Highly crosslinked polycyclooctyl-salen cobalt (III) for the hydrolytic kinetic resolution of terminal epoxides†
Michael G. C.
Kahn
and
Marcus
Weck
*
New York University and the Molecular Design Institute, New York University, New York, New York, 10003-6688, United States. E-mail: Marcus.weck@nyu.edu; Fax: +1 (212) 995 4895; Tel: +1 (212) 992 7968
First published on 18th November 2011
Abstract
A new support structure for Co(III)salen catalysts has been developed to improve the kinetics for the hydrolytic kinetic resolution (HKR) of terminal epoxides. The new support consists of a copolymer composed of crosslinked salen-containing cyclic oligomers. Previous studies show that higher molecular weight cyclic oligomers are more active HKR catalysts than lower weight oligomers. The crosslinking reaction forms high molecular weight oligomers using a similar support structure and in significantly greater synthetic yield making the presented protocol synthetically more viable.
Introduction
Asymmetric catalysis is a powerful tool for the modern organic chemist.1 Sub-stoichometric amounts of chiral catalysts can generate large amounts of optically pure compounds.2,3 Enantiopure terminal epoxides,2,4 in particular, are useful building blocks for organic synthesis because they can react with a wide variety of nucleophiles and other reagents such as radicals and Lewis acids with retention of stereochemistry.5 In 1997, Jacobsen et al. reported the use of a chiral cobalt salen complex for the first practical way to isolate enantiopure terminal epoxidesvia hydrolytic kinetic resolution (HKR).6,7 Later, Jacobsen and Blackmond elucidated that the HKR using Co-salen complexes has a second order rate dependence on catalyst concentration, consistent with a bimetallic mechanism, where the epoxide is activated by coordinating to a Co center while water forms a Co–OH species that attacks the activated epoxide.8 Various research groups have explored the synthesis of ligands supported on soluble or insoluble structures that place, after metallation, the metal centers in close proximity with the aim of increasing turnover frequencies.9–14 A drawback of small molecule catalysis is the separation of the catalyst from the reaction mixture which can cause problems15,16 since it requires, in most cases, arduous chromatographic separations.17 Supported catalysts simplify the process by allowing the facile recovery of the catalyst by filtration or precipitation, which can then be reactivated for subsequent use.12,15,18,19Our group developed the most active supported Co-salen catalyst for HKR to date (Fig. 1).13,14 The Co-salen complex is supported on macrocycles formed by the ring-expanding olefin metathesis of cyclooctene salen monomers using Grubbs' 3rd generation initiator.20 The ring-expanding reaction formed a distribution of ring sizes. We have demonstrated that the larger ring sizes proved to be superior HKR catalysts.21 Unfortunately, the product distribution of the metathesis reaction produced mainly dimers and trimers, which were determined to be less or not at all active.21 The yield of the large macrocycles, after removal of dimers and trimers, was only 12% making this strategy not commercially viable at this point. Therefore increasing the yield of the high molecular weight ligand is key to the success of this strategy. Here, we describe a cross-linking strategy to obtain new cyclooctene-based Co-salen supports that significantly increases the percentage of the highly active species.
 | ||
| Fig. 1 Cyclic octene-oligomeric supported salen catalyst. | ||
Our research design is based on monomer 2 which can act as a cross-linker when copolymerized with 3 to form copolymer 4 (Fig. 2). Compound 2 was synthesized as described in Scheme 1 by condensing two equivalents of aldehyde 11 with diamine 12. Monomer 3 was synthesized according to the literature.13
 | ||
| Fig. 2 Crosslinked cyclooligomeric supported salen 4. | ||
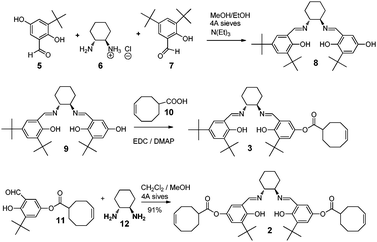 | ||
| Scheme 1 Synthesis of salen monomer and crosslinking agent. | ||
To determine if the feed ratio of 3 to 2 effects the molecular weight distribution of the resultant polymers, three sets of copolymers were synthesized using 3, 2 and Grubbs' third generation initiator. Polymerizations were conducted in 1,2-dichloroethane using molar feed ratios of compounds 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 of three eq. of 3 to one eq. of 2 as well as ratios of, 5:1 and 8:1 of 3 to 2 respectively. Monomer to initiator ratios were kept constant at 33:1. We first investigated all polymers using MALDI-TOF spectrometry. All copolymers, independent of feed ratio, contained dimers, trimers, and the more active tetramers-decamers and an even higher MW species. Next, we characterized all copolymers by gel-permeation chromatography (GPC). According to GPC, all copolymer samples show a high molecular weight species of approximately Mw = 9000 (Fig. 3–5). Unfortunately, these high molecular weight species could not be characterized by MALDI-TOF spectrometry. GPC traces of the three copolymers show the product distribution shifting toward the high molecular weight species (Fig. 3, 4 and 5 at approx. 17 min) as the mole fraction of 2 is increased. The amount of high molecular weight materials scale for the most part with the feed ratio except for the 3:1 copolymer sample that formed a disproportionably large amount of the high molecular weight material relative to the 5:1 and 8:1 samples.
:2 of three eq. of 3 to one eq. of 2 as well as ratios of, 5:1 and 8:1 of 3 to 2 respectively. Monomer to initiator ratios were kept constant at 33:1. We first investigated all polymers using MALDI-TOF spectrometry. All copolymers, independent of feed ratio, contained dimers, trimers, and the more active tetramers-decamers and an even higher MW species. Next, we characterized all copolymers by gel-permeation chromatography (GPC). According to GPC, all copolymer samples show a high molecular weight species of approximately Mw = 9000 (Fig. 3–5). Unfortunately, these high molecular weight species could not be characterized by MALDI-TOF spectrometry. GPC traces of the three copolymers show the product distribution shifting toward the high molecular weight species (Fig. 3, 4 and 5 at approx. 17 min) as the mole fraction of 2 is increased. The amount of high molecular weight materials scale for the most part with the feed ratio except for the 3:1 copolymer sample that formed a disproportionably large amount of the high molecular weight material relative to the 5:1 and 8:1 samples.


The crude copolymer mixtures could be separated by preparative size-exclusion chromatography (SEC) using Toyo pearl HW-49 resin and a 1:1 HPLC grade methanol and chloroform mixture with 0.1% triethylamine. This allowed for the removal of the less active dimers and trimers. SEC was also used to separate the tetramers-decamer fractions (4[A]) from the high molecular weight species (4[B]) (Fig. 6).
![GPC traces of high molecular weight 4[B] and tetramer-decamer fractions 4[A] of copolymers after preparative size exclusion chromatography.](/image/article/2012/CY/c1cy00290b/c1cy00290b-f6.gif) | ||
| Fig. 6 GPC traces of high molecular weight 4[B] and tetramer-decamer fractions 4[A] of copolymers after preparative size exclusion chromatography. | ||
MALDI-TOF spectrometry of fractions collected after SEC confirmed the complete removal of dimers and trimers. After the samples were metallated with cobalt (II) acetate·4H2O, the tetramers-decamers were detected with m/z consistent with the metallated copolymer. However, the metallated high-molecular weight species 4[B] was still not observed. Fig. 7 shows the MALDI-TOF MS for 4[A].
 | ||
| Fig. 7 MALDI-TOF MS of the tetramers-decamers copolymer. | ||
1H-NMR spectroscopy is a way to determine the 3:2 ratio, especially for the high molecular weight copolymers. Experiments were conducted to analyze the integrations of the t-butyl signals of the copolymers. The t-butyl resonances were used because they form sharp singlets and allow us to distinguish between 2 and 3. As an example, Fig. 8 shows the NMR peak assignment for the 8:1 copolymer 4[B]. Only 3 contains the α t-butyl resonances at 1.16 ppm. Both, 3 and 2, have resonances at 1.31 and 1.33 ppm from the β and γ t-butyl groups. In the homopolymer, the β and γ t-butyl groups integrate to 18 hydrogens. However, in the copolymers the sum of the β and γ t-butyl groups integrate to more than 18 due to the contribution of the t-butyl groups from 2. By taking the difference of the β and γ integrals from 18, the 3:2 ratio of the copolymers can be calculated. A higher proportion of 2 is incorporated into the high molecular weight material than the low MW material. This difference scales with the feed ratio of 2.
![Partial NMR spectrum (region from 1.1 to 1.4 ppm) of the t-butyl region to determine the 3 : 2 ratio of 4[B].](/image/article/2012/CY/c1cy00290b/c1cy00290b-f8.gif) | ||
| Fig. 8 Partial NMR spectrum (region from 1.1 to 1.4 ppm) of the t-butyl region to determine the 3:2 ratio of 4[B]. | ||
After SEC, the low molecular weight copolymers were determined to have a molecular weight of approximately 4500–5000 by GPC and the PDIs ranging from 1.2–1.3 (see Table 1). The molecular weights determined by GPC are significantly larger than determined by MALDI-TOF, m/z = 2600–5400. The MALDI-TOF data is more reliable as it is a direct measurement as opposed to GPC where the MWs are calculated versuspoly(styrene) standards. As seen in Table 1, the MWs of high and low molecular weight copolymers were similar independent of feed ratio. The main difference between the copolymers is the increasing amount of crosslinking agent 2 incorporated into the copolymers according to NMR spectroscopy measurements.
| Materiala | Mn b | Mwb | PDIb | MALDI-TOFc | Oligomersb,c | 3:2 ratiod |
|---|---|---|---|---|---|---|
| a Based on initial feed ratio. b determined by GPC. c degree of polymerization determined by MALDI-TOF stated as lower limit. d determined by 1H-NMR spectroscopy. | ||||||
| 8:1 low MW |
4000 | 4500 | 1.24 | 2.6–5.4 K | Tetramersb | 3.0:1 |
| 8:1 high MW |
10000 |
15000 |
1.51 | — | Heptamersc | 2.6:1 |
| 5:1 low MW |
4000 | 5000 | 1.15 | 2.6–4.0 K | Tetramersb | 2.4:1 |
| 5:1 high MW |
9000 | 13000 |
1.45 | — | Heptamersc | 1.5:1 |
| 3:1 low MW |
3500 | 4500 | 1.25 | 2.6–4.5 K | Tetramersb | 1.9:1 |
| 3:1 high MW |
9000 | 14000 |
1.57 | — | Heptamersc | 1.3:1 |
To test the activity of these supported ligands for the HKR of terminal epoxides, 4[A] and 4[B] were metallated with cobalt acetate in analogy to literature procedures.13 The cobalt (II) adducts were oxidized aerobically using acetic acid and then dried thoroughly to afford the catalytically active cobalt (III) species as a brown solid. Kinetic studies were undertaken with epichlorohydrin, hexene oxide, and allyl glycidyl ether. All experiments were conducted at room temperature with a cobalt loading of 0.01 mol% based on ICP-MS cobalt content.
The high molecular weight 4[B] and tetramers-decamers 4[A]copolymers exhibited similar activity for the HKR of epichlorohydrin, completing the kinetic resolution with an enantiomeric excess of 99% in 108 min (Fig. 9). The original system using 1 took 150 min, which demonstrates the enhanced activity of 4[A] and 4[B] when compared to the original mixture of 1. While the highest molecular weight species of 1 (the pentamer-decamers) reached 99% ee in 80 min, the yield of the pentamer-decamer fraction of 1 was only 3% compared to 33% yield recovered from the high molecular weight and tetramers-decamers copolymers.21
![HKR of epichlorohydrin at room temperature and 0.01 mol% catalyst loading. Low molecular weight copolymer 4[A] (●) and high molecular weight copolymer 4[B] (▲).](/image/article/2012/CY/c1cy00290b/c1cy00290b-f9.gif) | ||
| Fig. 9 HKR of epichlorohydrin at room temperature and 0.01 mol% catalyst loading. Low molecular weight copolymer 4[A] (●) and high molecular weight copolymer 4[B] (▲). | ||
Similarly, the kinetic plots for the HKR of hexene oxide of both the high molecular weight 4[B] and tetramers-decamers copolymers 4[A] had almost identical activity, indicated by the 99% ee within 120 min (Fig. 10). The kinetics of this reaction are comparable to the original system (1), which was surprising since no rate increases were observed with 4[B] and 4[A]. The HKR of allyl glycidylether catalyzed by 4[B] and 4[A] showed a 2-hour induction period in which little conversion was taking place. Stirring the epoxide-catalyst solution for two hours followed by the addition of water to initiate the reaction eliminated the induction period. This has been observed previously.13,18 It is thought that stirring the catalyst in the epoxides allows the epoxide enough time to properly coordinate to the metal center and for the catalyst molecules on the support to acquire the proper conformation and geometry for a cooperative bimetallic reaction. The kinetics show 96% ee after 11 h, which is similar to 1 (Table 2).
| Catalyst | % yielda | Epoxyhexane b | Epichlorohydrin b | Allylglycidylether b |
|---|---|---|---|---|
| a % yield reported as recovered ligand before metallation. All reactions were conducted at 0.01 mol% loading (based on Co content) and 0.6 mol% H2O at 20 °C. b Length of time to reach 99% ee reported in minutes. | ||||
| 4[A], 4[B] | 33 | 120 min | 108 min | 660 min |
| 1 n = 2–10 | 96 | 120 min | 150 min | 720 min |
| 1 n = 5–10 | 3 | 78 min | 80 min | 300 min |
![HKR of hexene oxide at room temperature, 0.01 mol% catalyst loading. Low molecular weight copolymer 4[A] (●) and high molecular weight copolymer 4[B] (▲).](/image/article/2012/CY/c1cy00290b/c1cy00290b-f10.gif) | ||
| Fig. 10 HKR of hexene oxide at room temperature, 0.01 mol% catalyst loading. Low molecular weight copolymer 4[A] (●) and high molecular weight copolymer 4[B] (▲). | ||
Surprisingly, there is little difference between 4[B] and 4[A] in terms of their HKR activity. Both samples were more active than the mixture of original system but not as active as the larger pentamers-decamers of 1 isolated by SEC. A possible explanation for this could be solubility. The crosslinked copolymers could be less soluble and more time could be required for the material to swell and become fully solubilized in the epoxide solution, making the metal centers more accessible for the catalytic reaction. The crosslinked catalyst reported in this contribution is among the most active and selective HKR catalysts in the literature. An important feature of this study is that the ligand can be synthesized in much greater yield compared to the original system. The purification of ring sizes using preparative SEC is a simple protocol and highly effective to separate ring sizes of different molecular weight.
Conclusions
In conclusion, we have synthesized crosslinked salen copolymers and tested their HKR activity for terminal epoxides. The copolymers could be separated by molecular weight using size-exclusion chromatography. Both the high molecular weight copolymers and medium-sized macrocycles performed similarly in terms of their HKR activity. This simplifies the purification because these species no longer need to be separated from each other because they display similar kinetics. Additionally, after removal of dimers and trimers the chemical yield of the crosslinked ligand was three times that of the original system thereby significantly increasing the yield of the more active catalyst. The copolymer catalyst showed similar activity for allyl glycidyl ether and hexene oxide but the kinetics were improved for epichlorohydrin compared to the original homopolymer.Acknowledgements
We are thankful to the Department of Energy Office Basic Energy Sciences through Catalysis Contract No. DEFG02-03ER15459 for financial support of this work.References
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS.
- T. D. Machajewski and C.-H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352–1374 CrossRef CAS.
- K. B. Sharpless, C. H. Behrens, T. Katsuki, A. W. M. Lee, V. S. Martin, M. Takatani, S. M. Viti, F. J. Walker and S. S. Woodard, Pure Appl. Chem., 1983, 55, 589–604 CrossRef CAS.
- (a) R. E. Parker, Chem. Rev., 1959, 59, 737–799 CrossRef CAS; (b) K. Venkatasubbaiah, X. Zhu, E. Kays, K. I. Hardcastle and C. W. Jones, ACS Catal., 2011, 1, 489–492 CrossRef CAS.
- M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936–938 CrossRef CAS.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307–1315 CrossRef CAS.
- L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360–1362 CrossRef CAS.
- J. M. Ready and E. N. Jacobsen, Angew. Chem., Int. Ed., 2002, 41, 1374–1377 CrossRef CAS.
- J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 2687–2688 CrossRef CAS.
- X. Zheng, C. W. Jones and M. Weck, Chem.–Eur. J., 2006, 12, 576–583 CrossRef.
- H. Yang, L. Zhang, W. Su, Q. Yang and C. Li, J. Catal., 2007, 248, 204–212 CrossRef CAS.
- X. Zheng, C. W. Jones and M. Weck, J. Am. Chem. Soc., 2007, 129, 1105–1112 CrossRef CAS.
- X. Zhu, K. Venkatasubbaiah, M. Weck and C. W. Jones, ChemCatChem, 2010, 2, 1252–1259 CrossRef CAS.
- N. Madhavan, C. W. Jones and M. Weck, Acc. Chem. Res., 2008, 41, 1153–1165 CrossRef CAS.
- C. Baleizao and H. Garcia, Chem. Rev., 2006, 106, 3987–4043 CrossRef CAS.
- C. S. Gill, K. Venkatasubbaiah, N. T. S. Phan, M. Weck and C. W. Jones, Chem.–Eur. J., 2008, 14, 7306–7313 CrossRef CAS.
- S. Jain, K. Venkatasubbaiah, C. W. Jones and R. J. Davis, J. Mol. Catal. A: Chem., 2010, 316, 8–15 CrossRef CAS.
- C. W. Jones, Top. Catal., 2010, 53, 942–952 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS.
- Y. Liu, J. Rawlston, A. T. Swann, T. Takatani, C. D. Sherrill, P. J. Ludovice and M. Weck, Chem. Sci., 2011, 2, 429–438 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cy00290b |
| This journal is © The Royal Society of Chemistry 2012 |