these catalysts is in the following order 17%ZrO2/4%Ga2O3/MCM-41 > 17%ZrO2/4%La2O3/MCM-41 ≈ 17%ZrO2/4%Al2O3/MCM-41 > 17% ZrO2/4%In2O3/MCM-41 > 17%ZrO2/MCM-41. The presence of Al, Ga, In and La ions or the presence of small aggregates of Al2O3, Ga2O3, In2O3 and La2O3 in interaction with the support generates Lewis acidity to the surfaces, which are expected to play important roles in the condensation reaction. It is interesting to note that the order for the condensation activity of the supported metal oxide catalysts (ZrO2/Ga2O3 > ZrO2/Al2O3 > ZrO2/La2O3 > ZrO2/In2O3-ZrO2) is quite similar to that for the acid strength of the catalysts. This indicates a close relationship between the acid strength and the catalytic activity of the supported metal oxides. The vapour phase synthesis of 2-MQ is reported by Venu Gopal and Subrahmanyam191 over zeolite catalysts from 1,2-phenylenediamine and 1,2-propanediol (Scheme 22).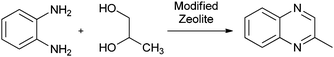 | ||
| Scheme 22 Synthesis of 2-methylquinoxaline over zeolites. | ||
The cyclization reaction of 1,2-phenylenediamine and 1,2-propanediol for the synthesis of 2-methylquinoxaline was performed over various zeolites such as HY, LaHY, PbHY, CuHY and CrHY zeolites. The catalytic activity towards the yield of 2-MQ is in the order of PbHY > MnHY > CrHY > LaHY > HY > CuHY. The medium acidic Mn and Pb-modified HY zeolite catalysts have shown high yield of 2-MQ. The yield of 2-MQ 77.0% and 93.2% conversion of 1,2-phenylenediamine were obtained over a 3 wt% PbHY zeolite. The low yield of 2-MQ over an La-modified HY zeolite is because of its high acidity, which resulted in the formation of side products like polyalkyl quinoxalines. A decreased yield of 2-MQ over CuHY may be due to the reductive nature of Cu ions, which causes the formation of dihydro and tetrahydro-2-MQ.
23. Zeolite beta catalyzed vapour phase synthesis of 2-methyl and 4-methylquinoline
In recent years several new methods have been reported for synthesis of 2,3-substituted quinolines in the liquid phase using transition metal-based Lewis acid catalysts, such as palladium, ruthenium, rhodium and iron complexes and aldehydes or alcohols in the presence of CO.192–195 A number of attempts to synthesize quinolines in the vapour phase were also successful.196–200 The gas-phase quinoline synthesis was reported by McAteer et al.,196 Ln(OTf)3 and Sc(OTf)3 were used to promote three component coupling reactions between aldehydes, amines, and dienes or alkenes. These Lewis acid catalysts were active in imine formation and successive imino Diels–Alder type reactions. High yields of poly-substituted tetrahydroquinolines were reported.201,202 Aniline reacted with crotonaldehyde over an aluminosilicate catalyst at 450 °C to yield mainly 4-methylquinoline with 2-methylquinoline as only a minor by-product (Scheme 23).203 Campanati et al. found that the reaction of 2-ethylaniline with ethylene glycol over an acid-treated commercial K10-montmorillonite catalyst yielded mainly 2-methyl-8-ethylquinoline.199 | ||
| Scheme 23 Synthesis of 2MEQ and 4MEQ over zeolites. | ||
Brosius et al. reported the synthesis of methyl substituted quinolines using aniline and acetaldehyde over zeolite catalysts. The reaction between molecular fluorine and H-ZSM-5 at room temperature afforded a nonframework fluorinated species, which are weakly attached to the lattice.200 Brosius et al. obtained octahedral Al3+ species upon NH4F impregnation. NH4F impregnation followed by calcination at temperatures above 500 °C on H-ZSM-5 leads to the formation of Si–F bonds and dealumination causing formation of extra-framework aluminium species.204,205
24. Synthesis of 3-isopropylindole over modified HY zeolites
Indole and its derivatives are widely used as intermediates in the synthesis of anti-inflammatory agents, dyes, and antibiotics.206–208 They also have biological activity as an antidiuretic, hypertensive, stimulant, muscle relaxant, inhibits the respiration in cancer cells, heart stimulant, tuberculostat and also plant growth regulators. N-Alkylation of indole with n-propanol and isopropanol in the presence of aluminum alkoxide and Raney Nickel catalyst was reported by Botta et al.209N-Alkylation of indole with benzyl halide in the presence of NaOH and over the catalyst Bu4NHSO4 under phase transfer conditions was reported by Bourak and Gallo.210 Venu Gopal and Subrahmanyam reported211 the vapour phase alkylation of indole with 2-propanol and methanol over modified zeolite molecular sieves (Scheme 24). | ||
| Scheme 24 Synthesis of 3-isopropylindole over zeolites. | ||
They used 1:6 mole ratio of indole and 2-propanol. The excess 2-propanol was used because of the formation of side product. Also because of the fact that the indole system contains π-electrons and their activation is easier than 2-propanol, excess 2-propanol (alkylating agent) is required. The acidic strength of the modified zeolites is in the order of LaHY ≥ CeHY > HY > CrHY ≥ CuHY > HZSM-5(30). The catalytic activity with respect to the yield of 3-isopropyl indole is in the order of HZSM-5(30) < CuHY < HY < CrHY < LaHY < CeHY. The low activity of HZSM-5(30) than HY is due to the lower pore size. This might be due to diffusion restriction of 3-isopropylindole through the pores of HZSM-5(30). Whereas, in the case of vapor phase alkylation of indole with methanol, La and Ce modified HY zeolite catalysts afforded high yield compared to other catalysts studied. The maximum yield (33.6%) of 3-methyl indole was obtained over a 3 wt% CeHY zeolite catalyst at 300 °C. It was also found that the catalytic activity increased with the increase of total acidity of the catalyst.
25. Beckmann rearrangement of cyclohexanone oxime over mesoporous SiMCM-41 and AlMCM-41 catalysts
Caprolactam is manufactured by Beckmann rearrangement of cyclohexanone oxime with oleum or sulfuric acid as a reaction medium and cyclohexanone oxime is produced by the reaction between cyclohexanone and hydroxylamine (only one exception is the Toray PNC process). The Dutch State Mines (DSM) industry has developed the hydroxylammonium phosphate-oxime (HPO) process to make oxime without co-production of ammonium sulfate, in which hydroxylamine was used in its phosphate form. Caprolactam is the monomer of Nylon-6 fibers and resins and liquid phase Beckmann rearrangement of cyclohexanone oxime is carried out industrially over concentrated sulfuric acid as the catalyst. Even though the process is highly selective, it has some disadvantages like the production of large amounts of low value ammonium sulfate, corrosion and environmental pollution caused by the use of fuming sulfuric acid. To overcome these problems, many researchers have attempted to carry out vapour phase Beckmann rearrangement using solid acid catalysts like mordenite,212,213 silica–alumina,214 zeolite Y,212 HNaY,215 TS-1,216 ZSM-11, ZSM-5,217 TS-2,218 MCM-22,219 silicoaluminophosphate,220 WOx/SBA-15,221 Nb-MCM-41,222 TS-1223 and H-beta.224,225 Chaudhari et al.226 reported the rearrangement of cyclohexanone oxime to ε-caprolactam over Si-MCM-41 and H-Al-MCM-41 with different Si/Al ratios (Scheme 25). | ||
| Scheme 25 Synthesis of caprolactam over zeolites from cyclohexanone oxime. | ||
The major product was ε-caprolactam. The conversion of cyclohexanone oxime was in between 70 and 100% and showed consistent results with time on stream up to 5 h over all the catalysts at all the temperatures between 300–390 °C. The transformation of the oxime was rapid and attained almost 100% at temperatures >350 °C. The decrease in selectivity at higher temperatures was due to the decomposition of the ε-caprolactam on the surface of catalyst and increase of side reactions.227 The selectivity to ε-caprolactam was more with Al-MCM-41 catalysts than Si-MCM-41. The selectivity was increased with increase in the Al content. The best yield (87.6%) of ε-caprolactam was obtained over the catalysts with a Si/Al ratio of 14. It seems that the silanol groups present on Si-MCM-41 were not sufficiently acidic to catalyse the rearrangement.
26. Conclusions
The present review is intended to demonstrate the possibilities in the synthesis of N-heterocyclic compounds in which zeolite catalysts can be employed. It also illustrates the special features afforded by these shape selective acid catalysts, such as thermal stability, controlled variability, reusability and eco-friendly nature, because of which these catalysts are most sought after in green chemistry, and emphasizes the future importance of such catalyst systems. Numerous economically interesting organic reactions, which were hitherto not feasible because of low activity, selectivity and life of the catalyst, can now be commercially exploited using zeolites. It has now become easier to change from homogeneous catalyzed processes to heterogeneous catalysis in order to improve environmental and economical benefits.Acknowledgements
The authors thank Council of Scientific and Industrial Research (CSIR), India, and V. V. Krishna Mohan Kandepi would like to thank Prof. Beatriz Royo, Instituto de Technologia Química e Biológica da Universidade Nova de Lisboa, Portugal, for her consistent and invaluable cooperation during his postdoctoral period.References
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30. By permission of Oxford University Press Search PubMed.
- D. W. Breck, Zeolite Molecular Sieves, Wiley, New York, 1974 Search PubMed; A. Dyer, An Introduction to Zeolite Molecular Sieves, John Wiley & Sons, Chichester, 1988 Search PubMed.
- M. W. Anderson and J. Klinowski, Nature, 1989, 339, 200 CrossRef CAS.
- N. Oku, S. Matsunaga and N. Fusetani, J. Am. Chem. Soc., 2003, 125, 2044 CrossRef CAS.
- Z. S. Song, M. Zhao, R. Desmond, P. Devine, D. M. Tschaen, R. Tillyer, L. Frey, R. Heid, F. Xu, B. Foster, J. Li, R. Reamer, R. Volante, E. J. Grabowski, U. H. Dolling and P. J. Reider, J. Org. Chem., 1999, 64, 9658 CrossRef CAS.
- A.-H. Li, S. Moro, N. Forsyth, N. Melman, X.-D. Ji and K. A. Jacobsen, J. Med. Chem., 1999, 42, 706 CrossRef CAS.
- B. Vacher, B. Bonnaud, F. Funes, N. Jubault, W. Koek, M.-B. Assie, C. Cosi and M. Kleven, J. Med. Chem., 1999, 42, 1648 CrossRef CAS.
- D. Craig and G. D. Henry, Tetrahedron Lett., 2005, 46, 2559 CrossRef CAS.
- F. Palacios, C. Alonso, G. Rubiales and M. Villegas, Tetrahedron, 2005, 61, 2779 CrossRef CAS.
- M. C. Bagley, C. Glover and D. Chevis, Synlett, 2005, 649 CrossRef CAS.
- A. Robin, K. Julienne, J. C. Meslin and D. Deniauld, Tetrahedron Lett., 2004, 45, 9557 CrossRef CAS.
- L. Ghosez, E. Jnoff, P. Bayard, F. Sainte and R. Beaudegnies, Tetrahedron, 1999, 55, 3387 CrossRef CAS.
- H. Bönnemann and W. Brijoux, Adv. Heterocycl. Chem., 1990, 48, 177 CrossRef.
- I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS.
- G. Zeni and R. C. Larock, Chem. Rev., 2004, 104, 2285 CrossRef CAS.
- U. Kameshwari, C. S. Swamy and C. N. Pillai, Stud. Surf. Sci. Catal., 1994, 84, 1959 CrossRef.
- S. E. Golunski and D. Jackson, Appl. Catal., 1986, 23, 1 CrossRef CAS and references cited therein.
- I. M. Liepinya, M. K. Sile, A. A. Avots and I. Ya. Lazdinsh, React. Kinet. Catal. Lett., 1984, 26, 123 CrossRef CAS.
- E. F. V. Scriven, J. E. Toomey Jr and R. Murugan, Pyridine and pyridine derivatives, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. J. I. Kroschwitz and M. Howe-Grant, John Wiley, New York, 4th edn, 1996, vol. 20, pp. 641–679 Search PubMed.
- M. P. Malveda with T. Kälin and A. Kishi, Chemical Economic Handbook, SIR consulting Today-News letter, February-2011.
- S. J. Kulkami and M. Subrahmanyam, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1991, 30, 1041 Search PubMed.
- A. V. Rama Rao, S. J. Kulkarni, R. Ramachandra Rao and M. Subrahmanyam, Appl. Catal., A, 1994, 111, L101 CrossRef CAS.
- F. J. Van der Gaag, F. Louter, J. C. Oudejans and H. van Bekkum, Appl. Catal., 1986, 26, 191 CrossRef CAS.
- T. Miyake, K. Noguchi and K. Imamura, British Patent, 1579473, 1980 Search PubMed.
- Y. Minato and S. Yasuda, U. S. Patent, 3932421, 1976 Search PubMed.
- D. Feitler, W. Schimming and H. Wetshtein, U. S. Patent, 4675410, 1987 Search PubMed.
- Y. Kusunoki, H. Okazaki, Y. Sato and E. Sano, U. S. Patent, 3781292, 1973 Search PubMed.
- T. Matsumoto and A. Tamano, U. S. Patent, 3417092, 1968 Search PubMed.
- C. W. Hargis, U. S. Patent, 3829428, 1974 Search PubMed.
- Y. Liu, H. Yang, F. Ji, Y. Zhang and Y. Li, Chem. Eng. J., 2008, 136, 282 CrossRef CAS.
- R. Ramachandra Rao, S. J. Kulkarni, M. Subrahmanyam and A. V. Rama Rao, 11th National Symposium on Catalysis, Hyderabad, India, 1993 Search PubMed.
- K. S. K. Reddy, I. Sreedhar and K. V. Raghavan, Appl. Catal., A, 2008, 339, 15 CrossRef CAS.
- S. J. Kulkarni, R. Ramachandra Rao, M. Subrahmanyanm and A. V. Rama Rao, Appl. Catal., A, 1994, 113, 1 CrossRef CAS.
- K. S. K. Reddy, I. Sreedhar and K. V. Raghavan, Can. J. Chem. Eng., 2011, 89, 854 CrossRef CAS.
- K. S. K. Reddy, I. Sreedhar and K. V. Raghavan, Appl. Catal., A, 2008, 339, 15 CrossRef CAS.
- K. S. K. Reddy, I. Sreedhar, S. Venkateshwar and K. V. Raghavan, Catal. Lett., 2008, 125, 110 CrossRef CAS.
- K. S. K. Reddy, I. Sreedhar, K. V. Raghavan, S. J. Kulkarni and M. Ramakrishna, Int. J. Chem. React. Eng., 2008, 6, A29 Search PubMed.
- S. Yasuda and N. Abe, Jpn. Kokai Tokkyo Koho, 1986, 61, 265 Search PubMed.
- F. J. Van der Gaag, F. Louter, J. C. Oudejans and H. van Bekkum, Appl. Catal., 1986, 26, 191 CrossRef CAS.
- S. J. Kulkarni, R. Ramachandra Rao, Y. V. Subba Rao, M. Subrahmanyam and A. V. Rama Rao, Appl. Catal., A, 1996, 136, L1 CrossRef CAS.
- R. Ramachandra Rao, N. Srinivas, S. J. Kulkarni, M. Subrahmanyam and K. V. Raghavan, Appl. Catal., A, 1997, 161, L37 CrossRef CAS.
- Y. Wakatsuki and H. Yamazaki, Synthesis, 1976, 26 CrossRef CAS.
- T. S. Sirlibaev, V. G. Kalyadin and A. Akramkhodzaev, Uzb. Khim. Zh., 1989, 6, 40 Search PubMed.
- N. Srinivas, V. Radha Rani, S. J. Kulkarni and K. V. Raghavan, J. Catal., 2002, 208, 332 CrossRef CAS.
- N. Srinivas, S. J. Kulkarni and K. V. Raghavan, Catal. Commun., 2002, 3, 521 CrossRef CAS.
- H. Sato, S. Shimizu, N. Abe and K. Hirose, Stud. Surf. Sci. Catal., 1994, 84, 1951 CrossRef CAS.
- A. E. Chichibabin, Bull. Soc. Chim. Fr., 1937, 4, 1831 Search PubMed; R. Graf and W. Langer, J. Prakt. Chem., 1938, 150, 153 CrossRef CAS.
- R. L. Frank, J. R. Blegen, R. J. Dearborn, R. L. Myers and F. E. Woodward, J. Am. Chem. Soc., 1946, 68, 1368 CrossRef CAS.
- K. V. V. Krishna Mohan, K. S. K. Reddy, N. Narender and S. J. Kulkarni, J. Mol. Catal. A: Chem., 2009, 298, 99 CrossRef.
- N. Narender, P. Srinivasu, S. J. Kulkarni and K. V. Raghavan, J. Catal., 2001, 202, 430 CrossRef CAS.
- C. H. McAteer and E. F. V. Scriven, in Heterocyclic Synthesis in Fine Chemical through Heterogeneous Catalysis, Wiley-VCH, Weinheim, 2001, vol. 275 Search PubMed.
- J. R. Calvin, R. D. Davis and C. H. McAteer, Appl. Catal., A, 2005, 285, 1 CrossRef CAS.
- N. Narender, K. S. K. Reddy, K. V. V. Krishna Mohan and S. J. Kulkarni, Catal. Lett., 2009, 130, 367 CrossRef CAS.
- Ullman's Encyclopedia of Industrial Chemistry, VCH, Weinheim, 1993, vol. A22 Search PubMed.
- R. Ramachandra Rao, S. J. Kulkarni, M. Subrahmanyam and A. V. Rama Rao, Appl. Catal., A, 1996, 142, L191 CrossRef CAS.
- E. P. Popodopoulos, A. Jarrar and C. H. Issidorides, J. Org. Chem., 1996, 31, 615 CrossRef.
- M. Subrahmanyam and A. R. Prasad, Appl. Catal., 1990, 65, L5 CrossRef CAS.
- G. Kamalakar, S. J. Kulkarni and K. V. Raghavan, Microporous Mesoporous Mater., 1999, 29, 283 CrossRef CAS.
- M. F. Siddiqui and A. I. Levey, Drugs Future, 1999, 24, 375 CrossRef.
- Y.-G. Lim, J.-B. Kang and Y. H. Kim, J. Chem. Soc., Perkin Trans. 1, 1996, 2201 RSC.
- Y.-G. Lim, Y. H. Kim and J.-B. Kang, J. Chem. Soc., Chem. Commun., 1994, 2267 RSC.
- S. J. Kulkarni, Stud. Surf. Sci. Catal., 1998, 113, 151 CrossRef CAS.
- S. J. Kulkarni, R. Ramachandra Rao, Y. V. Subba Rao, M. Subrahmanyam and A. V. Rama Rao, Appl. Catal., A, 1996, 136, L1 CrossRef CAS.
- R. Ramachandra Rao, N. Srinivas, S. J. Kulkarni, M. Subrahmanyam and K. V. Raghavan, Appl. Catal., A, 1997, 161, L37 CrossRef CAS.
- S. Shimizu, N. Abe, A. Iguchi, M. Dohba, H. Sato and K. Hirose, Microporous Mesoporous Mater., 1998, 21, 447 CrossRef CAS.
- V. Radha Rani, N. Srinivas, M. Radha Kishan, S. J. Kulkarni and K. V. Raghavan, Green Chem., 2001, 3, 305 RSC.
- D. Venu Gopal, N. Srinivas, B. Srinivas and M. Subrahmanyam, Green Chem., 2001, 3, 65 RSC.
- V. Radha Rani, M. Radha Kishan, N. Srinivas, S. J. Kulkarni and K. V. Raghavan, Catal. Commun., 2005, 6, 71 CrossRef.
- Y. V. Subba Rao, S. J. Kulkarni, M. Subrahmanyam and A. V. Rama Rao, J. Org. Chem., 1994, 59, 3998 CrossRef CAS.
- P. Biginelli, Gazz. Chim. Ital., 1893, 23, 360 Search PubMed.
- G. C. Rovnyak, S. D. Kimall, B. Beyer, G. Cucinotta, J. D. DiMarco, J. Gougoutas, A. Hedberg, M. Malley, J. P. McCarthy, R. Zhang and S. Moreland, J. Med. Chem., 1995, 38, 119 CrossRef CAS.
- K. S. Atwal, G. C. Rovnyak, S. D. Kimball, D. M. Floyd, S. Moreland, B. N. Swanson, J. Z. Gougouta, J. Schwartz, K. M. Smillie and M. F. Malley, J. Med. Chem., 1990, 33, 2629 CrossRef CAS.
- H. Cho, M. Ueda, K. Shima, A. Mizuno, M. Hayashimatsu, Y. Ohnaka, Y. Takeuchi, M. Hamaguchi, K. Aisaka, T. Hidaka, M. Kawai, M. Takeda, T. Ishihara, K. Funahashi, F. Satah, M. Morita and T. Noguchi, J. Med. Chem., 1989, 32, 2399 CrossRef CAS.
- J. J. Baldwin, D. A. Claremon and D. E. McClure, US Pat. 4609494, 1986 Search PubMed.
- J. J. Baldwin, S. M. Ptizenberger and D. E. McClure, US Pat. 4675321, 1987 Search PubMed; K. S. Atwal, US Pat. 4684656, 1987 Search PubMed.
- C. O. Kappe and S. F. Falsone, Synlett, 1998, 718 CrossRef CAS.
- J. Lu, Y. Bai, Z. Wang, B. Yang and H. Ma, Tetrahedron Lett., 2000, 41, 9075 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, R. Srinivas, C. Venugopal and T. Ramalingam, Synthesis, 2001, 1341 CrossRef CAS.
- F. Bigi, S. Carloni, B. Fraullanti, R. Maggi and G. Sarton, Tetrahedron Lett., 1999, 40, 3465 CrossRef CAS.
- D. J. Brown, The Pyrimidines, Wiley, New York, 1962, vol. 12, p. 440 CrossRef CAS; For a review, see: C. O. Kappe, Tetrahedron, 1993, 49, 6937 CrossRef CAS.
- V. Radha Rani, N. Srinivas, M. Radha Kishan, S. J. Kulkarni and K. V. Raghavan, Green Chem., 2001, 3, 305 RSC.
- M. G. Kulkarni, S. W. Chavhan, M. P. Shinde, D. D. Gaikwad, A. S. Borhade, A. P. Dhondge, Y. B. Shaikh, V. B. Ningdale, M. P. Desai and D. R. Birhade, Beilstein J. Org. Chem., 2009, 5(4) DOI:10.3762/bjoc.5.4.
- V. Radha Rani, N. Srinivas, M. Radha Kishan, S. J. Kulkarni and K. V. Raghavan, Green Chem., 2001, 3, 305 RSC.
- K. Segawa, S. Mizuno, M. Sugiura and S. Nakata, Stud. Surf. Sci. Catal., 1996, 101, 267 CrossRef CAS.
- R. A. Budnik and M. R. Sandner, Eur. Pat. 0158319, 1985 Search PubMed.
- W. Weiczorrek, Polym. Paint Colour J., 1988, 178, 827 Search PubMed.
- A. Belhekar, T. K. Das, K. Chaudhari, S. G. Hegde and A. J. Chandwadka, Stud. Surf. Sci. Catal., 1998, 113, 195 CrossRef CAS.
- N. Srinivas, D. Venu Gopal, B. Srinivas, S. J. Kulkarni and M. Subrahmanyam, Microporous Mesoporous Mater., 2002, 51, 43 CrossRef CAS.
- Y. S. Bhat, J. Das, S. Ali, B. D. Bhatt and A. B. Halgeri, Appl. Catal., A, 1996, 148, L1 CrossRef CAS.
- P. R. Reddy, M. Subrahmanyam and S. J. Kulkarni, Catal. Lett., 1998, 54, 95 CrossRef CAS.
- K. Grohe, Chem. Br., 1992, 28, 35 Search PubMed.
- K. Nagaiah, A. Sudhakar Rao, S. J. Kulkarni, M. Subrahmanyam and A. V. Rama Rao, J. Catal., 1994, 147, 349 CrossRef CAS.
- K. Grohe, Chem. Br., 1992, 28, 34 Search PubMed.
- W. Hammersctimidt, A. Baiker, A. Wokaun and W. Pluhr, Appl. Catal., 1986, 20, 305 CrossRef.
- V. Radha Rani, N. Srinivas, S. J. Kulkarni and K. V. Raghavan, J. Mol. Catal. A: Chem., 2002, 187, 237 CrossRef CAS.
- R. Vultier, A. Baiker and A. Wokaun, Appl. Catal., 1987, 30, 167 CrossRef CAS.
- A. Baiker, D. Monti and Y. S. Fan, J. Catal., 1984, 88, 81 CrossRef CAS.
- S. J. Kulkarni, M. Subrahmanyam and A. V. Rama Rao, Indian. J. Chem., Sect. A: Inorg., Phys., Theor. Anal., 1993, 32A, 28 CAS.
- A. V. Rama Rao, S. J. Kulkarni, R. Ramachandra Rao and M. Subrahmanyam, Appl. Catal., A, 1994, 111, L101 CrossRef CAS.
- M. Subrahmanyam, G. Muralidhar, P. K. Varma, K. V. H. Prasad, J. S. Yadav and A. V. Rama Rao, Ind. Patent, 435/DEL/91, 1991 Search PubMed.
- M. Subrahmanyam, G. Muralidhar, S. J. Kulkarni, V. Viswanathan, B. Srinivas, J. S. Yadav and A. V. Rama Rao, Ind. Patent, 281/DEL/92, 1992 Search PubMed.
- K. V. V. Krishna Mohan, N. Narender and S. J. Kulkarni, Microporous Mesoporous Mater., 2007, 106, 229 CrossRef CAS.
- M. Radha Kishan, N. Srinivas, K. V. Raghavan, S. J. Kulkarni, J. A. R. P. Sarma and M. Vairamani, Chem. Commun., 2001, 2226 RSC.
- M. Radha Kishan, V. Radha Rani, M. R. V. S. Murty, P. Sita Devi, S. J. Kulkarni and K. V. Raghavan, J. Mol. Catal. A: Chem., 2004, 223, 263 CrossRef CAS.
- The Chemistry of Heterocyclic Compounds “The Pyrazines”, ed. G. B. Barlin, John Wiley and Sons, New York, 1982 Search PubMed.
- M. Subrahmanyam, S. J. Kulkarni and B. Srinivas, React. Kinet. Catal. Lett., 1993, 49, 455 CrossRef CAS.
- C. Y. Watson, W. J. D. Whish and M. D. Threadgill, Bioorg. Med. Chem., 1998, 6, 721 CrossRef CAS.
- A. W. White, R. Almassy, A. H. Calvert, N. J. Curtin, R. J. Griffin, Z. Hostomsky, K. Maegley, D. R. Newell, S. Srinivasan and B. T. Goldings, J. Med. Chem., 2000, 43, 4048 CrossRef.
- A. Thiemermann, J. Bowes, F. P. Myint and J. R. Vane, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 679 CrossRef.
- F. G. Sariano, L. Virag, P. Jagtab, E. Szabo, J. G. Mabley, L. Liaudet, A. Morton, D. D. Hoyt, K. G. K. Murthy, A. L. Salzman, G. J. Southan and C. Szabo, Nat. Med. (N. Y.), 2001, 7, 108 Search PubMed.
- G. J. Abdelkarim, K. Gertz, C. Harms, J. Katchanov, U. Dirnagal, C. Szabo and M. Endres, Int. J. Mol. Med., 2001, 7, 255 CAS.
- R. W. Pero, B. Axelsson, D. Siemann, D. Chaplin and D. Dougherty, Mol. Cell. Biochem., 1999, 193, 119 CrossRef CAS.
- P. B. Page, G. A. Rassias, D. Barros, D. Bethell and M. B. Schilling, J. Chem. Soc., Perkin Trans. 1, 2000, 3325 RSC.
- W. Herz and L. Tsai, J. Am. Chem. Soc., 1955, 77, 3529 CrossRef CAS; W. M. Whaley and T. R. Govindachari, Org. React., 1951, 6, 151 Search PubMed.
- A. Thomas, S. Prathapan and S. Sugunan, Microporous Mesoporous Mater., 2005, 84, 137 CrossRef.
- B. Thomas, U. R. Prabhu, S. Prathapan and S. Sugunan, Microporous Mesoporous Mater., 2007, 102, 138 CrossRef CAS.
- A. Corma, V. Fornes, F. W. Melo and J. Herrero, Zeolites, 1987, 7, 559 CrossRef CAS.
- K. J. Chao and J. Y. Chern, J. Phys. Chem. B, 1989, 93, 1401 Search PubMed; K. Gaare and D. Akporia, J. Phys. Chem. B, 1997, 101, 48 CrossRef CAS.
- K. J. Chao and J. Y. Chern, J. Phys. Chem. B, 1989, 93, 1401 CAS.
- K. Gaare and D. Akporia, J. Phys. Chem. B, 1997, 101, 48 CrossRef CAS; A. E. Hirschler, J. Catal., 1963, 2, 428 CrossRef.
- A. E. Hirschler, J. Catal., 1963, 2, 428 CrossRef CAS.
- A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Compr. Heterocycl. Chem. II., 1996, 2, 207 Search PubMed and references cited therein.
- G. W. J. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045 RSC.
- J. J. Li and G. W. Gribble, Palladium in Heterocyclic Chemistry, Pergamon, Amsterdam, 2000 Search PubMed.
- F. Diederich and P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, Weinhein, 1998 Search PubMed.
- J. Tsuji, Palladium Reagents and Catalysts, John Wiley & Sons, Chichester, 1995 Search PubMed.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef.
- T. Sakamoto, Y. Kondo and H. Yamanaka, Heterocycles, 1988, 27, 2225 CrossRef CAS and references cited therein.
- A. Arcadi, S. Cacchi and F. Marinelli, Tetrahedron Lett., 1992, 33, 8317 Search PubMed.
- H. C. Zhang, H. Ye, A. F. Moretto, K. K. Brumfield and B. E. Maryanoff, Org. Lett., 2000, 2, 89 CrossRef CAS.
- M. C. Fagnola, I. Candiani, G. Visentin, W. Cabri, F. Zarini, N. Mongelli and A. Bedeschi, Tetrahedron Lett., 1997, 38, 2307 CrossRef CAS.
- R. C. Larock and E. K. Yum, J. Am. Chem. Soc., 1991, 113, 6689 CrossRef CAS.
- R. C. Larock, E. K. Yum and M. D. Refvik, J. Org. Chem., 1998, 63, 7652 CrossRef CAS.
- K. R. Roesch and R. C. Larock, Org. Lett., 1999, 1, 1551 CrossRef CAS.
- T. Jeschke, D. Wensbo, U. Annby and S. Gronowitz, Tetrahedron Lett., 1993, 34, 6471 CrossRef CAS.
- H.-C. Zhang, K. K. Brumfield and B. E. Maryanoff, Tetrahedron Lett., 1997, 38, 2439 CrossRef CAS.
- K. B. Hong, C. W. Lee and E. K. Yum, Tetrahedron Lett., 2004, 45, 693 CrossRef CAS.
- N. Batail, A. Bendjeriou, L. Djakovitch and V. Dufaud, Appl. Catal., A, 2010, 388, 179 CrossRef CAS.
- N. Batail, M. Genelot, V. Dufaud, L. Joucla and L. Djakovitch, Catal. Today, 2011, 173, 2 CrossRef CAS.
- X. Sheng, J. Gao, L. Han, Y. Jia and W. Sheng, Microporous Mesoporous Mater., 2011, 143, 73 CrossRef CAS.
- J. Michalik, M. Narayana and L. Kevan, J. Phys. Chem., 1985, 89, 1553 Search PubMed.
- W. M. H. Sachtler and F. A. Cacalcanti, Catal. Lett., 1991, 9, 261 CrossRef CAS.
- L. Djakovitch and K. Koehler, J. Am. Chem. Soc., 2001, 123, 5990 CrossRef CAS.
- S. Y. Chou, Heterocycles, 2003, 60, 1095 CrossRef CAS.
- A. Mobilio, L. G. Humber, A. H. Katz, C. A. Demerson, P. Hughes, R. Brigance, K. Conway, U. Shah, G. Williams, F. Labbadis, B. D. Lange, A. Asselin, J. Schmid, J. Newburger, N. P. Jensen, B. M. Weichiman, T. Chau, G. Neuman, D. D. Wood, D. V. Engen and N. J. Taylor, J. Med. Chem., 1988, 31, 2211 CrossRef.
- S. J. Garden, R. B. Da Silva and A. S. Pinto, Tetrahedron, 2002, 58, 8399 CrossRef CAS.
- J. S. Yadav, B. V. S. Reddy, A. D. Krishna, A. R. Reddy and A. V. Narsaian, Lett. Org. Chem., 2008, 5, 455 CrossRef CAS.
- H. Kotsuki, M. Teraguchi, N. Shimomoto and M. Ochi, Tetrahedron Lett., 1996, 37, 3727 CrossRef CAS.
- V. Khedkar, A. Tillack, M. Michalik and M. Beller, Tetrahedron, 2005, 61, 7622 CrossRef CAS.
- X. Sheng, J. Gao, L. Han, Y. Jia and W. Sheng, Microporous Mesoporous Mater., 2011, 143, 73 CrossRef CAS.
- A. Khorshidi and K. Tabatabaeian, J. Mol. Catal. A: Chem., 2011, 344, 128 CrossRef CAS.
- M. A. Zeligs, J. Med. Food, 1998, 1, 67 CrossRef CAS.
- G. M. Andrus and K. Arffman, US Patent, 5895787, 1999 Search PubMed.
- G. M. Andrus and K. Arffman, US Patent, 5895787, 1999 Search PubMed.
- J. J. Michnovicz and H. L. Bradlow, in Food Phytochemicals for Cancer Prevention 1: Fruits and Vegetables, ed. M. T. Huang, T. Osawa, C. T. Ho and R. T. Rosen, American Chemical Society, Washington, DC, 1994, pp. 282 Search PubMed.
- X. Ge, S. Yannai, G. Rennert, N. Gruener and F. A. Gares, Biochem. Biophys. Res. Commun., 1996, 28, 153 CrossRef.
- W. E. Loub, L. W. Wattenberg and D. W. Davis, J. Natl. Cancer Inst., 1975, 54, 985 CAS.
- J. J. Michnovicz and H. L. Bradlow, Proc. R. Soc. Edinburgh, 1989, 12, 1571 Search PubMed.
- M. Karthik, A. K. Tripathi, N. M. Gupta, M. Palanichamy and V. Murugesan, Catal. Commun., 2004, 5, 371 CrossRef CAS.
- P. Andy, J. Garcia-Martinez, G. Lee, H. Gonzalez, C. W. Jones and M. E. Davis, J. Catal., 2000, 192, 215 CrossRef CAS.
- P. J. Kunkeler, D. Moeskops and H. van Bekkum, Microporous Mesoporous Mater., 1997, 11, 313 CAS.
- M. Karthik, C. J. Magesh, P. T. Perumal, M. Palanichamy, B. Arabindoo and V. Murugesan, Appl. Catal., A, 2005, 286, 137 CrossRef CAS.
- Z.-H. Ma, H.-B. Hana, Z.-B. Zhoua and J. Nie, J. Mol. Catal. A: Chem., 2009, 311, 46 CrossRef CAS.
- S. H. Zee and C. S. Chen, J. Chin. Chem. Soc. (Taipei), 1974, 21, 229 CAS.
- H. Singh, R. Sarin and K. Singh, Heterocycles, 1986, 24, 3039 CrossRef CAS.
- K. Suda and T. Takanami, Chem. Lett., 1994, 1915 CrossRef CAS.
- J. N. Denis, H. Mauger and Y. Vallee, Tetrahedron Lett., 1997, 38, 8515 CrossRef CAS.
- M. Otake, I. Fukushima and S. Fujita, Japan Patent, 1190667, 1989 Search PubMed.
- J. Y. Johnson and G. W. Johnson, GB Patent, 1312463, 1973 Search PubMed.
- P. J. N. Meyer and J. M. Penders, European Patent, 0037603 A1, 1981 Search PubMed.
- U. Koehler and H. Siegel, US Patent, 5101045, 1992 Search PubMed.
- K. Hatada and Y. Ono, Bull. Chem. Soc. Jpn., 1977, 50, 2517 CrossRef CAS.
- K. Hatada, M. Shimada, Y. Ono and T. Keii, J. Catal., 1975, 37, 166 CrossRef CAS.
- M. Bergfeld and G. Wiesgickl, US Patent, 5478950, 1995 Search PubMed.
- Y.-S. Yoon, H. K. Shin and B.-S. Kwak, Catal. Commun., 2002, 3, 349 CrossRef CAS.
- M. Otake, I. Fukushima and S. Fujita, Japan Patent, 1186864, 1989 Search PubMed.
- Y.-S. Yoon, H. K. Shin and B.-S. Kwak, Catal. Commun., 2002, 3, 349 CrossRef CAS.
- M. Otake, I. Fukushima and S. Fujita, Japan Patent, 1186864, 1989 Search PubMed.
- G. W. H. Cheeseman and R. F. Cookson, in Condensed pyrazines, the chemistry of heterocyclic compounds, Wiley, New York, 1979, vol. 35, p. 6 Search PubMed.
- A. R. Katritzky and A. J. Boulton, in Adv. Heterocyclic Chemistry, Academic press, New York, 1978, vol. 22, p. 368 Search PubMed.
- S. Antoniotti and E. Donach, Tetrahedron Lett., 2002, 43, 3971 CrossRef CAS.
- R. S. Robinson and R. J. K. Taylor, Synlett, 2005, 1003 CAS.
- S. A. Raw, C. D. Wilfred and R. J. K. Taylor, Org. Biomol. Chem., 2004, 2, 788 CAS.
- C. Venkatesh, B. Singh, P. K. Mahata, H. Ha and H. Junjappa, Org. Lett., 2005, 7, 2169 CrossRef CAS.
- M. M. Heravi, S. Taheri, K. Bakhtiari and H. A. Oskooie, Catal. Commun., 2007, 8, 211 CrossRef CAS.
- M. M. Heravi, S. Taheri, K. Bakhtiari and H. A. Oskooie, Catal. Commun., 2007, 8, 1341 CrossRef CAS.
- A. Dong, G. Kai, F. Zhenghao, Z. Xinli and L. Zuliang, Catal. Commun., 2007, 9, 317 CrossRef.
- T.-K. Huang, R. Wang, L. Shi and X.-X. Lu, Catal. Commun., 2008, 9, 1143 CrossRef CAS.
- A. Kumar, S. Kumar, A. Saxena, A. De and S. Mozumdar, Catal. Commun., 2008, 9, 778 CrossRef CAS.
- S. Ajaikumara and A. Pandurangan, Appl. Catal., A, 2009, 357, 184 CrossRef.
- D. Venu Gopal and M. Subrahmanyam, Catal. Commun., 2001, 2, 219 CrossRef.
- Y. Watanabe, N. Suzuki and Y. Ohsuji, Tetrahedron Lett., 1981, 22, 2667 CrossRef CAS.
- Y. Watanabe, N. Suzuki, Y. Tsuji, S. C. Shim and T. Mitsudo, Bull. Chem. Soc. Jpn., 1982, 55, 1116 CrossRef CAS.
- Y. Watanabe, Y. Tsuji and J. Shida, Bull. Chem. Soc. Jpn., 1984, 57, 435 CrossRef CAS.
- Y. Watanabe, K. Takatsuki, S. C. Shim, T. Mitsudo and Y. Takegami, Bull. Chem. Soc. Jpn., 1978, 51, 3397 CrossRef CAS.
- C. H. McAteer, R. D. Davis and J. R. Calvin, WO9703051, 1997 Search PubMed.
- P. R. Reddy, K. V. Subba Rao and M. Subrahmanyam, Catal. Lett., 1998, 56, 155 CrossRef CAS.
- B. M. Reddy and I. Ganesh, J. Mol. Catal. A: Chem., 2000, 151, 289 CrossRef CAS.
- M. Campanati, P. Savini, A. Tagliani and A. Vaccari, Catal. Lett., 1997, 47, 247 CrossRef CAS.
- M. Campanati, A. Vaccari and O. Piccolo, Catal. Today, 2000, 60, 289 CrossRef CAS.
- H. J. Uebel, K. K. Moll and M. Mühlstädt, J. Prakt. Chem., 1970, 312, 263 CrossRef CAS.
- S. Kobayashi, H. Ishitani and S. Nagayama, Chem. Lett., 1995, 423 CrossRef CAS.
- H. Ishitani and S. Kobayashi, Tetrahedron Lett., 1996, 37, 7357 CrossRef CAS.
- R. Brosius, D. Gammon, F. Van Laar, E. v. Steen, B. Sels and P. Jacobs, J. Catal., 2006, 239, 362 CrossRef CAS.
- N. A. Sánchez, J. M. Saniger, J.-B. d'Espinose de la Caillerie, A. L. Blumenfeld and J. J. Fripiat, Microporous Mesoporous Mater., 2001, 50, 41 CrossRef.
- R. Le Van Mao, T. S. Le, M. Fairbairn, A. Muntasar, S. Xiao and G. Denes, Appl. Catal., A, 1999, 185, 41 CrossRef.
- S. K. Pur and R. G. Bryant, Zeolites, 1996, 16, 118 CrossRef.
- B. Elvers, S. Hawkins and W. Russell, in Ulmann's Encyclopedia of Industrial Chemistry, 1989, vol. 14, p. 167 Search PubMed.
- M. Botta, F. De. Angelis and R. Nicolettti, J. Heterocycl. Chem., 1979, 16, 501 CrossRef CAS.
- M. Bourak and R. Gallo, Heterocycles, 1990, 31, 447 CrossRef CAS.
- D. Venu Gopal and M. Subrahmanyam, Catal. Commun., 2001, 2, 305 CrossRef.
- P. S. Landis and P. B. Venuto, J. Catal., 1966, 6, 245 CrossRef CAS.
- W. K. Bell and C. D. Chang, European Patent, 056698, 1985 Search PubMed.
- P. B. Venuto and P. S. Landis, Adv. Catal., 1968, 18, 259 CrossRef CAS.
- A. Aucejo, M. C. Burguet, A. Corma and V. Fornés, Appl. Catal., 1986, 22, 187 CrossRef CAS.
- A. Thangaraj, S. Sivasanker and P. Ratnasamy, J. Catal., 1992, 137, 252 CrossRef CAS.
- H. Sato, K. Ishii, K. Hirose and Y. Nakamura, Stud. Surf. Sci. Catal., 1986, 28, 755 CrossRef CAS.
- J. S. Reddy, R. Ravishankar, S. Sivasanker and P. Ratnasamy, Catal. Lett., 1993, 17, 139 CrossRef CAS.
- A. Dahlhoff, U. Barsnick and W. F. Hölderich, Appl. Catal., A, 2001, 210, 83 CrossRef.
- T. D. Conesa, R. Mokaya, Z. Yangb, R. Luque, J. M. Campelo and A. A. Romero, J. Catal., 2007, 252, 1 CrossRef CAS.
- A. Bordoloia and S. B. Halligudi, Appl. Catal., A, 2010, 379, 141 CrossRef.
- M. Anilkumar and W. F. Holderich, J. Catal., 2008, 260, 17 CrossRef CAS.
- R. Palkovits, W. Schmidt, Y. Ilhan, A. E-Şenatalar and F. Schüth, Microporous Mesoporous Mater., 2009, 117, 228 CrossRef CAS.
- M. A. Camblor, A. Corma, H. García, V. Semmer-Herlédan and S. Valencia, J. Catal., 1998, 177, 267 CrossRef CAS.
- Y. Zhang, Y. Wang and Y. Bu, Microporous Mesoporous Mater., 2008, 107, 247 CrossRef CAS.
- K. Chaudhari, R. Bal, A. J. Chandwadkar and S. Sivasanker, J. Mol. Catal. A: Chem., 2002, 177, 247 CrossRef CAS.
- P. S. Singh, R. Bandopadhyay, S. G. Hegde and B. S. Rao, Appl. Catal., A, 1996, 136, 249 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Proposed zeolite catalyzed reaction mechanisms. See DOI: 10.1039/c2cy00162d |
| ‡ Present address: Ecologic Technologies Pvt. Ltd, Plot No. 56, Road No. 5, ALEAP Industrial Area, Pragathi Nagar, Kukatpally, Hyderabad-500072, India. E-mail: krizmohan@yahoo.com; Tel: +91-8008803531 |
| This journal is © The Royal Society of Chemistry 2012 |