Nanomaterials design and tests for neural tissue engineering
Gloria A. A. Saracinoab, Daniela Cigogniniab, Diego Silvaab, Andrea Capriniab and Fabrizio Gelain*ac
aCenter for Nanomedicine and Tissue Engineering, A.O. Ospedale Niguarda Cà Granda, Milan, 20162, Italy. E-mail: gelain@mit.edu; Fax: +39 02 6444 3258; Tel: +39 02 6444 3245
bBiotechnology and Biosciences Department, University of Milan-Bicocca, Milan, 20126, Italy. Fax: +39 02 6448 3314; Tel: +39 02 6448 3312
cIRCCS Casa Sollievo della Sofferenza Opera di San Pio da Pietrelcina, San Giovanni Rotondo 71013, Italy. E-mail: f.gelain@css-mendel.it; Fax: +39 0882 410 346; Tel: +39 0882 410 931
First published on 18th September 2012
Abstract
Nanostructured scaffolds recently showed great promise in tissue engineering: nanomaterials can be tailored at the molecular level and scaffold morphology may more closely resemble features of extracellular matrix components in terms of porosity, framing and biofunctionalities. As a consequence, both biomechanical properties of scaffold microenvironments and biomaterial–protein interactions can be tuned, allowing for improved transplanted cell engraftment and better controlled diffusion of drugs. Easier said than done, a nanotech-based regenerative approach encompasses different fields of know-how, ranging from in silico simulations, nanomaterial synthesis and characterization at the nano-, micro- and mesoscales to random library screening methods (e.g. phage display), in vitro cellular-based experiments and validation in animal models of the target injury. All of these steps of the “assembly line” of nanostructured scaffolds are tightly interconnected both in their standard analysis techniques and in their most recent breakthroughs: indeed their efforts have to jointly provide the deepest possible analyses of the diverse facets of the challenging field of neural tissue engineering. The purpose of this review is therefore to provide a critical overview of the recent advances in and drawbacks and potential of each mentioned field, contributing to the realization of effective nanotech-based therapies for the regeneration of peripheral nerve transections, spinal cord injuries and brain traumatic injuries. Far from being the ultimate overview of such a number of topics, the reader will acknowledge the intrinsic complexity of the goal of nanotech tissue engineering for a conscious approach to the development of a regenerative therapy and, by deciphering the thread connecting all steps of the research, will gain the necessary view of its tremendous potential if each piece of stone is correctly placed to work synergically in this impressive mosaic.
 From the left to the right: Fabrizio Gelain, Diego Silva, Daniela Cigognini, Andrea Caprini and Gloria A. A. Saracino | Gloria A. A. Saracino graduated from the University of Salerno in 1999 with a BSc in chemistry. She obtained her PhD in chemical sciences from the Univeristy of Naples Federico II in 2002 under the supervision of Prof. Vincenzo Barone. She is a postdoctoral research fellow working with Dr Fabrizio Gelain in the Centre for Nanomedicine and Tissue Engineering at the Niguarda Cà Granda Hospital in collaboration with the laboratory of Nanomedicine at the University of Milano-Bicocca. Her research interests focus on the study of the self-assembling peptides by computational methods. |
Daniela Cigognini received her Bachelor's degree in Medicine and Surgery from the University of Milano-Bicocca in 2004. She obtained her PhD in cell therapy for spinal cord injury from the University of Milano in 2009. She is a postdoctoral research fellow working with Dr Fabrizio Gelain in the Centre for Nanomedicine and Tissue Engineering at the Niguarda Cà Granda Hospital in collaboration with the laboratory of Nanomedicine at the University of Milano-Bicocca, where she has been studying the effect of novel nanostructured scaffolds in animal models of spinal cord injury. Her research interests include stem cell-based therapies and tissue engineering for nervous tissue repair. |
Diego Silva received his BSc in Industrial Biotechnology from the University of Milano Bicocca in 2010. He has been working under the supervision of Dr Fabrizio Gelain as scientist specialist in the Centre for Nanomedicine and Tissue Engineering at the Niguarda Cà Granda Hospital. He also collaborates with the laboratory of Nanomedicine at the University of Milano-Bicocca. He deals with synthesis, purification and physicochemical characterization of nanostructured materials. |
Andrea Caprini graduated from the University of Milan in 2000 with a BSc in Molecular Cell Biology. He obtained his PhD from the European School of Molecular Medicine (IFOM-IEO Campus and University of Milan) under the supervision of Professor Elisabetta Dejana. He is a postdoctoral research fellow working with Dr Fabrizio Gelain in the Centre for Nanomedicine and Tissue Engineering at the Niguarda Cà Granda Hospital in collaboration with the laboratory of Nanomedicine at the University of Milano-Bicocca. His research interests include the evaluation of the biological effect of self-assembling peptides on cell behaviours. |
Fabrizio Gelain is scientific vice-director of the ‘Center for Nanomedicine and Tissue Engineering” at Niguarda Ca’ Granda Hospital in Milan and head of the Nanomedicine Unit at the “Casa Sollievo Della Sofferenza-MENDEL” IRCCS institute in Rome. He was awarded a PhD in bioengineering by the Polytechnic of Milan in 2005 and worked at the Massachusetts Institute of Technology and at The Lawrence Berkeley National Lab. His research group activity is focused on developing and characterizing new functionalized self-assembling biopolymers, electrospun matrices and other nanotechnology-derived scaffolds for slow drug release and cell transplantation therapies in nervous system injuries like spinal cord contusion, peripheral nerve transection and stroke. |
1. Introduction
Growing knowledge of stem cell biology and the development of cellular therapies offer new therapeutic options for many diseases, but, in the case of nervous system injuries, cellular transplantation could not be sufficient to regenerate the damaged nervous tissue. As a matter of fact, while several preclinical studies reported the benefit of implanting foetal tissue, embryonic stem cells or neural stem cells (NSC) into the degenerating mature central nervous system (CNS), the use of cell therapy alone was inadequate for certain nervous system injuries or pathologies, where cavities or gaps need to be filled with a physical support granting transplanted cell engraftment and cytoarchitecture restoration. Hence, currently, there are no effective therapies available for the majority of nervous system traumas like, for example, important loss of peripheral nerve tracts, spinal cord injury (SCI) and stroke, which still lead to severe neurological deficits. To improve the outcome of cell therapy in these nervous system pathologies, significant efforts are necessary to improve our understanding of stem cell biology and to achieve an optimal microenvironment both promoting functional integration of the transplanted cells and enhancing reparative processes of the damaged tissue.In the last decade the use of nanomaterials, which are materials with constituent dimensions less than 100 nm, slowly but constantly infiltrated the field of regenerative medicine, yielding unexpected results and wonderful promises. This was mostly related to the revolutionary approach of overturning the classic “top-down” design typical in tissue engineering, where bulk materials were usually processed with a number of various techniques (e.g. solvent casting, melt molding, high pressure extrusion) down to the accuracy of the micron or sub-micron scale.1 In a few words, in the 80′s and 90′s the minimum reproducible and “tunable” features of the scaffolds to be implanted were some orders of magnitude above the molecular “accuracy” typical of the biological tissues to be regenerated.2 On the other hand, in the last decade the regenerative medicine field has extensively adopted nanotech-based approaches, comprising molecular design, deep investigations of the biomaterials at the nano-, micro- and mesoscales, and their molecular/mechanical interactions with the biological component of the scaffolds and/or with host tissues.
A drawback of the new powerful technology is the mandatory need for highly cross-disciplinary projects that pay attention to all the multiple facets involved in the design, production and testing of the sophisticated nanostructured scaffolds: much like an assembly line, with the scaffold being the new “product”, going forward through the process by means of various teams strictly interacting both forward and backward in the line. Obviously, this strategy requires additional resources and strong interactions among several disciplines. Systematic and highly multi-disciplinary investigations are rare in the literature, but necessary to produce novel scaffolds featuring topography, stiffness, bioabsorption time and multiple biofunctionalizations customized for the tissue to be regenerated.
The purpose of this review is therefore to illustrate most of the technologies involved in the complex process of developing a nanofibrous scaffold for neural tissue engineering. In other words, the main aim is to give the reader an overview of the promising synergies arising by the recent advances in each discipline involved (computational chemistry, biochemistry, rheology, phage display, stem cell biology, neurology) showing how they have already brought important discoveries that will likely produce successful therapies in the near future. A quick scheme of the logical process followed in this approach, and, consequently, in this review, is depicted in Fig. 1. An important advantage of such a process is the continuous feedback of each step to the previous ones, aiming at evincing critical issues, allowing us to refine the scaffold design principles more precisely and to save time between scaffold ideation and its final testing in vivo. In Table 1 are reported examples of critical issues triggering corrective actions in the previous step.
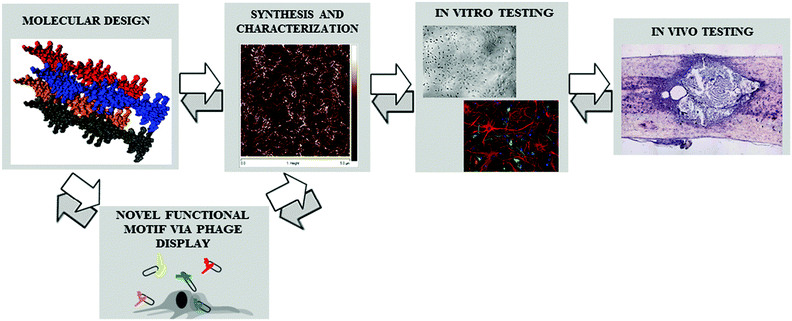 | ||
| Fig. 1 Scheme of the multi-facet approach aimed at developing nanostructured scaffolds for tissue engineering: continuous feedback among the various steps allows a synergic integration of the different disciplines involved. | ||
| Step | Feedback | Corrective action | Affected step | Ref. |
|---|---|---|---|---|
| Synthesis and characterization | Assessed morphology of the designed biomaterial in response to different environmental conditions | Validation of the theoretical framework (e.g. force field) to ameliorate the predictive potential of targeted simulations | Molecular design | 4–6 |
| Novel functional motif via phage display | Effect of functional motifs on nanostructure stability and scaffold mechanical stiffness | 7 | ||
| In vitro testing | Design/characterization of chemical modifications and use of protocols yielding scaffolds with accessible functional motifs and appropriate mechanical properties | Synthesis and characterization | 8 | |
| Seeded cell growth and differentiation in response to chemical and biomechanical properties of the tested scaffold | ||||
| Various degrees of cellular adhesion to differently functionalized scaffolds | Alternative choice of functional motifs and/or additional phage display screenings | Novel functional motif via phage display | 9 | |
| In vivo testing | Transplanted cell engraftment into the host tissue. | Development of in vitro models mimicking either three-dimensional tissue microenvironments or pathophysiological changes following tissue injury | In vitro testing | 10 |
The development of a new scaffold for neural tissue engineering application should arguably follow some crucial steps: (1) the choice of material to be used, (2) design of the scaffold via computational investigations, (3) identification (via phage display or other methods) of one or more bioactive motifs to be added to the scaffold, (4) synthesis and purification of the material, (5) morphometrical and biomechanical characterization of the scaffold, in order to confirm or disprove what predicted in computational simulations, (6) standardized tests in two- and three-dimensional cell cultures or organotypic culture systems, to evaluate in vitro the biological properties of the scaffold and set the best conditions to promote cell survival and differentiation, and lastly, (7) implantation of the matrix in animal models of CNS or peripheral nervous system (PNS) injuries in order to validate the feasibility and the putative benefits of the developed material.
Protein-based biological systems, such as cells and tissues, display hierarchical structures across multiple scales conferring them unique properties. A deep understanding of nature's design principles (e.g. the cross-scale relations between molecular details, structural hierarchies and mechanical properties) is the base for a more targeted “bottom-up” design and development of de novo biologically inspired materials. The continuous advance and deep integration of material science and experimental and computational biology allow us to move towards such goals, passing through a new emerging field called ‘materiomics’,3 which is the study of material structures, properties and function at different scales, from nano to macro. This integrated view of the material is referred to as ‘materiome’ (in analogy with ‘genome’ in genomics). Methods used to study the materiome include multi-scale simulation methods (e.g. molecular dynamics and continuum methods), multi-scale experiments (e.g. AFM, optical tweezers, etc.) and a combination of the above-mentioned techniques. The many scientific fields grouped under the materiomic banner have to deal with the complexity of tissue engineering but also with nanoscience and nanotechnology.
Among many developed nanofibrous constructs, we will review two categories of scaffolds: the electrospun and hydrogel-based scaffolds, which have been widely investigated in tissue engineering and feature great potential for nervous tissue repair.
Firstly, before discussing the details of each section, we will classify and quickly describe the most used materials available for nanostructured scaffolding.
2. Natural and synthetic materials for nanostructured scaffolds
A large number of synthetic soft scaffolds are commonly used in neural tissue engineering; these structures are made of various materials through different production techniques. The combined choice of both allows the production of fibers, wires, rings, belts and tubes.The complex interaction between neural cells and their natural microenvironment passes through cell receptors and the surrounding extracellular matrix (ECM), which displays specific chemical–physical properties. In order to reproduce these cell–matrix interactions, researchers focused their attention on the development of tailored natural and synthetic supports.
In the field of nerve tissue engineering, electrospun guidance channels and hydrogels, two promising categories of scaffolds, can be made of natural, synthetic or the combination of both materials.
Natural polymers are easily obtained from natural sources: they are reabsorbable and contain specific signals for cell adhesion, allowing cell infiltration, but they may induce immunological and inflammatory responses due to the presence of pathogens or undefined components, which are hard to safely eliminate during purification. Their undefined composition and, consequently, batch-to-batch variability make it difficult to define what component, and to what extent, has influenced the effect displayed by the natural material: as a consequence, the results obtained may be affected by low reproducibility. In comparison to natural polymers, synthetic materials permit us to more easily and precisely adjust key parameters of the scaffold (architecture, porosity, stiffness, degradation rate, etc.) and to add functionalizations; in particular, the addition of functional motifs to a synthetic peptide may improve the cell–scaffold interactions and, consequently, overcome the main issue linked to the use of synthetic materials, i.e. the lack of recognition signals and poor biocompatibility.
2.1 Polymers used for electrospun scaffolds
Electrospinning is a variant of the electrospray process and allows us to obtain a nanofibrous scaffold from a polymeric liquid solution.Both mechanical and “biological” properties of the electrospun nanofibrous scaffolds are related to the chemical properties of the polymer used for the electrospinning process, as exposed below.
Electrospinning is applicable to a wide variety of polymers that can be derived from natural sources or synthesized, and allows the production of nanofibrous scaffolds or conduits presenting different porosity, shape and fiber alignment; in addition, electrospinning allows the incorporation of ECM proteins and growth factors into the scaffold.
One of the most used biopolymers in electrospinning is collagen, one of the major components of the ECM and connective tissue. The combination of biocompatibility, low immunogenicity and tunable mechanical strength makes collagen ideal for testing cell growth, migration and for tissue engineering therapies.11
Another important natural protein used for the production of electrospun scaffolds is silk fibroin, usually produced by silkworms and spiders. It is characterized by good biocompatibility, good oxygen/water permeability and biodegradability. The production of silk fibroin-based scaffolds generally requires toxic spinning solvents such as hexafluoro-2-propanol, hexafluoroacetone, formic acid and poly(ethylene oxide): thus accidental residues of spinning solvents may impair their overall cytocompatibility. Chen and co-workers attempted at their removal by using only aqueous solution.12 Another particular property of silk fibroin is the possibility of changing the conformation of the fibrous structure from random coiled to β-sheets after water vapor13 or methanol treatment:14 this transition improves the mechanical strength and its cellular compatibility.
Polysaccharides are also being used to form nanofibrous scaffolds via electrospinning. Chitosan is a polysaccharide composed of D-glucosamine and N-acetyl-D-glucosamine that exhibits interesting physicochemical properties such as its solid-state structure and its dissolving state conformation. Generally chitosan is blended with polyethylene glycol, however Ohkawa and colleagues described the electrospinning of pure chitosan and the role of different solvents during the electrospun process. The work showed that only by using trifluoroacetic acid (TFA) as a solvent it is feasible to obtain the deposition of chitosan fibers onto the collector.15
Other polysaccharides such as alginate, dextran, cellulose and hyaluronic acid (HA) could be used for the production of non-woven nanofibrous scaffolds.16 Alginate is an anionic polysaccharide derived from brown seaweed; it is a linear copolymer composed by two building blocks: (1,4)-linked β-D-mannuronic acid and α-L-guluronic acid.17 Dextran is a bacterial polysaccharide constituted of D-glucopyranose residues linearly linked via α-1,6 bonds and branched via α-1,4.18 Cellulose, composed of (1,4)-linked β-D-glucose units, is extensively studied due to its abundance as a renewable source. HA, one of the main components of the connective tissue, has been widely used as well and it is formed by an alternate polysaccharide chain of (1,4)-linked α-D-gluconic acid and (1,3)-linked β-N-acetyl-D-glucosamine units.19
Biodegradable polyesters feature interesting mechanical properties but poor cell interactivity because of their hydrophobic component. An interesting compromise on this drawback could be the use of copolymers: macromolecules whose polymer chain contains monomers of two or more different species. The feasible attachment of a high-hydrophilic polymer to polyesters improves cell affinity while preserving the mechanical stiffness of polyesters. Copolymers allowed for the production of more resistant, uniform and biocompatible fibers.
Two of the most promising copolymers used for the production of electrospun conduits for peripheral nerve regeneration are poly(D,L-lactide-co-glycolic)acid (PLGA)21 and poly(ε-caprolactone-co-ethyl ethylene phosphate) (PCLEEP). PLGA is composed of lactide (L) and glycolide (G): the ratio of the two homopolymers determines the rate of degradation and the mechanical properties of the fibers.
Alternatively, electrospun scaffolds can be obtained via a polymer blend combining natural–natural, natural–synthetic or synthetic–synthetic polymers: this strategy is pursued to get improved biomechanical properties, durability and high cell affinity. As an example, the PLGA–PCL blend gave electrospun microstructures featuring improved mechanical stability, high permeability and good biocompatibility.22
2.2 Polymers used for hydrogels
Other biomaterials tested to mimic the physical–chemical properties of the ECM are hydrogels.23 Hydrogels are three-dimensional networks composed of a high percentage of water: their particular space-filling propensity, mechanical strength, scaffold topography, biodegradation rate and ability to promote cell adhesion make them ideal candidates for their usage in vivo.23Natural and synthetic gels can be classified into two classes: polymeric covalently cross-linked gels and self-assembled gels.24
In polymeric covalently bonded gels the monomer units are linked through covalent forces, usually giving rigid and less deformable gels. On the other hand, self-assembling describes the process ranging from disordered systems, composed of pre-existing monomers, to organized molecular structures; the process is driven by noncovalent interactions and leads to soft, usually randomly oriented, networks of nanofibers.
Natural gels have a moderate mechanical strength but their degradation rate, as well as their stiffness, can be modulated through chemical modifications. For example, capping of the N- and C-termini, or of reactive groups, is adopted to enhance the scaffold stability, while chemical cross-linking, by strengthening the nanofibrous network interactions, mainly increases its stiffness.
Alginate and collagen are low-cost and relatively biocompatible materials but present poor mechanical properties, thus they must be used in mixtures to improve the mechanical strength of the formed scaffolds.
Matrigel is certainly the most famous natural substrate used for cell cultures: it is an extract characterized by an undefined mixture of the natural compounds like laminin, collagen IV, entactin, nidogen and heparin sulfate.
Other commonly used polymers are polyethylene glycol (PEG), poly-N-(2-hydroxyethyl)methacrylamide (PHEMA) and poly-N-(2-hydroxypropyl)methacrylamide (PHPMA): their covalently cross-linked hydrogels are widely used in bioengineering because of their ability to support cell growth and mimic the ECM.25 Moreover, the degradation rate of their scaffold can be easily tuned by altering the chemistry of the cross-links within the polymer network.
However the cross-linking agents used to induce gelation are potentially toxic for cells; a way to bypass this problem is the development and use of self-assembling hydrogels.
There are several examples of nonpeptidic polymers used as hydrogelators. Among these, block copolymers have been widely studied in aqueous media to form versatile assembled structures at the molecular level. These amphiphilic block copolymers exhibit a sol–gel transition dependent on concentration, pH, temperature or ionic strength. For example poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO), a triblock copolymer, self-assembles into a hydrogel when placed at temperature above 20 °C. Other recently developed self-assembling gels are made of low-molecular-mass gelators (LMMGs): these gelators have gained interest in the field of biomaterials due to their low cytotoxicity. Lastly, self-assembling peptides (SAPs) constitute one of the most important classes of synthetic self-assembling hydrogels.
The first self-assembling sequence to be characterized was EAK16-II.26 EAK16-II is characterized by the alternation of polar and non-polar residues, allowing the formation of a double-beta sheet structure when peptides are solved in water. This assembling mechanism is similar to that found in amyloid proteins: under physiological conditions the extension of the double beta-sheets leads to the formation of nanofibers.
Following EAK16-II, other SAPs have been developed. The most studied SAPs belong to the RADA16 and KLDL12 families. These peptides are widely used as three-dimensional scaffolds for cells.27
These scaffolds may mimic the mechanical properties of the ECM and specific functional sequences can be added to the self-assembling sequence to improve cell adhesion.
The first functional sequence that was used is RGD. This pattern was found while studying integrins, proteins crucial for the phenomena of cell anchoring, differentiation and immune response.
Other important functional sequences have been derived from structural proteins such as collagen I and VI,28 laminin29 and fibronectin,30 or have been discovered by applying the phage-display technique (see Section 5.5 for details), such as the sequences BMHP1 (Bone Marrow Homing Peptide) and BMHP2.31 Usually the functional sequence is separated from the assembling core by a glycine spacer or a chemical linker. As functionalization of the assembling sequence is a critical step, in our research we studied the effect of different functional motifs on the self-assembling propensity of β-sheet forming peptides.7 We also investigated how the glycine spacer length influences the exposure of the functional motifs and the SAP's propensity to form nanostructured fibers.8
The addition of functional sequences led to the development of a second generation of SAPs, capable of promoting cell proliferation, differentiation and maturation.32 The length of the peptide chain is another important parameter playing a crucial role in the formation of β-sheet structures.33 Naska and co-workers have recently developed tetrapeptides that assemble to form β-sheet fibers. These fibers are able to generate an interconnected nanofibrous network clearly observable with microscopy techniques.
Vegners and co-workers reported for the first time a fluorenylmethoxycarbonyl (Fmoc)-protected dipeptide able to form hydrogels. The Fmoc moiety is widely used as a protecting group in solid phase peptide synthesis but, when linked to a short peptide sequence, may act like a hinge allowing the formation of nanofibers.34 For example, the dipeptide Fmoc-Phe-Phe assembles into a rigid hydrogel with mechanical properties suited for applications in tissue engineering; Saiani A. and co-workers have extensively characterized the gelation properties of glycine substituted Fmoc-Phe-Phe peptides.35 The characterization techniques used to define the self-assembling propensity of peptidic biomaterials will be discussed later (Section 4.2). Due to these properties, recently Orbach and colleagues inserted the functional motif RGD in a Fmoc-dipeptide, improving the biocompatibility of the material and extending the family of the aromatic Fmoc-dipeptides group.36
Another interesting use of Phe-Phe dipeptides was discovered by Adams J. D.37 In this work the dipetide was capped at the N-terminus with a different aromatic group. Using a naphthalene group instead of Fmoc, the naphthalene–diphenylamine solution self-assembles into a transparent β-structured scaffold when exposed to pH shifts. After the addition of a dansyl derivative the hydrogelation remained unchanged and, by monitoring the pre- and post-gelation fluorescence of the mixture, the dansyl derivative was demonstrated to be hosted in the peptidic scaffold. Interestingly this interaction resulted in an energy transfer between naphthalene and dansyl groups, showing their potential applications in light harvesting and molecular electronics. Nonetheless naphthalene and Fmoc aromatic compounds can be toxic and carcinogenic in certain forms,38,39 therefore their usage in conjunction with the above-mentioned peptides in in vitro and in vivo applications must be carefully assessed.
Other examples of SAPs without the alternation of hydrophilic and hydrophobic residues in their sequences are biotinylated peptides.40 Indeed we developed a group of biotinylated hierarchical SAPs with self-healing propensity and potential for tissue engineering applications.41
Another important class of SAPs are the peptide amphiphile molecules (PAs), designed by Stupp and colleagues.42 Typical components of PAs are an alkylic tail and a peptidic head forming β-sheets. PAs are similar to surfactant-like peptides designed by Zhang and his group, but PAs contain an hydrophobic alkyl tail, whereas surfactant-like peptides have an hydrophobic amino acid sequence instead.43
Different functional epitopes can be bonded to PAs in order to increase biocompatibility.44 Functional motifs can be linked via linear or branched sequences: branched PAs allow for the use of different epitopes by improving their availability on the formed nanofibers surface.45
Interestingly, recently Stupp and co-workers obtained, by heating a PA solution, a large array of aligned nanofibers forming a strongly birefringent liquid:46 this may have important applications in the regeneration of the nervous system, where re-establishment of the tissue's original spatial cytoarchitecture is crucial for an efficient functional recovery.
PAs contain unnatural alkyl parts combined with natural peptidic sequences: other SAPs made of unnatural structures are those designed by J. P. Schneider. These SAPs are composed of two identical short sequences linked by a sequence containing a dextrorotatory valine:47 a peptide solution is characterized by un-ordered molecular structures at low pH but when the pH is shifted to 9 self-assembling gives β-hairpins further aggregating into highly ordered nanostructures. This arrangement leads to the formation of nanofibers and, subsequently, of hydrogels.
Similar peptides, designed as metal-responsive hydrogelators by Pochan D. J. and Schneider J. P., are used to demonstrate self-assembling promoted by zinc ions (Zn2+):48 they also tested chirality as a design tool to control and tune the mechanical properties of hydrogels.49
The same researchers have recently published detailed characterization studies on β-hairpin peptide gels,50,51 demonstrating, via rheological and structural characterization, their shear thinning behaviour and their potential as cell carriers.51
Synthetic peptides are also used to mimic natural assembling sequences found in proteins: Hartgerink J. D. and co-workers have used this approach in order to produce several collagen mimetic peptides able to form triple helices and, successively, self-assembled hydrogels.52 Despite collagen being the most abundant protein in the human body, the folding and the fibrillization of this protein are not completely understood. Collagen mimetic peptides combine the possibility of studying the folding process of collagen with the ease of tuning their chemical composition to obtain self-assembled scaffolds for regenerative medicine applications. Detailed characterization studies on collagen mimetic peptides highlighted the crucial role of interactions among positively and negatively charged amino acids for first stabilization of adjacent helices and during fibril folding.53
The properties of complementary charged sequences have been studied by Y. B. Yu and co-workers: they designed and synthesized co-assembling peptide sequences characterized by mutual attraction and self-repulsion of peptide chains featuring opposite and similar net charges respectively.54
Various self-assembling peptides were synthesized and characterized by exploiting this design principle, using both natural amino acids and “non-natural” amino acids (ornitine instead of lysine) in order to obtain better control of scaffold formation. Further characterization studies have focused on the importance of the peptide chain length in hydrogelation,55 optimizing the peptide sequences to obtain scaffolds with desired rheological54 and diffusive56 properties.
Injectable peptide-based hydrogels can also be produced via recombinant DNA methods. Tirrel D. and co-workers described recombinant telechelic proteins expressed in E. coli: telechelic proteins with coiled-coil end-blocks can react with flexible polyelectrolyte mid-blocks yielding protein based hydrogels.57,58 Telechelic protein gels are characterized by a strong shear-thinning behaviour, a relaxation time dependent on the mid-block length and a near-to-instantaneous recovery of its stiffness. These rheological properties, and their importance in scaffold design, are described in Section 4.2.
For an exhaustive overview of protein engineering and its application in multifunctional material production please refer to the recent work published by Dimarco R. L. and Heilshorn S. C.59
J. D. Hartgerink and colleagues60 introduced multidomain synthetic peptides (MDPs) comprising a tri-block sequence (ABA): whereas A consists of a variable number of positively charged amino acids (lysine or glutamic acid) and the central block B is composed of alternating hydrophilic and hydrophobic residues (serine-glutamine and leucine respectively). MDP hydrogels can also be cross-linked in order to improve their rheological properties both chemically and enzymatically. A cysteine containing MDP is placed under mild oxidative conditions in order to trigger the formation of both inter- and intramolecular di-sulfide bonds.61 As to the enzymatic reaction, the lysyl oxidase is used to oxidize primary amines to aldehydes that can spontaneously react with other amines (via Schiff base reaction) or with other aldehydes (via aldol condensation) forming a linkage.62 The final product of both approaches is a cross-linked gel with increased stiffness.
MDPs can also be modified with a bioactive functional motif and with specific matrix-metallo-protease-2 sequences inserted to favour their enzymatic cleavage. Incorporation of both sequences improves the bio-compatibility of the scaffold, enhancing cell migration and spreading.63
Furthermore, Bing Xu's group linked the aromatic pyrene to vancomycin antibiotic, in order to combine hydrophobic interaction, π–π stacking and hydrogen bonding formation64 to obtain the first antibiotic-based hydrogel. In another work they obtained hydrogels from the enzymatic cleavage of an antineoplastic derivative.65 Moreover, they made use of enzymatic reactions to generate photoresponsive hydrogels66 and to promote the hydrolysis of self-assembling biomaterials.67 The same group has recently published an elegant work in which a nanofibrous hydrogel is used, via post-self-assembly cross-linking, to mimic the Belousov–Zhabotinsky reaction in order to obtain an oscillatory hydrogel displaying chemomechanical concentrical and spiral waves.68
Hybrid hydrogels can also be included among nanostructured materials: Kopeček J. and coworkers designed a hybrid hydrogel by combining a water soluble synthetic linear copolymer, obtained via radial polymerization of two monomers (N-(2-hydroxypropyl)-methacrylamide and N-(N′,N′-dicarboxymethylaminopropyl) methacrylamide (HPMA-DAMA)) and a folding protein domain (coiled-coil). Their system showed engineered volume-change properties as a result of temperature variation.69
Lastly, other promising self-assembling molecules are peptoids: oligomers of N-substituted glycine units that exhibit combined properties of natural peptide sequences and a tunable kinetics of delivery.70 Peptoids are appealing materials in nano-medicine because they feature the protein-binding tendency of peptides, high chemical versatility, stability of drug-like molecules and remarkable cell membrane permeability.
The submonomer method developed for peptoids is an easy synthesis method and allows us to investigate several substituted monomers in order to obtain sequences with ad hoc properties.
In Fig. 2 is compared the classical Fmoc solid phase peptides synthesis (A) and the submonomer method for the peptoids synthesis (B). The submonomer method consists of two chemical steps: the first one is the acetylation performed with a diimide-activated bromoacetic acid and the second step is the displacement of bromine using a primary amine (Fig. 2B).
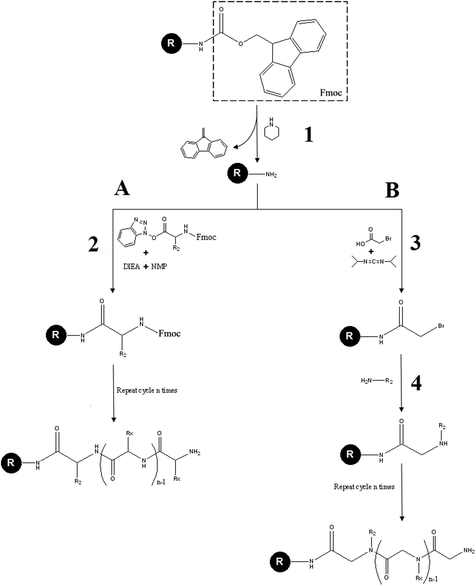 | ||
| Fig. 2 Comparison between the classical Fmoc peptide synthesis (A) and the submonomer peptoid synthesis (B). First the Fmoc protecting group of the solid resin is removed in a solution of piperidine (1). In the Fmoc peptide synthesis the primary amine of the resin reacts with the COOH-activated/NH2-protected amino acid in an activator base solution composed of N,N-diisopropylethylamine and N-methyl-2-pyrrolidone (2). In the submonomer peptoid synthesis the primary amine is subjected to an acetylation reaction by means of an haloacetic acid activated by N,N′-diisopropylcarbodiimide (3). Then the Br displacement, by means of another primary amine, gives rise to the formation of a peptoid bond (4). Both in (A) and in (B) repeated cycles of steps (2) and (3 and 4), respectively, lead to molecule elongation. | ||
Zuckermann R. N. and coworkers have developed a group of peptoids, describing their aqueous self-assembly into ordered nano-sheets, potentially usable as nano-structured scaffolds.71
To note, several of the above-mentioned SAPs have been serendipitously discovered and subsequently investigated at the molecular level and at the nano- and micro-scales. However, given the potential of the self-assembly phenomenon for tissue engineering applications, a deep understanding of the forces producing assembled molecular structures, as well as the supra-molecular arrangements yielding networks of nanofibers and scaffolds, is mandatory in order to predict the chemico-physical and biological properties of novel SAPs to be tailored for specific regenerative applications. In this way, in silico investigation is an unavoidable step in the previously mentioned assembly line.
2.3 Scaffold biodegradation for tissue engineering applications
In designing a scaffold for nervous tissue regeneration, the use of biodegradable materials is preferable. Ideally, the scaffold should be progressively replaced by the regenerating tissue, in order to last long enough to permit cell infiltration and support axon regrowth, but not to stay so long as to interfere with the ECM deposition, cell–cell interaction and reconnection of axons. In cell-based therapies, the scaffold should initially act like a barrier to the host environment, in order to warrant a user-tuned microenvironment for the transplanted cells, then the biomaterial should gradually degrade to permit engraftment of the transplanted cells into the host tissue.72 Moreover, the biomaterial must degrade into non-toxic products.73In vivo, biodegradation of materials occurs mainly through (1) bulk degradation via hydrolysis and (2) proteolytic degradation by enzymes. In the brain, the initial surface erosion of a hydrogel may be determined by infiltration of water, free radicals and secretion of esterases by immune cells. At later time points, when the compressive modulus of the hydrogel decreases, immune and glial cells infiltrate the scaffold and contribute to the proteolytic removal of the core of the hydrogel.74
Several approaches have been explored to temporally control material biodegradation. For example, it has been demonstrated that, considering a range of acetylation between 0% and 60%, a higher degree of acetylation75 (or a lower degree of deacetylation)76 increases the degradation rate of chitosan. However accelerating degradation can compromise the stiffness of a biomaterial, thus methods that allow us to control independently the degradation rate and elastic modulus have been investigated. For instance, the degradation rate of alginate hydrogels, which under physiological conditions are not hydrolytically nor enzymatically degraded, can be accelerated by increasing the oxidation degree,73 but oxidation also makes the gels less rigid. A method to solve this problem was proposed by Kong and colleagues: they showed that tuning the molecular weight distribution (MWD) of oxidized alginates allows the regulation of the degradation rate of gels without varying the number of oxidized uronic acids, thus limiting changes in the elastic modulus and swelling ratio.77
In addition to natural materials, synthetic polymers have been specifically designed to degrade in a controlled fashion. The degradability and biocompatibility of PEG hydrogels can be tuned by changing the length of hydrolytically degradable lactic acid units within the polymer crosslink, as assessed by Bjugstad and co-workers who implanted fast degrading (more lactic acid units), slow degrading (less lactic acid units) and nondegrading (no lactic acid units) PEG hydrogels in rodent brain tissue. Data indicate that both astrocytic and microglial responses to PEG hydrogels change as a function of degradability and contact time.74 Moreover the time scale over which neural cells extend processes throughout the PEG scaffold can be regulated by changing the degradation rate through incorporation of different hydrolytically degradable macromers.72
To achieve a specific degradation rate without altering the mechanical properties of the scaffold, oligopeptides that are sensitive to the enzymatic cleavage have been engineered into synthetic polymers. The resulting hydrogels are specifically degraded by targeted proteases involved in matrix remodeling such as matrix metalloproteases (MMPs), collagenases and plasmin. For example, genetically engineered protein polymers containing the RGD motif and two degradation sites for plasmin and MMPs,78 or multiple collagenase-sensitive domains,79 were cross-linked with PEG to enable cell adhesion and cell migration within the scaffold. RGD and MMP-2-specific cleavable substrates were also incorporated into SAPs (RADA16)80 and PAs81 allowing the formation of self-assembling biomimetic nanofiber networks with increased degradation rate.
Despite the above-mentioned promising results, further work needs to be done to precisely coordinate the scaffold degradation rate and new tissue formation rate while maintaining the initial biomaterial mechanical properties.
3. Computational investigation of self-assembling nanomaterials
Self-assembly is ubiquitous in nature at all scales and describes the spontaneous association and organization of multiple individual components into ordered structures without external direction.82 At the molecular level the self-assembly efficiency relies on the establishment of many weak and reversible interactions (hydrogen bonds, van der Waals, π–π and electrostatic interactions) among the components,83 with high numbers of these interactions triggering the formation of ordered supramolecular architectures. In nature this phenomenon gives rise to a range of highly performing and functional materials including silk, collagen, cellular organelles, bones and teeth. Hence the inspiration to develop self-assembled systems going from bi, tri-block copolymers, complex phospholipids,84 DNA,85 protein and peptide based materials.Even if each cited class of biomaterial follows specific self-assembly paradigms, in this section we focus mainly on the self-assembly of peptides and, to a lesser extent, on polymer and electrolyte hydrogels used in tissue engineering.
Self-assembling peptides (see Subsection 2.2.2) feature the ability to spontaneously form hierarchically organized aggregates comprising β-sheet or α-helix-rich structures and an unmatched versatility in scaffold biofunctionalization. In SAPs the monomer encodes most of the morphology of the assembled nanostructure: therefore an efficiently planned design depends on a deep understanding of the sequence-to-structure relationship. Computational methods have become increasingly useful in pursuing this goal thanks to the rapid evolution of high-performance computing and the development of parallelized algorithms. In computational experiments the study of SAPs forming β-sheet-rich amyloid-like structures can rely on a large list of achievements prevalently conducted to study amyloid peptides associated with human degenerative diseases. In fact, the self-assembling ability of peptides was considered to be a specific property of some sequences responsible for the formation and deposition of amyloids, highly ordered assemblies, observed in body organs and tissues of patients affected by a variety of diseases. In this context several theoretical methods have been used to back experiments for understanding the self-assembly phenomena, aiming at gaining feasible “tools” preventing the formation of amyloids and/or destroying amyloids already formed. In addition to that, in the last few decades, it became widely accepted that the self-assembling ability is a general property of many other peptides, enabling scientists to use peptides as building blocks of nano-materials in different applications. The SAPs therefore provide us the opportunity to introduce different theoretical methods to deal with the computer-aided biomaterial design not only at the simple building block level, but also in terms of the hierarchical organization of such building blocks. In this section we will focus mainly on the application of various computational techniques to SAPs involved in human degenerative diseases and in tissue engineering. We will begin with the description of the β-rich cores that form in SAP amyloid-like nanostructures, followed by a schematic discussion of the assembly mechanism. Then we will overview computational methods, mainly molecular dynamics, recently emerged as valuable tools to shine light on empirical data and on proposed molecular models at different length scales. We will report the recent computational achievements in the study of amyloid-like SAPs following, in our description, the increasing complexity associated with the different steps of the assembly pathway. Lastly, we will provide a quick overview of computational studies developed for polymers and polyelectrolyte hydrogels.
3.1 Self-assembling peptides: nanostructure and aggregation mechanism
The protein/peptide's ability to self-assemble into highly organized fibrillar aggregates, known as amyloids, has long been considered to be a peculiarity of proteic sequences involved in degenerative conformational diseases like Alzheimer's, Parkinson's and prion's.86Regardless of the protein sequence or length, amyloid fibrils share a structural organization known as cross-β structure with β-strands and β-sheets, respectively, perpendicular and parallel to the fibril axis.87 This common core of cross-β structures, likely suggesting a similar aggregation mechanism,88,89 is characterized by X-ray diffraction (XRD) patterns with meridional reflections at 4.7 Å (inter-strand distance), equatorial reflections at 8–11 Å (inter-sheet distances) and by apple-green birefringence upon staining with Congo Red solution (see Section 4.2 for details about characterization methods).
Today it is accepted that the propensity to assemble into ordered cross β-structures, in line with the “generic hypothesis” of amyloid formation,90 is an inherent characteristic of polypeptide chains, whose sequences modulate the response degree to the environment. Under specific environmental conditions (i.e. in solutions at a defined concentration, solvent, pH, salt concentration, temperature, etc.), aggregation kinetics and the resulting aggregate morphologies are influenced by charges, steric hindrance and the hydrophobicity/hydrophilicity profiles91 of the primary sequence of residues. The self-assembling ability of polypeptide chains, together with the ease of synthesis and functionalization (see Subsection 2.2.2), made very popular their use as building blocks of nano-structured materials adopted in different fields of material science, including tissue engineering. In this context the need arises to gain a deep knowledge of SAPs at different length scales associated to the different assembly stages. In Fig. 3 we propose a scheme of the most largely recognized polypeptide aggregation pathway.
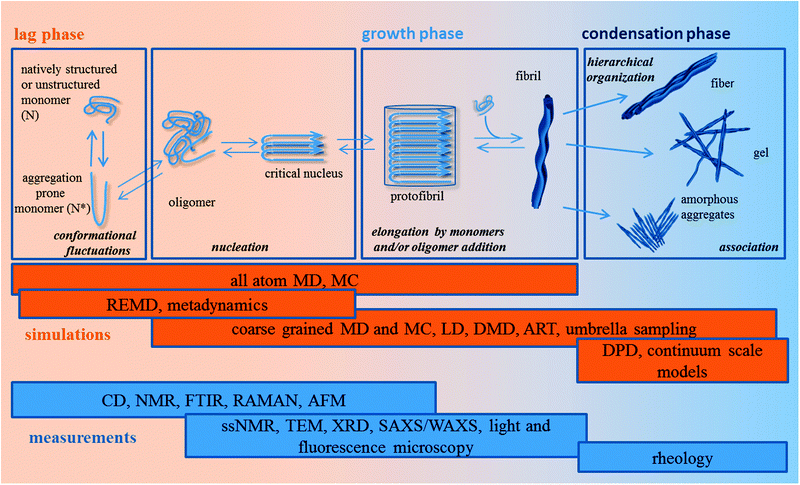 | ||
| Fig. 3 Schematic illustration of the self-assembly mechanism and computational/experimental techniques available to investigate different aggregation levels. | ||
Before going through the different aggregation steps we introduce a theoretical tool widely adopted in the study of protein folding and aggregation: the free energy landscape. Complex biomolecular systems (protein, peptides and even more complex aggregates) have typically access to a wide spectrum of conformations. Such a dynamical behaviour, essential to carry out biological functions, can be represented by means of a free energy surface (FES) or landscape (FEL). The ‘free energy landscape’ (Fig. 4) is obtained by plotting the free energy values, associated with the different conformations of the system, as a function of a couple of reaction coordinates. These last ones are abstract coordinates, often spatial coordinates, relevant in describing the system's kinetics and its dynamic properties. The landscape looks like a rugged surface with a complex topography characterized by the presence of valleys and passes: each point on the landscape corresponds to a configuration. Knowledge of the free energy minima (valleys) population and of free energy barriers (passes) provides information, respectively, about the relative stability of the states of the system (thermodynamic information) and about transition rates between these states (kinetic information).
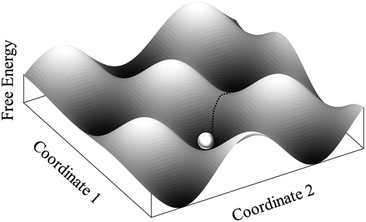 | ||
| Fig. 4 Free energy surface explored by a system during a simulation. | ||
At the beginning of the polypeptide aggregation pathway there is a lag phase. During the lag phase, starting from a monomer conformational spectrum dictated by its sequence and environmental conditions, nucleation occurs: oligomers form and, when a ‘critical’ size is reached, relax into stabilized nuclei. This phase is highly influenced by the folding landscape of the monomer. Indeed, the monomeric polypeptide chains conformationally fluctuate on their rugged free energy landscape92 and transiently populate an ensemble (N*) of aggregation-prone conformations influencing the oligomer formation and the morphology of the aggregates.
In the growth phase, the nuclei grow and elongate via monomer93 and/or oligomer94 addition to form protofibrils and fibrils. In the condensation phase aggregate–aggregate association steps lead to the formation of ordered fibers and scaffolds, e.g. fibrils bundling into fibers and, fibers association into scaffolds. In this last phase the formation and precipitation of amorphous aggregates may also occur.
In other words, the aggregation mechanism is a multistep process going through the formation of various transient species. These transient states are rather difficult to characterize by empirical measurements: a deeper understanding of the aggregation phenomena requires to penetrate as much as possible into the molecular details of the process.
In this scenario, molecular simulations (see Section 3.2) provide a valid complementary tool to nuclear magnetic resonance (NMR) and XRD experimental techniques to understand biological processes. Moreover, understanding the energetic profile and the mechanisms of the aggregation processes between proteins represents a fundamental theoretical challenge and has significant importance in biomaterial science as well as in other fields.
3.2 Computational approaches
Computer simulations enable us to obtain accurate structural and thermodynamic properties of molecular systems, as well as their time-dependent behaviours, through the generation of representative molecular configurations (conformational sampling).Given a system formed by N particles a configuration is defined by 3N spatial coordinates and 3N momentum components: each configuration corresponds to a point in the 6N dimensional space called ‘phase space’, namely the space collecting all possible configurations of the system. The accuracy in predicting the system properties within a simulation is directly linked to the simulation ability to ‘sample’ the phase space. An ad hoc simulation protocol can be designed by combining different sampling algorithms and structural models, depending on the chemico-physical properties and on the self-assembly phase we are interested in. The sampling algorithms and the structural models mainly used to deal with the self-assembling pathway of biopolymers are described below and, more schematically, in Tables 2 and 3.
| Sampling algorithms | Description | Theory Ref. | Applications Ref. |
|---|---|---|---|
| Monte Carlo (MC) | System conformations are generated by random moves of the structural units one at a time, and accepted by following the Metropolis criterion | 95, 96 | 4, 108, 112, 131, 173 |
| Molecular Dynamics (MD) | System conformations are generated by integrating Newton's equations of motion. Particles interact by means of continuous potentials. The system evolves with time | 97 | 123, 124, 134, 154, 156, 162, 164, 168–171 |
| Replica Exchange Molecular Dynamics (REMD) and Replica Exchange Monte Carlo (REMC) | A series of parallel MD or MC simulations at different temperatures are performed. Then, based on the Metropolis criterion, configurations can swap among adjacent temperatures | 101 | 122–124, 153 |
| Metadynamics | The simulation is driven by a history-dependent potential along a selected number of ‘slow’ degrees of freedom, the collective variables (CVs) | 104 | |
| Discrete Molecular Dynamics (DMD) | The simulation is driven by collision events modelled by means of discontinuous step-function potentials | 97 | 132, 133 |
| Langevin Dynamics (LD) | Friction and random terms are introduced in Newton's equations to take implicitly into account the solvent viscosity | 103 | 109, 110, 134, 166 |
| Umbrella Sampling and Steered Molecular Dynamics (SMD) | Molecular dynamics in which bias potentials are introduced to drive the system from a thermodynamic state to another one along a reaction coordinate | 102 | 151, 154 |
| Activation–Relaxation Technique (ART) | The configurational space is reduced to a network of local minima connected by activated states | 114 | 144 |
| Structural-coarsening model | Structural units | Description | Ref. |
|---|---|---|---|
| All-atom | Atom | Each atom is explicitly considered | 134–138, 154, 156, 168 |
| Caflisch model | Backbone atoms and side-chains | Four spherical backbone beads and six spherical side-chain beads of hydrophobic and hydrophilic nature | 109 |
| Shea model | Sub-residue interaction centres | Two interaction centres for backbone and one interaction centre for side-chains | 110 |
| PRIME model | Backbone atoms and side-chains | Three spheres for backbone and one for side-chain | 111, 132 |
| OPEP force field | Backbone atoms and side chains | One bead for each backbone atom and a specific bead for all side chains | 114, 137, 138, 144, 145 |
| Stedall model | Monomer | Each half-peptide is represented by a linear array of n interacting sites | 112 |
| Tridimensional lattice | Monomer | Each peptide chain consists of a number of beads confined to the vertices of a cube: the beads can be hydrophobic, hydrophilic or neutral | 108 |
| Amphiphilic model | Monomer | Three kinds of monomer units are considered: hydrophobic, peptidic and epitope | 4 |
| Tube model | Polypeptide chain | The polypeptide chain is represented by a tube | 115, 139 |
| Cuboid model | One or more peptides | Each peptide is modelled by a cuboid characterized by three parameters of the energy surface | 107 |
| DPD | Group of atoms or volume of fluid | Polymer chains are modelled by beads composed of groups of atoms | 116, 171 |
Among the plethora of strategies, used to understand and predict the molecular properties of a system, we will get into the details of two of the most used approaches: the stochastic approach of the Monte Carlo method and the deterministic approach of molecular dynamics.
In the Monte Carlo (MC) method,95 the conformational sampling is obtained by randomly and/or systematically moving a single atom (or a molecule) or by rotating one or more bonds. The generation of configurations is biased, by means of the Metropolis criterion,96 toward low energy configurations, facilitating an accurate estimation of the system thermodynamic properties. The Metropolis criterion states that the probability of occurrence of a given conformation is proportional to the Boltzmann factor, exp(−ΔE/KBT), associated with the difference ΔE of potential energy between the new configuration and its predecessor.
In molecular dynamics (MD) simulations97 the system conformations are obtained by means of the numerical integration of Newton's equations of motion over a given period of time. The interactions between atoms are described by means of pairwise additive potentials. The solvent effect can be taken into account both explicitly and implicitly. In the implicit modality the solvent effect can be incorporated into the energy function98 or described with the generalized Born model.99 MD simulations with full-atom resolution solutes embedded in explicit water are widely used to explore the conformational fluctuations of monomers and small oligomers at constant temperature. They are also used to test β-sheet models stability, providing insights into effects of net charge, amino acid and solvent compositions, and population of aggregation-prone conformations N*.100 To overcome the conformational sampling limits of conventional MD simulations, arising from the difficulty to escape from energy holes distributed over the rough energy landscapes of peptides, various research groups introduced enhanced MD sampling methods.
The replica exchange molecular dynamics (REMD)101 have been extensively used to simulate the large conformational changes related to the modelling of misfolded proteins. In this method a series of MD simulations (or replicas) of the system run in parallel at different increasing temperatures and configurations can exchange between neighbouring replicas according to an energy based Metropolis-type probability of swapping. The obtained extended conformational sampling provides a better description of the thermodynamic properties of the system, however, taking the system evolution out of a real “physical” time. The replica exchange modality can be applied also to MC simulations (REMC).
To overcome the sampling problem and provide a free energy profile along a reaction coordinate, the umbrella sampling method102 can be used with both Monte Carlo and molecular dynamics simulations. The name ‘umbrella’ derives from the application to the system of a bias, an additional energy term, to connect energetically separated regions in the phase space. The bias action pulls the system from one thermodynamic state to another in one simulation or in different simulations (windows) covering intermediate steps. The sampling in each window can be improved by replica exchange methods. A related approach named steered MD is used to model the effect of tensile-mechanical forces applied to the simulated system, mimicking the action of an atomic-force microscope cantilever.
Very fast alternatives to traditional molecular dynamics are the Langevin Dynamics (LD) simulations and the Discrete Molecular Dynamics (DMD). Langevin Dynamics relies on the Langevin equation of motion103 that is obtained by introducing a friction term in Newton's equation of motion. This allows us to incorporate the implicit viscosity of the solvent and to regulate temperature as a thermostat. Discrete Molecular Dynamics97 are collision-driven molecular dynamics, sampling much wider regions of conformational space, longer time scales, and larger systems. In DMD simulations particles interactions are treated by discontinuous step-functions of the inter-particle distance, rather than by continuous pair potential used in traditional MDs. This implies that bodies' trajectories evolve via discontinuous jumps in the momenta of the system at discrete interaction times. The solvent is modeled implicitly by including the hydrophobic interactions among nonpolar side chains. Backbone hydrogen bonding is modeled in explicit detail.
In the recently developed metadynamics104 an history-dependent potential is introduced to drive the system sampling along a selected number of ‘slow’ degrees of freedom (called collective variables, CVs) and to increase the occurrence of events that would be rare for the currently accessible time scale in standard MD simulations. The challenge is to find the appropriate CVs along which the process of interest is observable. In other words, the initial, intermediate and final states of the process have to appear clearly defined with the selected CVs.
Dealing with self-assembly at a bigger scale like in protofibrils and fibrils, statistical mechanics105 and coarse-grained modelling are generally used. The increasing time and structure dimensions to be covered can be reached abandoning the all-atom description and using simplified models retaining only the essential physical properties of the system.
A large variety of differently simplified coarse-grained models106 has been applied to protein aggregation. The ‘coarse-grained’ approach integrates a large number of degrees of freedom into a fewer interaction potentials chosen with the help of detailed knowledge of the system.
In bead models, amino acid atoms are grouped into interacting centers (beads or grains): the lower is the beads number, the less time-expensive is the simulation and the more extendable is the simulation time. Furthermore, the accuracy and transferability of the force fields, used to describe the interactions among beads, decrease with the ‘coarsening’: indeed condensing the system dynamic behavior in a smaller number of parameters makes these force fields highly system specific.
In the cuboid model107 identical cuboid units are used to represent a single peptide or a small oligomer without considering conformational transitions. Simulations are performed with the Monte Carlo procedure and use only three types of cuboid interactions: strong attraction along the intra-sheet hydrogen-bonding direction, weak attraction along the inter-sheet direction and repulsion along the direction parallel to the cuboid unit.
In another simple representation, chains of amino acid beads, which can be only hydrophobic, hydrophilic and neutral, are projected on a tridimensional lattice.108 This representation shows a relatively low portability in real tridimensional structures but, combined with the Monte Carlo algorithm, allows us to elucidate the roles played by the different interactions as a function of their relative weights in the aggregation mechanism.
Conformational flexibility modulated by a dihedral potential term is instead considered in the coarse-grained models of Caflisch109 and Shea.110 In the first model peptide units are represented with four spherical backbone beads and six spherical side-chain beads (hydrophobic or hydrophilic) and interact via van der Waals and electrostatic interactions. In the second one each residue is represented with one and two interaction centers, respectively, for the side-chain (hydrophobic, polar, positively or negatively charged) and the backbone.
The PRIME model,111 giving an intermediate-resolution description of a system, is an off-lattice model, where each amino acid is represented with four spheres (three for the backbone and one for the side chain), while geometric constraints, hydrogen bonds and hydrophobic interactions are modeled through a combination of hard-spheres and square–well interactions by using discontinuous molecular dynamics. The PRIME model has been recently extended to all 20 amino acids.111
A coarse-grained model for PAs has been developed by incorporating only the basic chemical structures necessary to differentiate hydrophobic peptide and bioactive epitope (which is also of amino acidic nature) units within each monomer.4
The self-assembly of the so-modeled PA molecules has been simulated by an alternate sequence of Monte Carlo and stochastic simulation steps.
Stedall and co-workers112 described two SAP sequences designed by the Woolfson group113 by means of coarse-grained rigid rods, each one made of a half peptide and represented by a linear array of n interaction sites. During Monte Carlo simulations, the half peptide can interact through specific and unspecific pair wise interactions using a limited number of parameters.
The Activated Approach allows a rapid exploration of the configurational space reducing it to a network of local minima connected by activated states. In the activation–relaxation technique (ART)114 the system passes the local minima through a well-defined transition point connecting two adjacent basins. The coarse-grained protein is represented with a bead per backbone atom and a specific bead per side chain, while energetics are described by the optimized potential for efficient peptide-structure prediction (OPEP).114 The generated trajectories are possible physically but not dynamically, because the physical time is not considered.
In the ‘tube model’ used by Auer and colleagues,115 the polypeptide chain is represented by a tube whose finite thickness accounts for the volume occupied by the backbone atoms and whose symmetry is broken only by hydrogen bonds, hydrophobic interactions are treated in a pair-wise additive manner. The simplicity of this model, neglecting sequence-specific interactions, allowed us to calculate the nucleation barriers involved in the aggregation of peptides and proteins into characteristic cross-β-structures of amyloid fibrils.115
Among the multi-scaled approaches, dissipative particle dynamics (DPD) have to be mentioned as a valid tool to study soft condensed matter including polymers. The DPD technique, introduced by Hoogerbrugge and Koelman116 to simulate dynamics and rheological properties of isothermal fluid systems, is a stochastic mesoscopic simulation method analysing time and dimensional scales greater than MDs. The system is composed of beads representing groups of atoms or a volume of fluid interacting via simplified pairwise conservative, dissipative and random forces. In this approach polymer chains are modeled by introducing spring forces tying adjacent beads.
Atomistic and molecular dynamics provide insights into the biomechanics of proteic materials at the molecular level. These aspects of understanding are connected to their macroscopic mechanical properties in a multi-scale simulation approach. According to this approach small scale considerations are used to derive averaged equations for a much larger scale so as to pass through different hierarchical levels of organization of the material. The required up-scaling work is known as homogenization: at its end the material can be described with the continuum theory,117 where Representative Elementary Volumes (REV) are introduced and defined as the smallest volumes of the material that can be described as continuous homogeneous media. By “connecting” multiple REV it is then possible to describe chemical and mechanical properties of whole scaffolds and compare them with empirical data (Section 4.2) crucial for transplanted cell survival and scaffold engraftment (Sections 5.2 and 5.4).
3.3 Following the self-assembly pathway: from monomers to fibers
Recently, Lin and Shell122 performed simulations of peptide folding for 142 short peptides: they compared the obtained predictions of the aggregation propensity with empirical predictors. Conformational samplings, performed with REMD simulations in implicit solvent, have been used to compute a number of single-peptide metrics, whose roles, in terms of the peptide aggregation propensity, have been assessed by means of statistical analyses. Regardless of a poor predictive efficiency likely due to their single-peptide basis, the obtained trends are consistent with the phenomenological predictive approaches in terms of the hydrophobic surface area and the number of exposed charged amino acids.
The last considerations introduce the issue of the connection between the monomer conformations sampled by simulations and their aggregation ability, but, most importantly, the high polymorphism at the nanoscale exhibited by assemblies. This polymorphism is closely related to different disease states in vivo as well as different functions and mechanical properties of the assembled structures.
The conformational change of the monomer is a necessary step toward aggregation, and molecular dynamics simulations allow for observing the formation of β-sheet-rich aggregation-prone conformations. This is the case of peptides belonging to the most flexible portion of the prion protein, the H1 peptide or the larger 82–146 fragment.123,124
Nevertheless, the correlation between sampled monomer conformational fluctuations and aggregation propensity cannot be direct.89,125 For β-sheet-rich native monomers, explicit links between the aggregation propensity and external perturbations (pH, punctual mutations) emerge from simulations. But in the case of monomers encountering great conformational changes, like highly flexible peptides, a central role is played by intermediates,126 in accordance with the protein folding landscape approach.92 In such a perspective, it has been showed that analysing monomer conformational spectra N*100,108,127 can provide insights into the aggregation propensity of the sequence and can justify different morphologies of aggregates, highlighting the monomeric seeds from which they can take shape. This is due to the fact that: (1) the N* conformations have a great overlap with the structures of the monomers in the fibril; (2) the probability of populating the N* conformations is directly linked to the ease of aggregation; (3) the free energy difference between N* and the native or unfolded states modulates the aggregation kinetics.
The results obtained by these theoretical works, together with experiments, highlighted two main plausible scenarios for early events of fibrillation.89 In the first one, the aggregation prone structures N* are populated through partial unfolding of the native state or partial folding of the unfolded state (applicable to Aβ-peptides of Alzheimer's disease). In the second one, N* structures are lower in energy with respect to the native state ensemble, thus making the folded functional state metastable (applicable to the pathogenic form of the prion protein PrPSC).
Nonetheless, conformational changes being important for the aggregation driven by inter-peptide interactions, it is unlikely that the conformational dynamics of isolated peptides can fully explain variations both in deposition rates and in nanostructure morphologies.
As a first step toward aggregation, dimer formation has been simulated for different peptides with all-atom molecular dynamics both in explicit and implicit solvents.128–130 Hydrophobic and electrostatic interactions appeared to be the main actors in the dimer formation, while β-structures stabilization is reached mainly via inter-backbone hydrogen bonds. Hydrophobic interactions, in particular, play a fundamental role in the entropically unfavourable removal of structured waters between the monomers.
The cooperative effect of interactions among monomers leads to the stabilization of well-packed β-sheet structures from disordered oligomers composed of a critical number of chains (critical nuclei). Nevertheless, ordered conformations (β-sheets or β-barrels) may also be obtained in disordered oligomers smaller than the critical size.
Ordered structures have been observed by MC simulations of six Aβ(16-22) chains131 and DMD simulations of polyalanine132 and prion fragments.133 The effect of system size and peptide concentration over peptide aggregation has been studied on islets of amyloid polypeptide IAPP.134 However, the reduced system size, consisting of a handful of peptides, introduced artefacts responsible for the instability of aggregates, altering H-bonding formation, secondary structure content and other system properties strongly dependent on peptide concentration.
All-atom simulations of small oligomers are necessary to identify the driving force of protein aggregation.135,136 Nevertheless, some inherent limits, like finite system size, conformational space and time scale accessible to standard all-atom MD simulations, need to be overcome in order to study the general aggregation features of proteins. To this purpose great contributions come from coarse-grained methods in combination with all-atom ones.
Notably, coarse-grained and all-atom methods were combined in a two-step multi-scale approach to tackle the thermo-dynamical and structural properties of low molecular weight oligomers: the idea was to take advantage of the extended sampling efficiency of coarse-grained simulations and of the accuracy of full atomistic simulations in explicit solvent.137 The free energy surface of the 7-mer NHVTLSQ (β2m83-89 peptide) organized in β-barrel assemblies (whose structures have been predicted by coarse-grained OPEP simulations) has been constructed via REMD simulations. The study demonstrated that β-barrels are true free energy minima on the two and three dimensional free energy surfaces, albeit with a lower probability than amorphous aggregates.
The same multi-scale approach has been used to investigate aggregation and polymorphism of GNNQQNY oligomers and of systems made of 20 peptides,138 highlighting how aggregation is mainly triggered by the formation of dimer, trimer and/or tetramer seeds.
Auer and colleagues clearly observed the formation of ordered nuclei within amorphous aggregates, a “condensation reordering” mechanism, in simulations conducted with fully coarse-grained tube models on a representative system of 80 weakly hydrophobic 12-mer homopolymers.115,139
Inspired by kinetic experiments, Straub and Thirumalai envisioned two main mechanisms for the longitudinal growth mediated by monomer addition.142 In the first one-step mechanism the monomer, assuming a growth-competent conformation, encounters the fibril-end and deposits on it. In the second one there are two steps corresponding to the dock-and-lock of the monomer to the fibril end: the monomer locks with nonspecific modality to the fibril-end and, subsequently, the monomer–fibril complex undergoes structural rearrangement resulting in the monomer integration into the fibril.
The dock-and-lock mechanism has been clearly observed in extensive all-atom MD simulations, showing the addition of the unstructured monomers GNNQQNY (from the yeast protein Sup35) and GGVVIA (from the Aβ-peptide) to the end of their respective amyloid fibrils.143 In particular, the analysis of the locking step highlighted the importance of expulsion of water molecules and the stabilizing contribution of the H-bond network involving peptide backbones and side chains.
Simulations of Aβ(16–22) using both the ART and MD-OPEP methodologies144,145 showed that the monomer integration is reached through ‘reptation’ moves (analogous to a slithering snake), rearranging the network of H-bonds without the need for detachment.
In the work of Pellarin and Caflisch et al.,109 the attention is focused on the effect played on the elongation pathway by the intrinsic tendency of an amphipathic polypeptide chain to self-assemble. Multiple Langevin dynamics with a coarse-grained model allowed us to sample hundreds of fibril formation events: the simulations showed a heterogeneous set of elongation pathways, fostering the formation of a number of on-pathway protofibril intermediates. Moreover, the fibril longitudinal growth appears to be determined mainly by dock-and-lock monomer addition. On the other hand a mechanism of lateral growth is also observed: the formation of an ordered fibril occurs by template assembly of a previously deposited file of monomers on the lateral surface of a preformed protofibril. This mechanism is preferred when the β-aggregation propensity is low.
Computational studies, designed to investigate the topologies of large aggregates (spherical micelles, fibers, amorphous aggregates etc.…) formed by self-assembly, have been conducted on the amphiphile peptides developed by Stupp et al.146 (see Subsection 2.2.2 for details). A coarse-grained model of these PAs has been used by Velichko and co-workers4 (see Section 3.2 for details) to gain insights into the effects of the competition between hydrogen bonding and hydrophobic interactions on the formation of the molecular aggregates and to draw a phase diagram as a function of these competing interactions. Tsonchev and co-workers5 focussed instead on the effects of the pH and salinity on the morphologies of PA aggregates4 by means of Monte Carlo and molecular dynamics simulations. In this case the amphiphilic peptides have been modelled as strings of spheres of radii progressively decreasing from the hydrophilic heads. The simulation results, in combination with empirical data, allowed us to build a pH/salinity phase diagram of PAs and to explain it in terms of electrostatic and hydrophobic interactions.
That is the case of the GNNQQNY peptide of Sup35. The stability of differently sized aggregates, obtained from recently determined crystal structures, has been showed by means of a detailed MD study.147 In addition to that, this study highlighted the compatibility of steric zipper interactions with both flat and twisted β-sheets.
Bellesia and Shea135 proposed a protofibril model of the KFFE peptide consistent with electron microscopy experiments.148 They used Langevin dynamics on a full-atom model in explicit solvent to assess the relative stability of small double-layered protofibrils. Four different arrangements of two β-sheet tapes with parallel and antiparallel interlayer orientation were considered. Both hydrophobic and electrostatic interactions concurred to the stabilization of the fibril, with a greater contribution from the electrostatic interactions between lysine and glutamic acid. Phenylalanine contributed to the overall self-assembling propensity both with its high β-sheet propensity and favourable stacking interactions.
Nguyen and Hall developed a fibril model of the 48 poly-alanine peptide Ac-KA14K-NH2 in simulations based on the PRIME model.132 The obtained fibril mimicked the structural characteristics observed in experiments and contained about three-to-six β-sheets, each one showing multiple highly parallel peptides with tightly packed side-chains. The stability of the obtained fibrils has been studied over a wide range of temperatures to investigate the relative importance of hydrogen bonding and hydrophobic interactions in fibril stability. They demonstrated that amorphous aggregates, rather than fibrillar ones, are formed when the strength of the hydrophobic interactions between nonpolar side chains is comparable to the strength of the hydrogen bonds between backbones,
In another work, making use of structures obtained by solid state NMR, fibers as long as hundreds of nanometers, featuring two- and three-fold symmetric morphology, have been modelled for studying the Alzheimer peptide Aβ(1–40).149 To elucidate their size-dependence properties, atomistic simulations were performed to analyse energies, structural changes and H-bonds formation as a function of the number of layers and fibril length. Nanomechanical properties have also been explored by MD constant force experiments by applying compressive and tensile deformations.
Lastly, the application of theoretical methods to SAPs used in tissue engineering led to model filaments of differently functionalized peptides of the RADA family. In particular, nanofibers of RADA16-I peptides have been constructed by using the cross-β-structure proposed by Park and colleagues150 to assess how functional groups affect the stability of the filaments.7 All-atom MD simulations in implicit solvents provided indications of the roles played by hydrophobic and hydrophilic interactions: while hydrophilic functional groups showed a marked propensity to open the initial bilayer conformation, the hydrophobic ones provide stability to the β-sheet bilayer filament.
For example, Schor and Bolhuis151 used a multi-scale modelling approach combined with rare event simulations such as steered MD and umbrella sampling to elucidate the self-assembly mechanism of silk-based block copolymers into fibrils. Starting from the Claviceps N. silk sequence, full-atom structures of aggregates have been obtained by means of replica exchange molecular dynamics. The obtained structures showed alternate disordered and ordered crystal domains composed of poly-glycine-repeats and poly-alanine β-sheets respectively.152 The two different domains have been represented by a minimal coarse-grained model153 with beads connected via multi-linear springs in a serial arrangement and their interactions were modelled using parameters extracted directly from the all-atom simulations. The mechanical deformation was then analysed in response to different levels of strain and for different sizes of β-sheet nanocrystals.
Buehler and co-workers154 explored the nanomechanics of collagen microfibrils incorporating both the full biochemical details of the amino acid sequences and their molecular arrangement at the nanoscale. Starting from the amino acid sequence of human collagen and by means of homology modelling, the molecular arrangement of human collagen has been extrapolated from the structure of Rattus Norvegicus collagen. Then, the nanoscale model has been constructed using crystallographic information on the naturally occurring unit cell. MD simulations conducted on the obtained model allowed us to equilibrate the all-atom structures and to assess the mechanical properties of a collagen fiber by applying increasing constant mechanical stress.
Moreover, the natural polymers chitosan and chitin, both structural elements of the arthropoda exoskeleton, have been studied in computational investigations from the atomic to the nanoscale level. At the atomic level, a systematic MD study of conformational flexibility of chitosan and chitin in di-, tri- and tetra-saccharides has been performed leading to theoretical conclusions consistent with experimental data.155 In particular, in agreement with NMR data, chitin showed longer persistence lengths (i.e. the length over which the memory of the initial orientation of a polymer persists) than chitosan. In another work, a multi-scale approach has been used to study the mechanical properties of the multi-layered hierarchically organized lobster cuticle, composed of chitin and other various proteins, mineral nanoparticles and water.156 Being an inhomogeneous material, the determination of the overall elastic properties of the cuticle was obtained by means of an homogenization work integrating features at different scales, from the lowest (atomic) to the highest level (the cuticle tissue) of its hierarchic structure. For example, the elastic constant of chitin, obtained via molecular dynamics, was introduced in a continuum-scale model so as to find the elastic constants of chitin-protein fibers forming the cuticle.
3.4 Polymer and polyelectrolyte hydrogels
Hydrogels formed by water-soluble non-peptidic polymers cross-linked in a network are widely used in tissue engineering (see Subsection 2.2.2 for details). Cross-linking reactions can involve the formation of covalent bonds, giving rise to permanent hydrogels, or of physical interactions (ionic interaction, hydrogen bonding, etc.…), giving rise to physical hydrogels. Hydrogels swell up to several hundred times their dry mass after water imbibition and are classifiable as conventional hydrogels or stimuli responsive hydrogels whether their swelling capability can be unaffected or affected by physical (temperature, magnetic and electric fields, solvent composition, light, pressure), chemical or biochemical (pH, ions, molecular recognition events) stimuli.Uncharged polymer networks can be studied by the scaling approach,157 assuming that the polymer network behaves like a polymer solution under semi-dilute conditions forced by cross-links. Under the semi-dilute conditions gels may be represented by a collection of adjacent closely packed blobs, each blob being associated with a chain portion and having properties very similar to those of a single chain and, inside the blob, still behaving as an isolated self-avoiding walk.158
A modified scaling approach is instead required to treat charged polymers cross-linked in a network, i.e. the polyelectrolyte gels.159–161 In this case interactions between charges indeed lead to a behaviour qualitatively different from those of uncharged polymers. The crossover dilute/semi-dilute solution regime occurs at much lower polymer concentrations than in solutions of neutral chains; also, the osmotic pressure strongly depends on added salts and, in salt-free solutions, it exceeds the osmotic pressure of the neutral counterpart because it almost linearly increases with polymer concentration. This is mainly due to counterions dissociation from the polymer when the polyelectrolyte gel is dissolved in water. Counterions are free to diffuse in water but in the absence of salts, in order to fulfil electroneutrality requirements, remain confined inside the gel and the local osmotic pressure thus generated leads to the swelling of the network with a swelling ratio greater than their uncharged counterparts.
The possibility of developing hydrogels optimized for a particular application relies on the understanding of the stimuli–structure response relationship. Modeling and simulations allow us to unambiguously describe the polymer network and to systematically tune and assess the effect of various interactions on the hydrogel performance.
The complexity of the system can be reduced by performing single chain simulations to extrapolate insights into the polymer network behavior. This strategy has been adopted to investigate with MD simulations the conformational transitions and swelling of hydrogels made of poly(N-isopropylacrylamide) (PNIPAAm) cross-linked with N,N-methylenebisacrylamide (MBA).162 The work highlighted the potential of atomistic molecular dynamics of single polymer chains in predicting the conformation changes of PNIPAA hydrogels.
In molecular simulations of systems composed of multiple chains the hydrogel network is typically described as an ideal cubic163 or diamond164 structure.
On the other hand, more complex polymer networks have been preferentially studied adopting the bond fluctuation model165 with the Monte Carlo method, better suited to properly sample the configuration space. Recently, Langevin Dynamic Simulations have been used to study networks under different conditions mimicking realistic conditions of network formation.166 In this case three different types of chain particles interacting by classical potential energy functions are defined: the cross-linker (X), the middle (M) and the ending (E) unit. The modulation of the parameters within the interaction potential functions allows us to tune and describe the network formation phenomena in terms of natural response to the interactions ongoing within the system.
Even if simplified molecular models of polymeric networks and gels began to consolidate in the 80's,167 only in the last decade also fully atomistic MD studies of hydrated polymer networks appeared in the literature.
Full-atom MD simulations have been conducted to investigate the effect of water content on equilibrium structure and mechanical properties of poly(N-vinyl-2-pyrrolidone-co-2-hydroxyethyl methacrylate) (P(VP-co-HEMA)),168 evincing how such properties are mainly dominated by water or by the monomeric sequence (random or blocky) within the polymer chains. MD simulations gave also insights of the thermo-responsive behaviour of poly(vinyl alcohol)/poly(methacrylate-co-N-isopropyl acrylamide) in terms of modifications of the internal texture of the network,169 whereas temperature, hydration degree, residue composition and their sequences were set in agreement with empirical data in order to achieve a realistic representation of the system.
Full-atom MDs were fruitfully combined with incoherent neutral scattering methods in a theoretical/experimental approach to study the network structure, the local polymer dynamics in the network mobility and the polymer induced modification of water properties of poly-methacrylate/poly-vinylalcohol (PVM/PVA) hydrogels.170 Moreover, coarse-grained models of polyethylene grafted to non-adsorbing surfaces were adopted in simulations developed for polyethylene oxide and polyethylene glycol.171 Lastly, simulations using the Dissipative Particle Dynamic method to study the self-assembly, in different solvents, of di-block copolymers (methyl methacrylate-co-hydroxyethyl methacrylate-co-butyl methacrylate)-b-2-(perfluoroalkyl)ethyl methacrylate provided results in qualitative agreement with experimental characterization data.172
Computational methods provided a deeper understanding of the polyelectrolyte hydrogel topology formation, which is almost impossible to determine empirically. In MD simulations Mann et al.,6 starting from a diamond-like network model successfully used for hydrogels in good solvents, explored the set of structures assumed by polyelectrolyte gels in poor solvents. The different conformations assumed by the network have been collected and positioned in the parameter space of charge fraction (f) and Bjerrum length (lB), defined as the separation at which the electrostatic interaction between two unit charges is comparable to the thermal energy KBT (where KB is the Boltzmann constant and T is the absolute temperature in Kelvin). The obtained structure diagram of the equilibrium swelling conformations of charged hydrogels identified five regimes of similar structures: two subregimes of collapsed conformations for small f or large lB, pearl necklaces for moderate f and lB values, stretched structures for large f and moderate lB, and the “sausage” regime for larger lB. The observed regimes fit with the expected propensity of the macromolecules to self-interact rather than interact with molecules of the poor solvent, minimizing the surface contacts with the surrounding water and collapsing into a globular state.
Simulation techniques have been also applied to investigate structural properties of hydrogels composed of two interpenetrating networks. Crosslinking a polymer/polyelectrolyte (polymer I) into a pre-cross-linked polymer/polyelectrolyte (polymer II) is indeed a strategy used to improve the performance of highly swollen electrolyte hydrogels featuring poor mechanical properties. Edgecombe and Linse173 used MC simulations and a coarse-grained polymer model, composed of charged beads connected by springs, to systematically study single polymer networks and double interpenetrating polymer networks in terms of effects on structure, mechanics and swelling. In particular, they highlighted how the swelling capacity of the interpenetrated networks increased and decreased for, respectively, neutral and charged polymers.
Several efforts have been made to properly describe the influence of environmental stimuli.
pH and salt concentration effects on weak polyelectrolyte gels have been analysed by Longo et al.174 in a single theoretical framework, considering conformational degrees of freedom of the polymer, acid–base equilibrium, solution entropy as well as molecular interactions: they demonstrated a nonlinear response of the gel pH to changes of pH and salt concentration of the bath.174
Counterions and ions can also be explicitly considered in MD simulations applied to polyelectrolyte solutions, modelled as an ensemble of bead-spring chains of charged Lennard-Jones particles.175
Environmental stimuli effects on polyelectrolyte gels may also be considered via a coupled multi-field formulation in a finite element method framework. Indeed Wallmersperger et al.176 applied a chemo-electro-mechanical model to gels, placed in a solution bath under pH and salt concentration variations, taking into account dissociation reactions of the bound charges.
4. Synthesis and characterization of nanostructured scaffolds
In the scaffold synthesis step all the information collected through computational modelling, phage display studies and literature knowledge convoy to prepare the scaffolds for the in vitro and in vivo applications. However, before testing the scaffold in cell cultures or animal models, it is necessary to study the morphometrical and biomechanical properties of the novel material. The characterization is undoubtedly a crucial step in the development of natural and synthetic nanofibrous scaffolds for nervous tissue engineering: it is closely related to molecular modelling, confirming or disproving what predicted in computational simulations.4.1 Synthesis: scaffolds preparation
For natural scaffolds it is necessary to choose the most convenient natural source for the extraction of the compound and optimize the extraction process and the quality of the product. As previously exposed in Section 2, natural materials used to construct electrospun nanofibrous scaffolds and three-dimensional hydrogels are extracted from various natural sources such as insects, plants, crustaceans and bacteria.177 In the case of natural materials used for the production of electrospun scaffolds, once extracted the material should be purified and dissolved into the appropriate solvent to obtain the desired concentration and viscosity of the polymer solution.In the electrospinning process the formation of the nanofibers is due to a viscoelastic liquid jet elongation. The viscoelastic jet is derived from a polymer solution (or a melt) and subjected to an electric field generated by a high-voltage power supply. The electrified jet is continuously stretched by the electrostatic repulsion between the charged surfaces. A typical electrospinning setup is made of three components: the high-voltage power supply, the spinneret and the collector; a high-speed camera could also be used to monitor the process. The power supply charges the polymer solution at voltage above 200 V m−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 178 while the electric field is generated by alternating or direct (preferred) current.179 The spinneret is connected to a syringe pump, necessary to maintain a constant flow rate, and, at the other end, to a needle, to guarantee a thin polymer jet. At the end of the needle the polymer solution produces a viscous cone, the Taylor cone, which is under the effect of two electrostatic forces: the Coulombic force exerted by the high electric field and the repulsion of the charged surface. When the electrostatic repulsive force overcomes the surface tension of the viscous polymer solution the result is an electrified jet erupting from the spinneret, yielding fibers collected in non-woven mats. The fiber diameter is reduced by the whipping effect. The configuration of the collector is a crucial set-up to obtain a desired electrospun nanofibers orientation.180 A high-speed camera could be used to monitor the erupting jet from the spinneret to the collector: for example, Reneker and coworkers demonstrated the instability of the electrified jet via high-speed camera recordings and computational modelling simulations.181
178 while the electric field is generated by alternating or direct (preferred) current.179 The spinneret is connected to a syringe pump, necessary to maintain a constant flow rate, and, at the other end, to a needle, to guarantee a thin polymer jet. At the end of the needle the polymer solution produces a viscous cone, the Taylor cone, which is under the effect of two electrostatic forces: the Coulombic force exerted by the high electric field and the repulsion of the charged surface. When the electrostatic repulsive force overcomes the surface tension of the viscous polymer solution the result is an electrified jet erupting from the spinneret, yielding fibers collected in non-woven mats. The fiber diameter is reduced by the whipping effect. The configuration of the collector is a crucial set-up to obtain a desired electrospun nanofibers orientation.180 A high-speed camera could be used to monitor the erupting jet from the spinneret to the collector: for example, Reneker and coworkers demonstrated the instability of the electrified jet via high-speed camera recordings and computational modelling simulations.181
Electrospun nanofibers can be produced via either polymeric solutions182 or polymer melt.183 Electrospinning of polymeric solutions is preferred because it requires simpler equipment: nonetheless usual drawbacks are usage of expensive and hazardous solvents and a low rate of fiber production in the case of weak polymer concentration. On the other hand electrospinning of polymer melts is less frequent due to the higher viscosity of the melts in comparison with polymer solutions: indeed the higher viscosity of the melt usually results in microfibers rather than the nanofibers that could be obtained from polymeric solutions. Consequently the viscosity of polymer solutions or melts plays a crucial role in the fabrication of electrospun scaffolds: rheometry used for the control of this parameter will be discussed in the characterization Section (4.2).
Additionally, other parameters such as electrical conductivity and surface tension of solutions must be monitored in order to obtain uniform fibers with the desired dimensions.184 A common setback in electrospinning is the formation of polymer beads among the electrospun fibers: these beads alter the morphology and the mechanical properties of the formed fibers. This phenomenon can be avoided by modifying the collector, the voltage, the viscosity and especially the surface tension of the polymer solution. The electrostatic repulsion generated between charges on the jet surface increases the surface area promoting the formation of a jet rather than spherical beads; on the other hand, if the surface area decreases, the surface tension tends to convert the jet in beads. Another way to avoid the beads formation consists in increasing the viscosity of the solution: indeed it has been proved that the concentration of beads decreases when the viscosity increases.
The fiber diameter can also be tuned by changing the electrospinning set up: e.g. by varying the solution flow rate, the shape of the needle, the applied electric field or the distance between the collector and spinneret. Collector modifications and changes in the aforementioned parameters led to different electrospinning innovations for biomedical applications, such as aligned or patterned nanofibers, multilayered scaffolds and nanofibrous frames with tuned porosity. Another innovation is the core–shell nanofibrous scaffold in which the shell increases the strength of fibers in the inner lumen and/or protects the bio-active core. In this technique two different polymer solutions are co-electrospun without direct mixing; the formed fibers are characterized by the presence of a protective shell, generally composed of a synthetic and more resistant polymer, and a bioactive core, constituted by a natural polymer possibly loaded with a drug.185
In ionic cross-linking reaction, e.g. for chitosan and alginate, a mixture of cross linker ions are added to the biomaterial solution to stimulate the gel formation.187 Chemical cross-linking is obtained with several chemicals that, in the case of tissue engineering applications, must be biocompatible or easily removable. HA and its chemically modified variants are widely cross-linked through various chemicals.19 Chemical cross-linking and pH–temperature shift are two different ways to promote gelation of collagen. Unlike collagen, Matrigel gelation is caused by a temperature shift only.
Chemical cross-linking, solvent exchange and photo-polymerization are the preferred ways to obtain hydrogel scaffolds from synthetic material solutions.186
In the solvent exchange approach a water-insoluble polymer is dissolved in combination with a water-miscible and biocompatible solvent: when the mixture comes in contact with the tissue, the solvent diffuses out and the polymer precipitates, forming the solid polymeric scaffold. This approach is generally used for synthetic and hydrophobic materials like PLGA.188
Photo-polymerization requires the presence of a photo-initiator and the presence of UV-light to allow the sol–gel transition; the drawback of this technique was the bio-toxicity of most of used photoinitiators. Just recently the development of new and less harmful photo-initiators allowed the use of photopolymerization in several fields such as dentistry, electronic materials, surface coating and nanomedicine. For example Irgacure™ 2959, due to its high cytocompatibility, is the most common UV-activated initiator;189 recently some visible light initiators have been developed as well.190
In the case of peptide-based scaffolds synthesis Fmoc chemistry is widely used (Fig. 2A). Briefly, the Fmoc group, removable under basic conditions, protects the N-terminal end of the amino acid whereas different protective groups removable under acid conditions protect the amino acid side chains. The first amino acid of the sequence (C-terminal) is linked to a resin bead and, through a sequence of deprotection and activation steps, the peptide is elongated. The last step is the simultaneous removal of all the side chain protective groups and the cleavage of the completed peptide from the resin.
4.2 Characterization
In this section we will discuss several characterization techniques aimed to obtain structural, morphological and mechanical parameters of both electrospun scaffolds and hydrogels, discussing the most relevant techniques depending on the type of scaffold to be tested.The first step of characterization of a nanofibrous peptidic scaffold is generally done through a chromatographic analysis followed by a mass spectrometry check to monitor the quality of the synthesis and the purity of the materials. The next phase of the characterization process is aimed at defining the secondary structures of the scaffolds, identifying which types of bond-forces lead to the formation of the three-dimensional structures. Subsequently the morphological analysis of the formed nano- and microstructures can be carried out with different microscopy techniques. Once obtained information about structures and morphology the next step consists in the mechanical characterization of the assembled scaffold in order to obtain visco-elastic parameters of the biomaterials at the meso-scale. A scheme of an ideal characterization process is summarized in Fig. 5. The circular dichroism (CD) spectroscopy is used to obtain information about the secondary structure of proteins and polypeptides in very dilute solutions (0.02–0.06% w/v). Left (L) and right (R) circularly polarized lights combine to produce linearly polarized light: when this light encounters an optically active molecule, with a different absorption for one of two of β-sheet or random coil structures through the analysis of the Amide I (1700–1600 cm−1) and Amide II (1600–1500 cm−1) vibrational bands,191,192 detecting the presence of secondary structures (Fig. 5(b)). Moreover, FTIR analyses allow us to monitor the self-assembling kinetics: i.e. they permit the investigation of pH, time and temperature effects on the assembling kinetics. Information about the secondary structures of samples can be obtained using another spectroscopic technique: the RAMAN spectroscopy. RAMAN and FTIR spectroscopies are fast and not destructive techniques that require small sample volumes and allow the use of the same sample for both techniques, in order to compare directly the obtained information.8 Another spectroscopic characterization technique is NMR. In recent years NMR spectroscopy has been widely used for the characterization of amyloid fibrils and hydrogels.193 Solid state NMR and more precise magic-angle spinning (MAS-NMR) techniques give insights into the secondary structure of noncrystalline and insoluble samples; in particular, amyloid and hydrogel fibril structures exhibit high resolution spectra with sharp resonance lines. Thanks to these data related to the backbone conformations, the supramolecular organization of β-sheets (parallel or antiparallel) and amino acid side-chain interactions can be obtained. Combining data from these different spectroscopic techniques it is possible to develop full-atom models of molecular structures.194,195
In X-ray diffraction (XRD) secondary structures as well as inter-atomic distances of biomaterials can be detected. An incident beam passes through the sample and diffracted beams are collected on a film or a detector; the collected XRD patterns are usually radial-integrated in order to obtain more detailed information about its structure (Fig. 5a); the anisotropy of the collected rings, if present, gives hints about the anisotropy of the molecular structures of the sample itself. By analyzing the X-ray diffraction pattern and the radial integrated diffracted intensity graph it is possible to verify the presence of secondary structures such as α-helix, β-sheet (peak at 0.46 nm in Fig. 5(a)) or β-turn structures. In XRD experiments can also be detected van der Waals distances of packed amino acid side-chains and aromatic–aromatic (π–π) interactions, one of the most important driving force for the assembling of protein and peptides. Through the analysis of XRD patterns some recurring measurements can be attributed components of circularly polarized light (L or R), the intensity of one of two components is reduced. Secondary structures like α-helical, β-sheet or β-turn and random-coil conformations exhibit characteristic dichroic signatures: accordingly, CD spectra can give useful information about secondary structures of nanofibrous biopolymeric samples.41,105
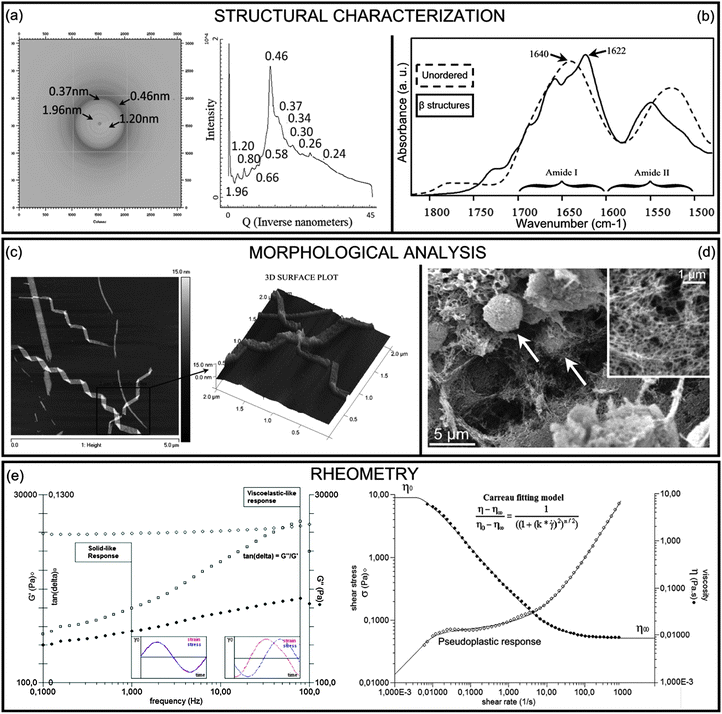 | ||
| Fig. 5 Structural characterization of nanomaterials: (a) X-ray diffraction pattern with a typical β-sheet main ring (4.6 Å distance) and its radial integrated diffraction pattern distance; (b) FT-IR spectra in the region of amide I, used for monitoring the assembling kinetics and the presence of recurring β-structures (peak at 1622 cm−1).44 (c) Morphological analysis of nanostructured scaffolds: 2D AFM image and 3D zoom plot (enlarged boxed field) of twisted ribbons of BMHP1-derived SAP. (d) SEM image of neural stem cells (arrows) embedded in a 3-D self-assembled scaffold. Inset: high-magnified field of the nanostructured scaffold. (e) Rheological characterization of a hydrogel scaffold (left): oscillatory frequency sweep test of an assembled hydrogel, G′, G′′ and tan δ are represented. Insets: in phase solid-like and out of phase viscoelastic-like responses of the tested scaffold at low and high frequencies respectively. Pre-assembling gel solution (right): graphical correlation of viscosity and shear stress as a function of the shear rate: a mathematical fitting model used for the extrapolation of η0 and η∞ is shown. | ||
Fourier transform infrared spectroscopy (FTIR) is also widely used to assess peptide secondary structures: advantage of FTIR is a less sensitive light scattering influence than CD, allowing greater concentrations of samples.
Attenuated total reflection (ATR) FTIR combines the use of an ATR crystal and infra-red spectroscopy in order to improve spectral reproducibility and minimize user-to-user spectral variation. FTIR spectroscopy allows us to monitor the presence to tertiary and quaternary structures of protein and, in the case of nanofibrous scaffolds, these measurements may provide indications up to the nanofiber scale.196–198
All the above described characterization techniques are meant to define, through the interpretation of spectra, the secondary structures of the measured samples. Other important information such as nanofibers shape, dimension and their interactions within the formed scaffold can be achieved by different microscopy techniques. The most important techniques to monitor the presence of nanostructures are atomic force microscopy (AFM), transmission electron microscopy (TEM) and scanning electron microscopy (SEM). AFM is preferred for the visualization of nanofibers (Fig. 5(c), but due to its limited scanning size, it is not ideal for the imaging of jellified hydrogels or electrospun scaffolds: in these cases TEM and SEM are preferred.
In AFM a piezoelectric mechanism makes a probe oscillate and move above the sample, drawing a topographic three-dimensional surface of the sample. The disadvantage of this technique is the dependence of the measurements on the used probe.8 Nevertheless AFM is a powerful tool thanks to the possibility of analyzing static dry samples and dynamic processes in liquid buffers.199 The high sensitivity of the AFM tip to attractive and repulsive forces has been exploited into a unique application: the measurement of the interactions between the tip and sample surfaces. This technique is called force microscopy (FM). In FM the probes are chemically modified (functionalized) in order to obtain a tip interacting only with specific molecular targets, allowing for the measurement of tip–sample force interactions.200 AFM can also be used for biomechanical measurements: e.g. the elastic modulus of nanofibers can be estimated by analyzing an atomic force curve obtained with a cantilever with a defined spring constant. The atomic force curve is a plot where the tip deflection voltage, measured on the photodiode, varies as a result of different piezo-displacements in the Z direction.201
Recently, technical innovations in the field of FM increased the speed analysis and the signal-to-noise ratio of this technique: for example, Bruker AXS (CA, USA) obtained a resolution of 10−9 N in force variation measurements. The so-called PeakForce™ quantitative nano-mechanical mapping was used for a high-resolution Young's modulus (E) measurement of different polymers: from soft gels (E ∼ 1 MPa) to hard polymers (20 GPa).202
More details about mechanical characterization techniques of nanomaterials will be discussed later in this section.
Other microscopy techniques such as TEM and SEM are more laborious and expensive but they give direct images of the sample at higher resolution: the maximum resolution for SEM is 1.5 nm while TEM resolution could be higher. The achievable resolution is strictly related to the nature of the sample and sample preparation. For inorganic, high conductive and crystalline materials TEM resolution could be as high as 1 Å. However, in the case of soft materials, imaging resolution remains in the order of a few nanometers due to low crystallinity of the sample and high inelastic scattering cross-section of electrons at lower accelerating voltage used for imaging.
Sample preparation for standard TEM analysis involves drying and staining steps that may generate artefacts that must be considered for the data interpretation. Cryo-TEM has been developed to prevent possible artefacts ascribable to dry stress coming from conventional TEM sample preparation.
Cryo-TEM is an evolution of TEM and allows us to visualize directly the nanostructured samples preserving the close-to-native structure of the particles, providing unique information not easily obtainable by other characterization techniques. Using a plunge-freezing method Cryo-TEM allows the revelation of size and shape of complex nano-structures in their near-native solvated state,203 avoiding the issue of TEM artefacts related to the preparation of dried samples.204
N. A. J. M. Sommerdijk and colleagues interestingly discussed the comparison between TEM and Cryo-TEM and the interpretation of images from both techniques:205 they outlined how Cryo-TEM potentially allows us to reconstruct shapes and internal structures of nanomaterials. Three-dimensional Cryo-TEM imaging is obtained by combining multiple projections of the same object from different scanning angles: this technique, also called electron cryo-tomography, is considered one of the most powerful characterization techniques in life sciences.
D. J. Pochan and coworkers have focused the importance of this critical technique in the characterization of assembled nanostructures,206 not forgetting to describe some artefacts that may be encountered in cryo-TEM sample preparation.206
Even if TEM data analysis is not model-dependent this technique is not strictly quantitative. Quantitative data are provided by “indirect techniques” like small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS). Data analysis in these two small-angle scattering (SAS) techniques, unlike TEM, relies on the chosen physical model. Therefore, the best practice is creating a physical model from TEM data in order to interpret SAS data. SAS techniques are suited for self-assembled materials and allow the investigation of nanostructures, with minimal sample preparation (e.g. diluting, drying and\or freezing), in a 10 to 500 nm resolution.
SANS and SAXS techniques allow the operator to obtain the similar structural information. Guilbaud J. B. and Saiani A. exhaustively discussed both SAS techniques, elucidating advantages and disadvantages of both in the characterization of self-assembled materials.207 SANS allows the experimentalist to use both hydrogenated and deuterated compounds. Deuterium (D) and hydrogen (H) have very different scattering lengths and by replacing H by D the user can manipulate the scattering densities of both solvent and sample. Replacing H atoms by D atoms can provide insights into the importance of H-bonds in the biomaterial structure while letting the thermodynamic properties of the sample unchanged. Furthermore, in SANS analysis of hydrogenated samples the scattering background has to be subtracted, thus quality of results is strictly dependent on the signal-to-noise ratio of the instrument. On the other hand, SAXS allows an experimental time scale out of reach for SANS but is likely to damage samples more than SANS, this is the case in particular for soft materials and/or long experiments. SAS techniques became very interesting when combined with other characterization studies: in various works SANS, SAXS, Rheo-SANS, rheometry and TEM are combined in order to obtain a detailed characterization of β-hairpin peptide hydrogelators50,208 and oppositely charged peptides.209
SEM is another heavily used instrument in research areas and industry: SEM has a lower resolution than TEM but a larger depth of field, thus allowing the analysis of larger amounts of the sample. The obtained images are a good representation of the three-dimensional sample and they are widely used to gain information about topography, morphology, composition and porosity of hydrogels and electrospun nanofibrous scaffolds (Fig. 5(d)).210
SEM is a valuable tool to measure the surface porosity of the sample but a swelling test is necessary to obtain detailed information about the real porosity of the sample. Porosity is an important parameter in the design of scaffolds because it affects the mechanical properties of the structure. The swelling test is widely used in the characterization of electrospun fibers or hydrogels, and it estimates the change in sample weight before and after immersion in a buffer solution. The choice of the buffer depends on the chemistry of the sample, while the mass increase is directly dependent on the porosity of the network.187,210,211 Indeed the swelling ratio of a hydrogel not only depend on the hydrophilicity of the functional groups but also on the three-dimensional architecture of the formed network. The introduction of pores decreases the resistance to the applied strain and, consequently, the quasi-static modulus and stress.210 These mechanical characteristics are obtained through the tensile strength test, a traction test mainly used for the characterization of the mechanical properties of electrospun scaffolds. In this experiment the instrument deforms the sample with a constant rate and calculates the stress–strain curve. Through the analysis of this curve it is possible to gain information about the ultimate stress until failure, the elastic modulus and the value of strain at rupture.212
An alternative instrument for the characterization of the visco-elastic behavior of nanofibrous scaffolds is the rheometer. There are various types of rheometers presenting different mechanisms of operation but the laminar flow rheometer is the most used one for the characterization of self-assembled soft materials. However a good number of issues in using rheometer on cross-linked gels may arise: indeed cross-linking reactions may not be compatible with the normal setup of the instrument. To perform rheological analyses of cross-linked biomaterials a specific setup, likely including micro-fluidic chambers and inert geometries, must be prepared.
An example is the rotational plate–cone rheometer, ideal for the characterization of small quantity of soft materials. This instrument is composed of a motor, a geometry, a lower plate and a transducer. The motor imposes a controlled rotation on the geometry, generating a stream flow in the sample that is placed on the lower plate of the instrument. The transducer measures the torque required to rotate the geometry and finally the instrument calculates the viscosity of the sample.
The plate–cone rheometer can work in oscillatory or flow modes in order to measure different properties of the sample. In a standard oscillatory experiment the instrument induces a sinusoidal shear deformation in the sample and measures the resultant stress response. In oscillatory mode it is crucial to apply a defined and sufficiently small strain in order to perform the experiment in the linear visco-elasticity region of the material, that is, where it exhibits a linear strain–stress relationship. When strain and stress responses are in phase the sample behaves like an ideal elastic solid: in the opposite situation the response of an ideal viscous liquid is in quadrature of phase (δ = π/2). An example of how the response of a material changes as a function of the stimulus frequency is given in the left graph in Fig. 5(e); in this figure are indicated both the perfect overlapping of stress and strain typical of a solid-like profile and the out of phase response typical of a visco-elastic like material. Soft materials, displaying intermediate responses between the two just mentioned, exhibit both in phase and out of phase contributions.
The main aim of rheometry is to study the mesoscopic properties of the materials, evincing parameters helpful for the characterization of their viscoelasticity. In the case of hydrogel characterization the storage modulus (G′), the loss modulus (G′′) and loss factor (tan δ) are usually considered: these parameters are calculated as a function of time, frequency or strain percentage.213G′ measures the deformation energy stored during the shear process (namely the stiffness of the material), on the other hand, G′′ indicates the dissipated energy (i.e. its liquid-like response). The comparison between G′ and G′′ gives an indication of the visco-elastic profile of the tested material: if G′′ is greater than G′ (tan δ > 1) the sample behaves like a viscous liquid, otherwise when G′ is higher than G′′ (tan δ < 1) the sample behaves like an elastic solid, which is the typical hydrogel profile.214
By monitoring the temporal trend of G′ and G′′ it is possible to monitor the gelation process, whereas by analyzing the two moduli as a function of strain, the linear viscoelastic region or the strain percentage before failure can be assessed. By measuring G′ and G′′ as a function of frequency the user can investigate the behavior of hydrogels at different time scales: high and low frequencies correspond to fast and slow time scales respectively. Typically hydrogels behavior is time-scale dependent: at high frequencies they resemble a solid scaffold while at low frequency values they produce liquid-like responses.213
On the other hand flow mode experiments provide other rheological parameters such as the real viscosity of a sample, i.e. its rheological properties dependence on the applied shear rate and on the timescale of the experiment.
The plot of the viscosity versus the shear rate allows the classification of the analyzed material into (1) ideal Newtonian fluids if the viscosity is constant and does not depend on the applied shear, (2) plastic and pseudo-plastic fluids when the viscosity decreases as a result of shear increase (shear thinning fluids), (3) dilatant fluids (shear thickening materials) if the opposite occurs. Most of the soft scaffolds belong to (2) their viscosity being dependent on the intensity of the applied shear.
Usually, a viscosity plot is subjected to a mathematical fitting with different models in order to extrapolate parameters of interests like infinite-rate viscosity, zero-rate viscosity and yield stress.
The shear-dependent behavior is an important parameter in the design of new scaffolds for tissue engineering. Shear thinning materials allow easier delivery via injection ensuring a viscosity decrease, as a result of shear increase, and a fast recovery of the original viscosity after transplantation. Indeed, rapid self-healing after injection allows for scaffold delivery and its localization within the site of injection, presumably corresponding, but not necessarily, to the site of injury.215
Shear thinning, re-healing properties and their biological importance in nanomedicine are treated in the Material Section 2.2.2 and discussed in detail in other interesting recent publications.50,54,208,209 Another rheometer, employed for the characterization of polymer melts, is the Melt Flow Index Rheometer (MFIR). In MFIR measurements samples are loaded in pellets or granulates, then heated up to the polymer melt temperature. Once the sample is melted a piston applies a controlled force causing the extrusion of the material. The Melt Flow Index (MFI), measured in grams of the sample extruded in 10 minutes, is related to the molecular weight of the polymer and corresponds to the inverse of the viscosity.216 Nonetheless this technique is not applicable to unstable components like peptides and does not provide the flow curve and related viscoelastic parameters (G′, G′′) of the melt.
Rheological properties and, in particular, stiffness, are certainly important parameters to design biomimetic supports, because they play a crucial role in the adhesion and differentiation of the seeded cells.217 More detailed information about the relationship between stiffness and cell behavior will be given in Section 5.2.
Lastly, another tool for the characterization of nanofibrous scaffolds is the thermogravimetric analysis (TGA). TGA is performed in a controlled atmosphere to monitor the continuous change of the sample mass; the result is a thermal decomposition curve that provides information about the thermal stability of the compound.212
5. In vitro models
In this section we will discuss the biomaterial properties to be tailored when designing a scaffold for a cell-based therapy of nervous tissue injuries.The phase of in vitro testing represents a fundamental step between the design/characterization phases and the in vivo experimentation: indeed it is useful for verifying if the novel scaffold affects the cell behaviour and how it exerts its effect. With respect to functionalization of the material, the second part of this chapter will discuss the use of the phage display technique to identify novel cell-adhesive short peptides, which, in turn, can be added to the scaffold.
Lastly, the information obtained through the in vitro models can provide useful indications about scaffold properties improving the survival and integration in vivo of the cellularized matrix, when implanted into the damaged tissue. Furthermore the in vitro experimentation represents also a screening phase before in vivo testing, in order to avoid scaffold cytotoxic effects, to reduce the number of animals and to lower experimental costs and timing.
5.1 Scaffold chemico-physical properties relevant for cell-based regenerative approaches
Organisms acquire their proper characteristics through the complex mechanisms of the embryonic development that determine the cell number and fate. In the adult organisms both the cell number and fate are always tightly controlled. Organs have the ability to respond to cell death by activating tissue-specific populations of undifferentiated cells called adult stem cells.218 Upon activation, adult stem cells can proliferate, thus allowing at the same time the maintenance of the pool of stem cells and the regeneration of the dead cell types.218 Therefore they have been subjected to intensive studies for regenerative medical approaches.219 However, a pure cellular strategy can be rarely applied; in other words the simple injection of cells in the sites of cell loss is very frequently not sufficient for the tissue regeneration. The main reason for this problem is that cell survival, proliferation and differentiation require an ad hoc microenvironment, which is disrupted or perturbed in damaged tissues. Physiologically, cell microenvironments are characterized by three parameters: the chemical composition, viscoelasticity and topography. All three characteristics are tissue-specific and they must be controlled to maintain the functional and anatomical characteristics of each organ. Therefore a successful regenerative approach lies also in the creation of biocompatible materials with tissue-specific physico-chemical properties suited for cell encapsulation and the related cell behaviour.5.2 The importance of biomaterial mechanical properties
Organs are characterized by mechanical properties strictly related to their cellular and extracellular compositions. A striking example is represented by the differences in mechanical properties between bones and nerves. Therefore the mechanical properties of the biomaterials must be considered and adequately tuned. Indeed it is known that they affect cell proliferation, migration and differentiation in vitro. This is driven by the binding of adhesion ligands to specific cell surface receptors that act as mechanical transducers between the material and the cell cytoskeleton, allowing cells to respond to the mechanical properties of the substrate.220,221 Very interestingly, in the case of stem cells this molecular crosstalk has been demonstrated to affect self-renewal,222 differentiation and gene expression.223 Indeed human adult mesenchymal stem cells are able to differentiate along neurogenic, myogenic, or osteogenic cell lineages simply according to the substrate elasticity.223 Concerning the nervous system, its cellular and extracellular heterogeneity leads to significative differences in the mechanical properties of specific districts, with a G′ around 3 kPa for the myelin-enriched spinal cord tissue224 and around 500 Pa for the laminin-enriched encephalon.217 Therefore the optimal matching between the mechanical properties displayed by different materials and specific neural environments is critical in influencing neural growth, morphology and differentiation on different materials. For example embryonic cortical-derived neural cells exhibit a stiffness-dependent cell survival when plated on polyacrylamide or fibrin gels, with a peak of neuronal vitality at 200 Pa.225 Moreover adult NSCs differentiation on variable moduli interpenetrating polymer networks displays a peak of neuronal differentiation around 500 Pa and of glial differentiation around 10 kPa.217 Also the neurite extension in dorsal root ganglia (DRG) cells has been demonstrated to inversely correlate with the mechanical stiffness of agarose gels,226 as well as for PC12 cells grown on photopolymerizable PEG hydrogels.227 The soft nature of nervous tissues potentially makes hydrogels ideal for use in these environments, considering also their biodegradability, flexibility and their poor inflammatory potential. However further work is needed to create an appropriate balance between material mechanical properties, material degradation and the related cell behaviour.5.3 Scaffold dimensionality: effect on seeded cells
The physiological properties of organs are not only determined by the presence of specific cell types and specific ECM components, but also by specific morphologies and tissue architectures. Frequently, tissues inside the same organ display heterogeneous architectures allowing cells to live in a bi-dimensional or three-dimensional fashion. For example, the ependymal cells monolayer of the nervous system is bi-dimensional while the neural network is three-dimensional. Therefore materials designed for cell-based regenerative approaches must allow the bi- or three-dimensional cell growth, survival and differentiation according to the physiological architecture of the tissue to be regenerated; moreover they must allow a proper dimensional cell remodelling and cell-based material degradation in order to favour the optimal integration of cells in the tissue of interest.However it is important to underline that many materials consist of microfibers with diameters similar or even bigger in size than cells. Therefore cells attached on these microfibers must be considered growing onto bi-dimensional “curved” substrates rather than in a true three-dimensional microenvironment. Furthermore the micropores between fibers are larger than cells, thus allowing a diffusion rate of biomolecules not comparable with the one characterizing physiological environments. For a true three-dimensional environment, fibers comprising the scaffolds must be much smaller than cells, thus creating a milieu in which cells are surrounded by the scaffold (see Fig. 5(d)) as in the native extracellular matrices. Numerous types of primary cells have been successfully grown in three-dimensional scaffolds, including cardiomyocytes and myoblasts on collagen,228,229 chondrocytes on SAP hydrogels230 and hepatocytes on Matrigel.231 Also embryonic stem cells have been cultured in true three-dimensional scaffolds without loss of totipotency.232 This indicates that the material architecture may modulate cell signaling and behaviour as well: indeed cell differentiation to specific phenotypes in vitro has been demonstrated to be enhanced when cells are cultured in three-dimensional scaffolds.233 Therefore it is essential to create scaffolds that can support a true “three-dimensional cell growth” according to the cell types to be encapsulated. Among materials suitable for cell-based nervous system repair, hydrogels provide a good equilibrium between mechanical and architectural properties because of their tunable softness and their nanostructure characterized by highly swollen, porous polymer networks; therefore hydrogels simulate key structural aspects of native extracellular matrices. Among hydrogels, SAP nanofibrous scaffolds have been proved to provide three-dimensional microenvironments very promising for neural cells survival and differentiation (Fig. 6). Indeed hippocampal primary neural cells have been successfully grown in three-dimensional SAP scaffolds.234 Interestingly, when encapsulated in these three-dimensional microenvironments, retinal cells not only survive and migrate but they also express retinal neuron-specific genes not expressed when cultured in two dimensions.235 Moreover, functionalized SAPs have been demonstrated to support the differentiation of adult NSCs in three-dimensional cell cultures.27,236
 | ||
| Fig. 6 (A) Three-dimensional culture of mouse NSCs cultured in a KLDL12-based scaffold, at 2 weeks from the induction of the differentiation. Neurons are marked with anti-βIII Tubulin antibody (green) and astrocytes with anti-GFAP antibody (red). In three-dimensional cultures neurons and astrocytes, forming a highly intermingled cellular network, more closely resemble the distribution of nervous cells in vivo than in standard 2D cell culture systems. Scale bar: 50 μm. (B) Graphical representation of a three-dimensional culture in a porous insert, where cells are embedded into the scaffold and immersed into the growth medium. | ||
5.4 The impact of substrate biochemical composition
The main requirement for the survival of cells is the presence of a material fostering their attachment; the lack of a substrate permissive for cell adhesion unequivocally leads to a particular form of cell apoptosis called anoikis,237 thus impeding any possible cell-based therapy developed for the regeneration of cavities formed in the injured tissues. Cell attachment onto materials can occur through nonspecific adsorption or molecule adhesion.238 Chitosan and poly-lysine are examples of, respectively, natural and synthetic materials that exhibit hydrophilic- and electrostatic-based neuronal cell adhesion.75,238,239 Despite cell binding, this molecule-unspecific phenomenon limits the control of the cell fate; therefore other biomaterials, especially natural materials, with more known biological molecular adhesive properties, have been investigated. Matrigel, a natural hydrogel, has been extensively used in vitro to study the proliferation and differentiation of many cell types.240,241 Alternatively biomaterials can be combined with purified or chemically synthesized components of specific extracellular matrices. Molecules such as laminin are known to promote cell differentiation, adhesion and migration in the CNS.242 Indeed materials pre-coated with laminin showed significant improvement in neural cell affinity and functional recovery.243 Moreover biomaterials coated with neuronal adhesion molecules, such as the molecule L1 that is expressed in growing axons and Schwann cells during development and regeneration,244 have been proposed as promising scaffolds. However pathogen transfer, batch variability and unsatisfactory purity are justified concerns limiting their usage. An alternative method is to functionalize biomaterials with short peptides (functional motifs) known to stimulate cell attachment. These short peptides can be synthesized and covalently attached to both natural and synthetic materials. Specific functions of several key extracellular proteins physiologically comprising the tissue microenvironments have been attributed to short peptide sequences; in the case of laminin the best characterized and used sequences are YIGSR,245 IKVAV,245 RNIAEIIKDI,246 RYVVLPR247 and RGD.248 Indeed RGD-functionalized PHPMA hydrogels (NeuroGel) and RGD-functionalized PEG polymers have been shown to be capable of promoting neural tissue repair and enhancing cell adhesion,78,249 respectively. Also RGD-containing HA scaffolds as well as fibrin gels modified with laminin were shown to increase neuronal cells proliferation.250,251 Peptide sequences that incorporate both YIGSR and IKVAV have been found to significantly increase neuronal adhesion.252 Moreover various electrospun nanofibrous tubes, made of natural, synthetic or biosynthetic materials and functionalized with bioactive molecules, have been demonstrated to be able to support the survival, proliferation and differentiation of neural cells.253 One of the most intriguing approaches in scaffold design for cell-based regenerative therapies is related to the use of functionalized SAPs. RADA16-I and RADA16-II are the most commonly used SAPs for neuronal cell culture.254–256 Several types of neuronal cells have been grown on RADA16 including rat PC12 cells, cerebellar and hippocampal primary cells and fresh neuronal cells prepared from neonatal rats.255 Their functionalization with bioactive sequences has been demonstrated to be a promising strategy for enhancing NSCs survival and differentiation. Indeed RADA16-based scaffolds with different functional motifs have been used for culturing and differentiating adult mouse NSCs.27 Moreover the incorporation of the IKVAV sequence in PA nanofiber gels has been shown to enhance the differentiation of neural progenitor cells (NPCs).254 Finally it has to be underlined that cell attachment to materials is related not only to the choice of ligand for the functionalization but also to ligand density and exposure. Indeed, by using modular biomimetic interpenetrating network hydrogels, NSC differentiation on substrates with varied functional motif and mixed functional motif densities has been demonstrated to be sensitive to a minimum RGD density.257 In another work we also showed how the length of a flexible Gly-spacer inserted between the self-assembling RADA16 sequence and the BMHP1 functional motif influenced the responsiveness of cultured NSCs, suggesting that a longer spacer, and consequently an enhanced exposure of BMHP1, has to be preferred for improved cellular interactions with the functionalized scaffold.8Many ongoing studies aim at optimizing the biomaterial choice, at discovering new cell-adhesive peptide sequences, at identifying the best cell-adhesive functional motifs and at selecting the proper functional motif concentrations.
These considerations highlight how, for a successful cell-based regenerative approach, is important not only the choice of proper cell type but also the choice of nanostructured biomaterial with physico-chemical peculiarities reproducing the properties of the tissue to be regenerated.
In an effort to identify novel and more effective cell-adhesive short peptides for the specific cell type to be cultured or transplanted, high-throughput screening techniques such as the phage display technique showed remarkable potential.
5.5 The phage display technique: an historical and technical overview
The phage display technique is a simple and powerful technique for the rapid identification of protein–protein interactions. This technique is based on the use of bacteriophages genetically modified to display proteins or peptides fused to their exterior coat proteins. The phage display technique gained a prominent position in studies aimed at identifying peptides displaying affinity for a given ligand. This is due to three key advantages: the direct link between displayed peptides/proteins and the bacteriophage genome, the possibility of displaying either short peptides or large proteins and the possibility of sorting out the interacting sequences according to their binding affinity for the target molecule. The general procedure of the technique is shown in Fig. 7. It can be divided into three steps: (1) the creation of the bacteriophage library and its amplification, (2) the bacteriophages selection from the library (a process called panning) and (3) the elution of bound bacteriophages for genome sequencing. The phage display technique is a simple and powerful technique for the rapid identification of protein–protein interactions. This technique is based on the use of bacteriophages genetically modified to display proteins or peptides fused to their exterior coat proteins. | ||
| Fig. 7 Schematic representation of the experimental steps of the phage display technique. Phage libraries are created by inserting a set of different sequences into the phage genome: each phage receives a single nucleotide sequence and exposes the new peptide on its surface. After amplification of the phage library in bacteria, the library is exposed to the target (single molecules, cells or even living tissues). In the latter step, called panning, phage displaying molecules that specifically bind to the target are recovered, while non-specifically bound and unbound ones are removed. The specifically bound bacteriophages are then amplified: the re-amplified phage populations can be subjected to further rounds of panning or processed for DNA sequencing. Eventually the selected amino acid sequences, derived from phage genome sequencing, can be searched in various protein databases to locate, if possible, the functional motif in other known protein sequences. | ||
Libraries are created by inserting the nucleic acid sequences, coding for the peptides or proteins of interest, in frame in one of the bacteriophage genes coding for the capsid proteins; libraries are then amplified by incubating bacteriophages with bacteria such as Escherichia coli.
The panning requires the physical contact between bacteriophages and the target; bacteriophages bound to the target molecule are retained while non-specific or unbound ones are washed off. Then the bound bacteriophages are eluted and re-amplified. The re-amplified phage population can, in turn, be subjected to more rounds of panning or processed for genome sequencing. The stringency of the result is directly proportional to both the number of rounds of panning and to the strength of the bacteriophage elution phase. This protocol can be easily adapted for targeting many types of molecules. Indeed the phage display technique has been successfully applied for therapeutic and basic research purposes not only to single molecules, but also to different cell types and even directly in vivo.
Intriguingly, liquid-crystalline film matrices made using M13 bacteriophages displaying RGD and IKVAV sequences have been recently shown to be capable of supporting neural proliferation and differentiation.271 In the optic of enhancing transplanted cell survival and differentiation, heptapeptides, identified by targeting neural precursor cells, have been used for the functionalization of SAPs and tested for their regenerative properties in spinal cord injuries.9
The phage display technique on NSCs has been also applied technique against single molecules has been demonstrated to be a powerful tool in other fields; for example libraries created using random peptides have been successfully used to identify the sites of protein–protein interaction, to develop reagents for animals immunization and to create vaccines.259 Libraries of enzymes or substrates with modified specificity or kinetics have been also created;260 moreover they have been used also to find sequences able to bind growth factors, therefore useful for the controlled entrapment and slow-release of cytokines from nanostructured scaffolds.261,262 Interestingly also peptides selectively binding inorganic surfaces such as iron oxide,263 metallic gold and chromium, as well as inorganic materials used to make semiconductors,264 have been identified with the phage display technique.
The phage display technique on cells has been extensively used for therapeutic purposes, especially in cancer research. An elegant example testifying the power of this approach was done by targeting the entire National Cancer Institute (NCI) panel of 60 cancer cell lines, thus identifying a plethora of peptides differentially binding specific cancer cells.265 Interestingly peptides found by the phage display technique on cells can be used also for the in vivo imaging of tumors in animal models.266
Due to the fundamental importance of angiogenesis in both tissue physiology and tumor progression, the phage display technique has been extensively applied also to endothelial cells, thus allowing the identification of many molecules differentially expressed by tumor endothelial cells.267
Another tissue subjected to intense phage display-based analyses is the nervous system. The phage display technique has been used to identify peptides able to bind primary motor neurons and DRG cells potentially useful in the treatment of motor neuron disease, neuropathy, and pain.268 It has also been successfully applied to cerebellar granule neurons in order to find peptides facilitating neuronal gene transfer.269 A for basic research purposes, allowing for example the identification of a previously unrecognized complex between the soluble protein netrin-4 and laminin γ1 subunit.272 Despite the clear strength of this technique in identifying functional motifs interacting with specific cell types, the major disadvantage is that the nature of the cell molecules recognized by bacteriophages cannot be directly identified; however this information may be obtained by applying classical biochemical approaches.272
A remarkable work has been done in targeting the endothelium to study the heterogeneity of physiological and tumor-related endothelial cells.273 Another tissue subjected to several in vivo phage display-based analyses is the cardiac tissue; indeed this application has been used to identify peptides targeting the ischemic myocardium274 and with potential for cardiomyocyte transduction in vivo.275 Moreover, a peptide able to detect apoptotic neuronal cells in the ischemic brain has been described.276 By applying this strategy to animal models of cancer, it has been possible to identify peptides targeting tumors particularly aggressive such as pancreatic carcinomas.277
The in vivo phage display can be applied also directly to humans; indeed this strategy led to the identification of peptide ligands homing to specific tissues, including bone marrow, skin, fat, muscle and prostate in a patient carrying the Waldenström macroglobulinemia.278
Finally the versatility of this approach is again underlined by the fact that it can be virtually applied to all animals, thus opening intriguing new medical scenarios; a very interesting example, the in vivo phage display has been applied to mosquitoes in order to find peptides blocking the malarial infection.279
Taken together, these scientific lines of evidence place the phage display technique as one of the most powerful tools for the identification of peptides able to bind specific molecules or cells; therefore it can be considered a cutting edge strategy in all the scientific fields in which these interactions must be analysed, ranging from material sciences to regenerative medicine. Indeed, SAPs (and other biomaterials) functionalization being a widely established technique (see Subsection 2.2.2 for details), multiple moieties can virtually be included in bioabsorbable scaffolds in order to tailor transplanted cell engraftment, host tissue reaction and slow drug delivery (Fig. 8).
 | ||
| Fig. 8 A RADA nanofiber interacting with growth factors by means of different functional motifs discovered by the phage display or other random library techniques. Glycine-spacers (white spheres) and functional motifs (purple, yellow, green and ice blue spheres) protruding from the β-sheet core of the RADA fiber (represented by ball and sticks: white, red and blue, respectively, for alanine, arginine and aspartic acid) may interact with cell membrane proteins and cytokines (grey and orange spheres) held by weak interactions. | ||
6. Nanostructured scaffolds for PNS and CNS injuries
In vivo experimentation constitutes the final step of the process we have described so far: indeed, regardless of the improved reliability of in vitro three-dimensional models, in vivo testing is still essential for validating the feasibility of the implantation method and the putative regenerative potential of the developed scaffold.In in vivo experiments for neural regeneration, the therapeutic effect of the implanted scaffold (possibly combined with cells and drugs) is usually evaluated through behavioural tests and electrophysiology, followed by histological analysis: overall these tests permit us to assess the functional recovery of animals, the response of the tissue to the damage (cavities formation, gliosis, inflammation, apoptosis, ECM deposition, demyelination, etc.), the reparative process (regrowth of axons, myelination, neural cell infiltration, etc.) and the integration of the scaffold, alone or seeded with cells, into the host tissue.
In this section we will discuss the application of electrospun matrices and hydrogel scaffolds to repair both PNS and CNS injuries, as schematized in Fig. 9. The coverage of hydrogel-based nerve guidance channels exceeds the focus of this review: for an analysis of such application the reader can refer to other publications.280,281
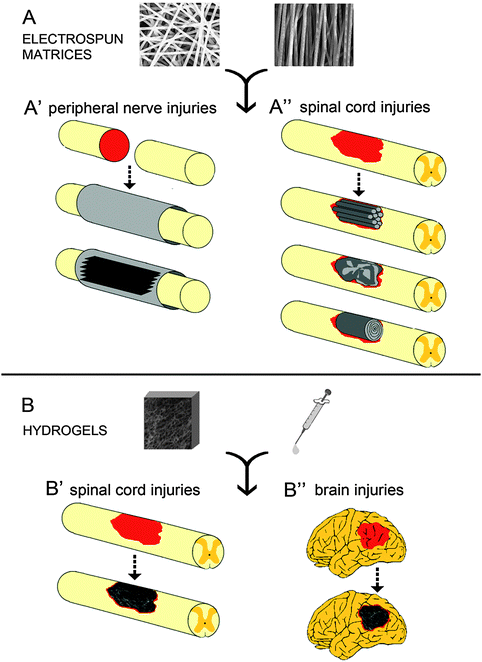 | ||
| Fig. 9 Electrospun fiber- and hydrogel-based scaffolds for neural tissue engineering. (A) Electrospun matrices, comprising either randomly or longitudinally oriented micro- and nanofibers, may have multiple layers featuring different fibers orientation. In peripheral nerve transections (A′) electrospun scaffolds serve as nerve conduits and/or as multiple films to be stacked into the nerve channel. On the other hand, electrospun scaffolds for spinal cord reconstruction (A′′) include multiple micro-channels or a single electrospun film that can be crumpled into the lesion area or rolled up to form a multilayered tubular construct. (B) Hydrogel-based scaffolds can be prepared as either preformed solid gel or viscous solution and, respectively, implanted or injected into the lesion site. Both strategies have been employed to fill cavities resulting from either spinal cord injuries (B′) or brain injuries (B′′), thus providing a nanostructured matrix for tissue regrowth. | ||
6.1 Nanostructured guidance channels for peripheral nerve injuries
Unlike the adult CNS, the PNS is a permissive environment for axon regeneration after injury. This different regenerative ability is exemplified by DRG neurons that project axons both in the CNS and PNS but they can only regenerate their peripheral branch.282However, the reparative process leads to a spontaneous reinnervation only when injury results in small gaps, moreover growing axons may reach the wrong target, thus not leading to a complete functional recovery. Clinically, the current gold standard for bridging large nerve gaps is nerve autograft, but this strategy is limited by tissue availability and donor site deficits. Allografts and xenografts represent a possible alternative, however bringing the risk of causing immunological rejections and carrying diseases.
Tubulization techniques with nerve guidance conduits (NGC) are another alternative to the autologous nerve graft. In the last three decades a number of different NGC-based therapies have been tested to direct axonal growth from the proximal to the distal nerve stump, to prevent the infiltration of ectopic fibrous tissue and to increase the concentration of the neurotrophic factors secreted by the injured nerve ends. NGCs have been also used to deliver cells and growth factors. Indeed, a conduit engineered for nerve regeneration should preferably be a tubular scaffold with an internal matrix delivering bioactive molecules and biomechanically supporting cells.283
The early NGCs were made of nondegradable materials, such as silicone,284 but the use of biodegradable polymers is preferable because of the advantage of eliminating a second surgery to remove the conduit.285 Another important parameter to consider when designing a nerve conduit is scaffold porosity, which should mimic the structure of the ECM (as described in Section 5.3) and be able to promote both cell adhesion and nutrients diffusion. These and other required properties, such as appropriate mechanical strength and compliance, can be virtually tuned by employing the electrospinning technique (for more details see Section 4.1). The versatility of this method makes electrospun nanofibers an interesting class of scaffolds for peripheral nerve tissue engineering. However, while a variety of in vitro investigations have demonstrated the potentiality of electrospun tubes to support cell attachment and proliferation, only a few in vivo studies, employing electrospun tubes to guide nerve regeneration, have been carried out to date. Thus we here resume the main results and issues emerging from these studies performed on animal models of peripheral nerve injury (Table 4). Experimental PNS injuries usually consist in a complete transection of rat sciatic nerves followed by removal of a nerve segment, in order to leave a gap between the two stumps of at least 10 mm.
| Biomaterial | Structure and fiber orientation | Addition of other components (cells, functionalization, cytokines) | Animal model | Ref. |
|---|---|---|---|---|
| OL = outer layer, IL = inner layer; DAc = deacetylation rate. | ||||
| Chitosan | Bilayered tube of chitosan film (OL) and chitosan random electrospun nano/microfiber mesh with a DAc of 78% or 93% (IL) | — | 10 mm sciatic nerve transection in rats | 76 |
| Chitosan | Bilayered tube of chitosan film (OL) and chitosan random electrospun nano/microfiber mesh with a DAc of 93% (IL) | YIGSR laminin-1 sequence | 10 mm sciatic nerve transection in rats | 286 |
| Chitosan | Bilayered tube of random (OL) and aligned (IL) electrospun nano/microfiber mesh with a DAc of 93% | — | 10 mm sciatic nerve transection in rats | 288 |
| PLGA | Monolayered tube of random electrospun fibers | — | 10 mm sciatic nerve transection in rats | 21 |
| PCLEEP | Bilayered tube of PCLLEP film (OL) and PCLLEP electrospun fibers (IL). Fibers were either longitudinally or circumferentially aligned | GDNF | 15 mm sciatic nerve transection in rats | 287 |
| PAN-MA | Electrospun fiber films stacked within a polysulfone nerve conduit. Fibers were either longitudinally or circumferentially aligned | — | 17 mm tibial nerve transection in rats | 289 |
| PLGA/PCL | Bilayered tube of PCL random electrospun fibers (OL) and PLGA/PCL electrospun fibers (IL) | — | 10 mm sciatic nerve transection in rats | 22 |
| PCL | Multilayered tube of random (OL) and aligned (IL) electrospun fibers | — | 10 mm sciatic nerve transection in rats | 253 |
The first study investigating the feasibility of nerve regeneration in vivo using electrospun NGCs was made by Ramakrishna's group in 2004.21 PLGA-made nanofiber conduits were implanted in a 10 mm gap of rat sciatic nerves and, after one month, they were shown not to cause an inflammatory response, to be biodegradable and to promote axonal regeneration.
Following investigations took advantage of the possibility offered by this technique to control nanofibers orientation and to produce tubes having a multi-layered structure. Bilayered tubes without aligned fibers were employed in Wang's group and in ours. In our study, we used conduits presenting a base of PCL electrospun microfibers, assuring mechanical stability and compliance, and a coating of smaller PLGA/PCL electrospun nanofibers, preventing invasion of ectopic cells and allowing nutrient diffusion, to regenerate a 10 mm nerve gap in a rat sciatic nerve model.22 Four months after surgery, the multi-scaled electrospun nerve conduits induced a functional reconnection of the two severed stumps. On the other hand, Wang's group tested the use of NGCs made of chitosan. In comparison to synthetic materials, natural polymers present the advantage of containing specific signals for cell adhesion but generally have a lower mechanical strength. Wang and colleagues developed through the electrospinning technique two different chitosan nano/microfibrous tubes with a deacetylation rate (DAc) of 78% or 93%, then they combined them with a not electrospun chitosan film tube to form bilayered tubes, and finally examined the effect in a 10 mm rat sciatic nerve model.76
The chitosan mesh tubes with a DAc of 93% were shown to have both sufficient mechanical strength to preserve tube space and good permeability for cell migration and nutrients diffusion; however the bilayered tubes appeared to be less permeable to nutrients than single-layered chitosan tubes with a DAc of 93%, leading to a delay in nerve regeneration and functional recovery. In parallel with these investigations, Wang and co-workers tested a similar chitosan bilayered tube that was functionalized with the YIGSR sequence linked to two different glycine spacers, in order to evaluate the role of the linkers in vivo.286 After implantation into a 10 mm sciatic nerve gap in rats, the bilayered chitosan tube, on which the YIGSR peptide and 6 glycines were added, exhibited efficacy similar to sciatic nerve isografts (i.e. sciatic nerves harvested from other rats) used as positive controls. As an improvement of cell adhesion and axon sprouting was observed in tubes containing the bioactive peptide with the major number of glycines, the authors suggested that, by augmenting the glycine spacer length, the peptide can increase conformational degrees of freedom, which in turn may promote its interaction with cells or axons.
In several in vitro studies, alignment of electrospun micro- and nanofibers has been demonstrated to direct the migration and proliferation of cells and provide guidance for neurite extension. Chew and colleagues tested the biological effect, in a 15 mm gap rat sciatic model, of NGCs containing an inner layer of PCLEEP (see Subsection 2.1.2) electrospun microfibers aligned longitudinally or circumferentially to the channel.287 These authors provided a topographic guidance for nerve regeneration and, in addition to that, introduced a biochemical cue by encapsulating the glial cell line-derived growth factor (GDNF) into the aligned electrospun fibers. After 3 months, animals treated with tubes with electrospun fibers (regardless of the fiber orientation) showed a higher content of myelinated axons and an improved electrophysiological recovery in comparison with animals treated with tubes without electrospun fibers. Moreover, the addition of GDNF improved nerve regeneration and functional recovery seen in animals receiving electrospun scaffolds only. Interestingly, as no significant differences were seen between longitudinally and circumferentially aligned electrospun fibers, the authors suggested that the enhancement of nerve regeneration provided by aligned electrospun fibers could be due to the increase of the total surface area available for cell ingrowth, rather than to the effect of contact guidance for axon growth.
The use of aligned electrospun fibers was also investigated by Wang and colleagues and Xia's group, who conducted, respectively, in 2008 and 2009 initial studies in a 10 mm sciatic nerve transection model using multi-layered NGCs with randomly oriented electrospun nanofibers in the outer layer and axially aligned electrospun nanofibers in the inner layer. The rationale for constructing such guidance channels consists in combining the positive nerve regenerative effect provided by aligned fibers with the mechanical strength of randomly oriented fibers. Wang and colleagues fabricated a bilayered chitosan nanofiber mesh channel and demonstrated the efficacy of longitudinally oriented fibers in promoting myelination and axonal maturation, as well as functional and electrophysiological recovery of sciatic nerve injured animals.288 Notably, nerve regeneration was inferior in chitosan mesh tubes without longitudinal nanofiber alignment. In a similar study, Xia's group reported the preliminary results of the implantation of multilayered PCL electrospun guides in the rat sciatic nerve transection model.253 Multilayered NGCs were shown to be capable of supporting nerve regeneration and of preserving quantity, quality and maturity of axons regenerating across the nerve injury, and these observations were also confirmed by electrophysiological and behavioural tests.
Kim and co-workers stacked 10 to 12 films made of either longitudinally aligned or randomly oriented electrospun PAN-MA (polyacrylonitrile-co-methylacrylate) microfibers into a polysulfone nerve conduit.289 The authors chose such structure in order to maximize the topographic directional cues for neurite outgrowth and Schwann cell migration while minimizing the cross-sectional space occupied by the polymer. Significantly, 16 weeks after implantation of the tubes across 17 mm tibial nerve gaps in rats, histological, electrophysiological and behavioural analyses revealed that aligned but not randomly oriented microfibers enhanced regeneration and improved functional outcomes. This study confirmed the importance of topographical cues, capable of promoting endogenous regeneration of nerves regardless of the presence of biochemical cues.
Altogether, these works demonstrate the feasibility of using electrospun micro- and nanofiber scaffolds to guide peripheral nerve regeneration and the possibility of successfully modifying their properties by choosing different polymeric biomaterials, mixing different polymers, including oriented fibers, adding bioactive molecules and fabricating multilayered constructs. However, the ability of these advanced constructs to enhance a regenerative process comparable to nerve autograft remains to be further investigated and probably improved by adding cells, specific biochemical cues and an internal matrix, for example a hydrogel. Additional investigations are also required to clarify the optimal material and structure to be used in NGCs.
6.2 Nanostructured scaffolds for CNS injuries
In contrast to the PNS, in the CNS a pathological insult, as trauma or stroke, usually leads to loss of function and permanent disabilities, making CNS injuries a relevant health and social issue. Indeed, even if some spontaneous reparative processes occur after a CNS injury, they are largely impeded by the hostile environment and glial scar formation, so axons fail to regrow and new precursor cells mainly differentiate into glial cells.290Over the past decades various therapeutic strategies have been proposed, including cell transplantation, scaffold implantation and drug delivery. The encouraging but not fully restorative beneficial effects that were achieved through several cell-based approaches have suggested that a combinatory strategy, making use of cells, biomolecules and scaffolds (necessary to fill the gap and support viability of the implanted cells) may be more effective. Hydrogels are promising biomaterials because their soft, highly hydrated structure, by mimicking the ECM, helps cells to survive; moreover most of hydrogel preparations can be delivered simply through direct injection into the lesion site. Even if an injectable hydrogel does not provide any spatial cue to “guide” the regrowing nervous tissue, scaffold injection may be preferred because CNS injuries often lead to a complex mixture of cavities and spared tissue that results very difficult to fill with a pre-formed scaffold. An injectable scaffold also permits us to minimize the risk of damaging the surrounding intact nervous tissue during implantation.177 For these reasons, besides discussing some important studies using pre-assembled hydrogels, in the present section we will focus our attention on works employing injectable hydrogels, which form solid structures in situ through various physical or chemical assembling processes (for more details see Section 4.1). A selection of relevant studies that tested the implantation of hydrogels or electrospun matrices in CNS injuries is reported in Table 5.
| Biomaterial | Gel state for delivery: injectable fluid or preformed gel | Addition of other components (cells, functionalization, cytokines) | Animal model | Ref. |
|---|---|---|---|---|
| Hydrogels | ||||
| Collagen | Preformed | DRG neurons | SCI: hemisection in rats | 293 |
| Collagen | Injectable | EGF or FGF2 | SCI: compression in rats | 294 |
| Collagen | Preformed | hBMSCs | TBI: cortical contusion in rats | 320 |
| Fibrin, fibronectin or a blend of fibrin/fibronectin | Injectable | — | SCI: dorsal transection in rats | 297 |
| Fibrin | Preformed | NPCs + NT-3 and PDGF | SCI: dorsal transection in rats | 296 |
| Fibrin | Injectable | BMSCs | TBI: cortical lesion in rats | 322 |
| Alginate | Preformed | — | SCI: dorsal transection in rats | 297 |
| Chitosan | Injectable | — | SCI: compression in guinea pigs | 299 |
| Methylcellulose | Injectable | — | TBI: cortical contusion in rats | 319 |
| Agarose | Injectable | BDNF | SCI: dorsal hemisection in rats | 300 |
| Hyaluronic acid | Preformed | Laminin, IKVAV or RGD sequences | TBI: cortical lesion in rats | 250, 316, 317 |
| Matrigel | Injectable | Human BMSC-derived Schwann cells | SCI: contusion in rats | 301 |
| Matrigel | Preformed | Human NSCs | SCI: hemisection in dogs | 302 |
| PEG and lactic acid units | Injectable | NT-3 | SCI: dorsal transection in rats | 303 |
| PGA | Preformed | NSCs | HI: ligature of the right common carotid artery in mice | 323 |
| HEMA | Preformed | — | SCI: complete transection in rats | 304 |
| PHPMA (NeuroGel) | Preformed | RGD sequence | SCI: complete transection in rats | 306 |
| PAs | Injectable | IKVAV laminin epitope | SCI: compression in mice and contusion in rats | 308 |
| SAPs (RADA16-I) | Injectable | BMHP1 motif | Contusion in rats | 310 |
| SAPs (RADA16-I) | Preformed | Schwann cells or NPCs | SCI: dorsal transection in rats | 309 |
| SAPs (RADA16-I) | Injectable | — | TBI: cortical lesion in rats | 324 |
| Electrospun matrices | ||||
| Electrospun PCL/PLGA tubes filled with SAPs | — | SAPs: BMHP1 motif + biomolecules (BDNF, CNTF, VEGF and chondroitinase ABC) | SCI: contusion in rats | 312 |
| Randomly folded electrospun polyamide fabric | — | D5 peptide derived from tenascin-C | SCI: dorsal hemisection in rats | 313 |
In this subsection we review the results recently achieved for spinal cord repair through the use of hydrogels and/or electrospun guidance scaffolds. In SCI research, the most commonly used animal model is rat, on which several lesion techniques have been developed to reproduce human injuries. Among these methods, complete transection, hemisection and contusion models are the most popular ones.291
Common natural materials utilized in SCI models to form hydrogel scaffolds include collagen, Matrigel, chitosan and fibrin. Collagen represents one of the first biomaterials used for scaffolding in experimental SCI. In 1995 a study performed by Joosten and co-workers compared the outcome of two methods of collagen scaffold preparation (as either a fluid or preformed solid gel) in a model of transection in rats: the authors showed that both methods resulted in a considerable reduction of the gliotic response, but only the fluid collagen gel induced the regrowth of damaged axons, whereas no axons were observed to enter the preformed solid collagen graft.292 Collagen hydrogels have been also proven to support survival of transplanted cells, such as DRG neurons,293 and delivery of growth factors, for example the recombinant human epidermal growth factor (rhEGF),294 inducing in both cases a therapeutic benefit to the injured spinal cord.
In SCI research, fibrin gels, alone or in combination with other materials, have been widely used to fill the lesion, improve tissue repair and deliver stem cells,295,296 showing great potential in nervous tissue regeneration. An interesting study compared the effect of four biomaterials, namely collagen, fibronectin, fibrin and a blend of fibrin/fibronectin, injected into the lesion cavities of the rat spinal cord: the best results in terms of biocompatibility, integration with the spinal cord tissue, the number of growing axons and ease in controlling the gelling process were obtained for the blend fibrin/fibronectin.297
Chitosan, alginate and agarose have been extensively investigated in a variety of tissue engineering applications, including spinal cord repair: alginate-based scaffolds were shown to promote axonal elongation,298 while chitosan scaffolds were employed to favour neuroprotection299 and agarose to locally deliver growth factors into the injured spinal cord tissue.300 Matrigel is another naturally derived material, which is often transplanted with cells in experimental SCI. For example, a mixture of Matrigel and Schwann cells was injected after spinal cord contusion in rats,301 while Matrigel seeded with human NSCs was transplanted into hemisections in dogs:302 in both studies the implantation of cells and Matrigel had beneficial effects on functional recovery and fate of transplanted cells, which were found to survive for several weeks and integrate into the damaged spinal tissue.
As discussed in Section 2, natural polymers, when compared to synthetic biomaterials, show some advantages, like the natural content of cell adhesion signals, but also some critical recovery and axon growth. Of note, this material polymerized under visible light.303
Several studies have also shown that the nanofibrous structure of synthetic hydrogels, by varying physical and chemical parameters, can be modified to enhance cell infiltration and tissue repair of the damaged spinal cord. For example positively charged HEMA hydrogels reduced cavity volume and promoted ingrowth of axons after implantation in a complete spinal cord transection.304 Interestingly, as the negatively charged HEMA hydrogels displayed a lower cell infiltration, the intensive ingrowth of cells was ascribed to the positive charge of the hydrogel.
A macroporous hydrogel of PHPMA (NeuroGel) was developed by Woerly's group, which after demonstrating its potential in promoting axonal regeneration in several SCI models,305 attempted to improve the achieved results by adding the RGD sequence: following a spinal cord transection, the scaffold induced tissue ingrowth in the lesion cavity, including angiogenesis and axon regeneration but, unfortunately, a comparative study with the non-functionalized PHPMA hydrogel was not performed.306 However PHPMA cannot gel in situ, making the implantation surgery invasive and hard to perform in irregularly shaped lesions. Injectability is, on the other hand, an important characteristic displayed by two recently introduced classes of synthetic polymers, PAs and SAPs.
The use of PAs as injectable scaffolds was investigated by Tysseling and colleagues, who injected PAs containing the laminin epitope IKVAV (IKVAV-PA) into the contused spinal cord of rodents.307,308 Of interest, the behavioural and histological improvements occurred only in animals receiving the PA containing the functional IKVAV motif, while they were not observed with the injection of PA displaying a randomized functional sequence, suggesting the critical importance of functionalizing a synthetic scaffold with a proper bioactive motif.
The role of SAPs in promoting spinal cord tissue regeneration was demonstrated by Guo and colleagues in 2007, and more recently by our group. Guo and colleagues transplanted RADA16-I alone or mixed it with either Schwann cells or NPCs into the transected spinal cords of rats, showing that scaffolds were permissive to gap bridging and capable of inducing robust migration of host cells, blood vessels and axons into the lesion.309 Even if this study did not prove that the beneficial effect of SAPs on spinal cord tissue was accompanied by an improved locomotor recovery of animals, a positive influence on motor functions was recently reported in our study. In this work, RADA16-I was functionalized with the motif BMHP1 (RADA16-4G-BMHP1) and injected immediately after drawbacks like the risk of inducing immunological and inflammatory responses, batch variability and a less ease in allowing scaffold physical modifications. For these reasons in the last decade many synthetic polymers, such as PEG, HEMA, PHPMA, SAPs and PAs, have been investigated.
A PEG-based hydrogel, composed of PEG and lactic acid units and mixed with neurotrophin-3 (NT-3), was injected into the transected spinal cord in rats, improving locomotor contusion in the rat spinal cord, showing 8 weeks later an improvement in stepping;310 moreover histology and RNA expression analysis showed that RADA16-4G-BMHP1 induced favourable reparative processes, such as matrix remodelling, and fostered nervous tissue ingrowth, resulting in an increased cellular infiltration and axon regeneration/sprouting. The BMHP1 sequence has been discovered via phage display (see Section 5.5) and proven to enhance in vitro NSCs survival and differentiation.27
Despite the simplified implantation approach offered by injectable hydrogels, as well as their ability to support axonal growth and to stabilize the lesion, these materials cannot provide a well-defined spatial guidance, thus leading to random axonal sprouting that likely fails to fully reconnect the damaged neural tracts. As a consequence, the combinatorial use of guidance channels and hydrogels has been investigated in the last years. In such engineered constructs, besides acting as lumen fillers favouring nervous tissue ingrowth, hydrogels are also frequently used as cell carriers and drug delivery systems. An interesting study compared the neuroregenerative effect of different matrices (collagen, fibrin, Matrigel and methylcellulose) and different neurotrophic factors (FGF-1 and NT-3) which were loaded into a large poly(2-hydroxyethyl methacrylate-co-methylmethacrylate) (pHEMA-MMA) hydrogel channel and implanted into the gap of rat complete spinal cord transections.311 In summary, both the matrix composition and neurotrophin content influenced the quantity and origin of regenerating axons, suggesting a possible use ad hoc of different combinations of matrices and growth factors on the basis of the spinal cord tract to be restored. Indeed most human injuries involve only partial areas of the spinal cord segments, thus requiring a therapy targeting specific nerve tracts only. Such a strategy, which should better fit with the clinical reality, could be achieved by using small tubes implantable even in narrow and irregular cavities, thus enabling reconnection of only specific spinal tracts. Recently a similar approach has been proven to be feasible by our group: one month after inducing a contusion injury in rats, we inserted numerous nanostructured microguidance channels within the chronic lesion and we obtained a significant functional and anatomical repair of spinal cord tissue.312 The conduits were produced by electrospinning a blend of PCL/PLGA and were filled with a hydrogel-based matrix (RADA16-I functionalized with BMHP1 motif) mixed with several biomolecules (brain derived neurotrophic factor –(BDNF), ciliary neurotrophic factor (CNTF), vascular endothelial growth factor (VEGF), and Chondroitinase ABC), showing the feasibility of mixing together different therapeutic strategies.
As discussed in the previous section dedicated to PNS injuries, nanofibers produced via electrospinning present ideal properties for neural tissue engineering in terms of porosity, surface/volume ratio, fibers organization, strength and flexibility. Meiners and colleagues explored the potential of polyamide electrospun nanofibrous constructs, alone or covalently modified with a bioactive peptide derived from the tenascin-C (D5 peptide), to facilitate axonal regrowth in dorsal hemisections in rats.313 The implants containing the functional motif provided a favourable environment to tissue regeneration, but the random orientation of nanofibers impeded a proper longitudinal growth of axons, thus the authors suggested the use of a new multilayered tubular construct, in which the nanofibers of each layer are oriented parallel to the axis of the spinal cord.
To conclude, many natural and synthetic materials, as well as different processing and delivery methods, displayed a therapeutic potential in the damaged spinal cord. However, as previously discussed, a successful therapy will probably need a combinatorial approach comprising a scaffold (promoting and spatially guiding nervous tissue ingrowth), the transplantation of cells (replacing lost tissues and/or providing a favourable regenerative environment), and the delivery of different biomolecules (anti-inflammatory agents, growth factors, modulators of inhibitory molecules, etc.).
Among naturally derived materials, HA-based hydrogels have been investigated in several studies. For example HA hydrogels functionalized with laminin316 or laminin-derived sequences, i.e. RGD250 and IKVAV,317 have been used for implantation in the cortex of rats after traumatic injury. The obtained results, confirmed by ultrastructural analysis with either TEM or SEM, showed that HA hydrogels modified with either laminin or laminin-derived peptides were infiltrated with cells, vessels and axons. These studies demonstrated that HA hydrogels modified with laminin or laminin-derived peptides promote further neurite extension and angiogenesis than unmodified HA scaffolds, nevertheless it remains to be clarified which of the chosen bioactive peptides may provide the best neuroregenerative effect.
In the context of stroke, an implant of HA hydrogel carrying an antibody to the Nogo-66 receptor (NgR) was tested in the brain lesion of rats following occlusion of the middle cerebral artery.318 The cross-linked HA hydrogel was modified with NgR antibody by covalently binding the Fc part of the antibodies to the HA polymer backbone. NgR binds to myelin-associated inhibitors of axon growth, thus the delivery of an antibody antagonizing the NgR effect supposedly promotes axon regeneration. In this study the hydrogel effectively delivered the NgR antibody and supported neural regeneration in the brain, leading to a better functional recovery in treated animals.
Methylcellulose was also demonstrated to be a good scaffolding material for implantation into the injured brain. Tate and colleagues produced various methylcellulose solutions with specific gelling behaviours by varying the polymer concentration and the solvent environment, then they tested in vivo a 2% methylcellulose formulation exhibiting low viscosity at 23 °C and forming a soft gel at 37 °C, thereby making possible the injection of the scaffold into the brain. After injection into the contused rat cortex, such methylcellulose-based hydrogel was shown to be biocompatible, even if it did not show a pro-regenerative effect.319
Collagen, fibrin and fibronectin have been combined with cells, mainly bone marrow stromal cells (BMSC), in various studies attempting to repair brain damage. Xiong and colleagues transplanted a cylindrical collagen scaffold seeded with human BMSCs into the lesion cavity of a cortical contusion in rats, and they obtained a reduction of the lesion volume, together with an improved functional outcome.320 Furthermore scaffolds composed of a fibronectin/collagen matrix seeded with NSCs have been directly injected into mouse brains after cortical contusion, showing an increase in survival and migration of cells in comparison to injection of cells alone.321 A fibrin matrix was also injected with BMSCs into the injured cortex of rats and was proven to enhance survival, migration and differentiation of the cells.322
In addition to natural materials, also synthetic materials have been employed in brain tissue engineering. Loh and colleagues249 demonstrated the potential of a combinatory approach making use of PHPMA-RGD hydrogels (NeuroGel) and genetically engineered cells in order to promote axon regrowth within tissue defects in the damaged brain. The hydrogel matrices were seeded with fibroblasts engineered to express BDNF and CNTF and were inserted into cavities made in the optic tracts of rats, showing the survival of large numbers of grafted fibroblasts for at least 8 weeks, leading to a significantly greater axon regrowth. However, the presence of a non-degradable hydrogel may become deleterious over long periods because of the risk of developing a foreign body reaction. Moving to bioabsorbable synthetic polymers, Park and co-workers323 developed a PGA-based scaffold seeded with NSCs and implanted it into the cavity resulting from HI injury in mice. The cellular matrix fostered the regrowth of cortical tissue, together with reducing inflammation and scar formation within the brain.
The use of self-assembling materials was also explored to repair the injured brain. In an important study, a solution of RADA16-I was injected into the severed optic tract in hamsters and was shown to create a permissive environment for axon growth, resulting in functional return of vision.324 More recently RADA16-I was employed by Guo and colleagues to reconstruct a cavity in the rat cortex after TBI:325 the scaffold was shown to be capable of reducing the inflammatory reaction, integrating with the host tissue and promoting the closure of the lesion cavity.
Finally, a recent study described the effects of scaffold architecture on the regeneration of rat cortical injuries. Even if this study did not employ a hydrogel matrix, the interesting results showed that porous PCL-based sponges presenting differently oriented channels and microgrooves induced both qualitatively and quantitatively different cellular infiltrations: for example, microgrooves enhanced cellular ingrowth and nerve fibers alignment when oriented in the direction of cell migration; moreover the orthogonally designed matrix, consisting of intersecting macrochannels and microgrooves, had a better total cellular infiltration than the matrix containing only longitudinally oriented channels and grooves.326
Overall, the implantation of nanostructured gel-based scaffolds, either alone or seeded with progenitor/stem cells, has been demonstrated to provide a neuroprotective effect and ameliorate the cognitive deficits in brain injuries. The proposed therapies however did not lead to a full restoration of lost functions: hence future in vivo studies need to be performed before translating cerebral tissue engineering research into the clinic.
Final remarks
In this document we gave an overview of the diverse phenomena, methods and techniques that have to be carefully considered in developing a nanostructured scaffold for regenerative purposes, in particular for neural tissue engineering. This methodology, in turn, should drive the experimenter to the choice of suitable scaffold synthesis and characterization methods and, most importantly, to the understanding of the phenomena involved in its three-dimensional nanostructure formation. Applications in in vivo studies provide also an idea of the current status of research and, most importantly, of the magnitude of the efforts that are still needed for an effective nervous tissue engineering therapy. Here follows an example of the multi-disciplinarity intrinsically involved in scaffold development: if a nanostructured scaffold is required to achieve cell engraftment and/or a specific protein interaction it will likely have to be biofunctionalized with one (or more) specific functional motif (discovered either via phage display or combinatorial chemistry in combination with high-throughput screenings), the proposed modification has to be corroborated by computational investigations prior to synthesis as well as by scaffold characterization, followed by in vitro experiments and eventually by in vivo experiments. Last but not least, crucial steps will be, if required, the choice of right cell type (or mixture of cell types) to be transplanted, cocktail of bioactive molecules to be delivered with the right kinetics, and animal model resembling the chosen nervous pathology.Therefore, even if most of the necessary basic science is likely now available, it is our opinion that the major task of regenerative nanomedicine will be to efficiently coordinate these several fields of investigation.
References
- S. Yang, K. F. Leong, Z. Du and C. K. Chua, Tissue Eng., 2001, 7, 679–689 CrossRef CAS.
- H. Park, C. Cannizzaro, G. Vunjak-Novakovic, R. Langer, C. A. Vacanti and O. C. Farokhzad, Tissue Eng., 2007, 13, 1867–1877 CrossRef CAS.
- S. Cranford and M. J. Buehler, Nanotechnology, 2010, 3, 127 CAS.
- Y. S. Velichko, S. I. Stupp and M. O. de la Cruz, J. Phys. Chem. B, 2008, 112, 2326–2334 CrossRef CAS.
- S. Tsonchev, K. L. Niece, G. C. Schatz, M. A. Ratner and S. I. Stupp, J. Phys. Chem. B, 2008, 112, 441–447 CrossRef CAS.
- B. A. F. Mann, K. Kremer, O. Lenz and C. Holm, Macromol. Theory Simul., 2011, 20, 721–734 CrossRef CAS.
- F. Taraballi, M. Campione, A. Sassella, A. Vescovi, A. Paleari, W. Hwang and F. Gelain, Soft Matter, 2009, 5, 660–668 RSC.
- F. Taraballi, A. Natalello, M. Campione, O. Villa, S. M. Doglia, A. Paleari and F. Gelain, Front. Neuroeng., 2010, 3, 1 CrossRef CAS.
- F. Gelain, D. Cigognini, A. Caprini, D. Silva, B. Colleoni, M. Donega, S. Antonini, B. E. Cohen and A. Vescovi, Nanoscale, 2012, 4, 2946–2957 RSC.
- M. Abu-Rub, S. McMahon, D. I. Zeugolis, A. Windebank and A. Pandit, Drug Discovery Today, 2010, 15, 436–443 CrossRef CAS.
- J. A. Matthews, E. D. Boland, G. E. Wnek, D. G. Simpson and G. L. Bowlin, J. Bioact. Compat. Polym., 2003, 18, 125–134 CrossRef CAS.
- C. Chen, C. B. Cao, X. L. Ma, Y. Tang and H. S. Zhu, Polymer, 2006, 47, 6322–6327 CrossRef CAS.
- B. M. Min, L. Jeong, K. Y. Lee and W. H. Park, Macromol. Biosci., 2006, 6, 285–292 CrossRef CAS.
- K. H. Zhang, X. M. Mo, C. Huang, C. L. He and H. S. Wang, J. Biomed. Mater. Res., Part A, 2010, 93A, 976–983 CAS.
- K. Ohkawa, D. I. Cha, H. Kim, A. Nishida and H. Yamamoto, Macromol. Rapid Commun., 2004, 25, 1600–1605 CrossRef CAS.
- K. Y. Lee, L. Jeong, Y. O. Kang, S. J. Lee and W. H. Park, Adv. Drug Delivery Rev., 2009, 61, 1020–1032 CrossRef CAS.
- N. Bhattarai and M. Q. Zhang, Nanotechnology, 2007, 18, 455601 CrossRef.
- H. L. Jiang, D. F. Fang, B. S. Hsiao, B. Chu and W. L. Chen, Biomacromolecules, 2004, 5, 326–333 CrossRef CAS.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS.
- D. N. Rockwood, J. Fromstein, K. A. Woodhouse, D. B. Chase and J. F. Rabolt, Abstr. Paper. Am. Chem. Soc., 2004, 228, U364 Search PubMed.
- T. B. Bini, S. J. Gao, T. C. Tan, S. Wang, A. Lim, L. B. Hai and S. Ramakrishna, Nanotechnology, 2004, 15, 1459–1464 CrossRef CAS.
- S. Panseri, C. Cunha, J. Lowery, U. Del Carro, F. Taraballi, S. Amadio, A. Vescovi and F. Gelain, BMC Biotechnol., 2008, 8, 39 CrossRef.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS.
- W. T. Truong, Y. Y. Su, J. T. Meijer, P. Thordarson and F. Braet, Chem–Asian J., 2011, 6, 30–42 CrossRef CAS.
- P. D. Dalton, L. Flynn and M. S. Shoichet, Biomaterials, 2002, 23, 3843–3851 CrossRef CAS.
- S. Zhang, C. Lockshin, A. Herbert, E. Winter and A. Rich, EMBO J., 1992, 11, 3787–3796 CAS.
- F. Gelain, D. Bottai, A. Vescovi and S. Zhang, PLoS One, 2006, 1, e119 Search PubMed.
- C. G. Knight, L. F. Morton, A. R. Peachey, D. S. Tuckwell, R. W. Farndale and M. J. Barnes, J. Biol. Chem., 2000, 275, 35–40 CrossRef CAS.
- J. P. Ranieri, R. Bellamkonda, E. J. Bekos, T. G. Vargo, J. A. Gardella, Jr. and P. Aebischer, J. Biomed. Mater. Res., 1995, 29, 779–785 CrossRef CAS.
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491–497 CAS.
- G. S. Nowakowski, M. S. Dooner, H. M. Valinski, A. M. Mihaliak, P. J. Quesenberry and P. S. Becker, Stem Cells, 2004, 22, 1030–1038 CrossRef CAS.
- S. Giudice, L. Benassi, G. Bertazzoni, M. Costi, A. Gelain, A. Venturelli, C. Bernardi, G. Gualdi, A. Coppi, T. Rossi, A. Giannetti and C. Magnoni, Toxicol. In Vitro, 2007, 21, 240–248 CrossRef CAS.
- S. Zhang, C. Lockshin, R. Cook and A. Rich, Biopolymers, 1994, 34, 663–672 CrossRef CAS.
- R. Vegners, I. Shestakova, I. Kalvinsh, R. M. Ezzell and P. A. Janmey, J. Pept. Sci., 1995, 1, 371–378 CrossRef CAS.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Chem. Commun., 2010, 46, 4267–4269 RSC.
- M. H. Dietz and B. A. Flaxman, Cancer Res., 1971, 31, 1206–1209 CAS.
- M. D. Ford, K. A. Delaney, L. J. Ling and T. Erickson, Clinical Toxicology, Saunders Elsevier, Philadelphia, 1st edn, 2001 Search PubMed.
- S. Matsumura, S. Uemura and H. Mihara, Mol. BioSyst., 2005, 1, 146–148 RSC.
- F. Gelain, D. Silva, A. Caprini, F. Taraballi, A. Natalello, O. Villa, K. T. Nam, R. N. Zuckermann, S. M. Doglia and A. Vescovi, ACS Nano, 2011, 5, 1845–1859 CrossRef CAS.
- H. G. Cui, M. J. Webber and S. I. Stupp, Biopolymers, 2010, 94, 1–18 CrossRef CAS.
- U. Khoe, Y. Yang and S. Zhang, Langmuir, 2009, 25, 4111–4114 CrossRef CAS.
- S. R. Bull, L. C. Palmer, N. J. Fry, M. A. Greenfield, B. W. Messmore, T. J. Meade and S. I. Stupp, J. Am. Chem. Soc., 2008, 130, 2742–2743 CrossRef CAS.
- D. A. Harrington, E. Y. Cheng, M. O. Guler, L. K. Lee, J. L. Donovan, R. C. Claussen and S. I. Stupp, J. Biomed. Mater. Res., Part A, 2006, 78A, 157–167 CrossRef CAS.
- S. I. Stupp, S. M. Zhang, M. A. Greenfield, A. Mata, L. C. Palmer, R. Bitton, J. R. Mantei, C. Aparicio and M. O. de la Cruz, Nat. Mater., 2010, 9, 594–601 CrossRef.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS.
- C. M. Micklitsch, P. J. Knerr, M. C. Branco, R. Nagarkar, D. J. Pochan and J. P. Schneider, Angew. Chem., Int. Ed., 2011, 50, 1577–1579 CrossRef CAS.
- K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS.
- C. Q. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- C. Q. Yan, M. E. Mackay, K. Czymmek, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Langmuir, 2012, 28, 6076–6087 CrossRef CAS.
- L. E. R. O'Leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef CAS.
- J. A. Fallas, J. Dong, Y. J. Tao and J. D. Hartgerink, J. Biol. Chem., 2012, 287, 8039–8047 CrossRef CAS.
- S. Ramachandran, Y. Tseng and Y. B. Yu, Biomacromolecules, 2005, 6, 1316–1321 CrossRef CAS.
- M. B. Taraban, S. Ramachandran, I. Gryczynski, Z. Gryczynski, J. Trewhella and Y. B. Yu, Soft Matter, 2011, 7, 2624–2631 RSC.
- Y. Feng, M. Lee, M. Taraban and Y. B. Yu, Chem. Commun., 2011, 47, 10455–10457 RSC.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS.
- B. D. Olsen, J. A. Kornfield and D. A. Tirrell, Macromolecules, 2010, 43, 9094–9099 CrossRef CAS.
- R. L. Dimarco and S. C. Heilshorn, Adv. Mater., 2012, 3923–3940 CrossRef CAS.
- H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468–12472 CrossRef CAS.
- L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS.
- E. L. Bakota, L. Aulisa, K. M. Galler and J. D. Hartgerink, Biomacromolecules, 2011, 12, 82–87 CrossRef CAS.
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223 CrossRef CAS.
- B. Xing, C. W. Yu, K. H. Chow, P. L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846–14847 CrossRef CAS.
- Y. Gao, Y. Kuang, Z. F. Guo, Z. H. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576–13577 CrossRef CAS.
- X. Li, Y. Gao, Y. Kuang and B. Xu, Chem. Commun., 2010, 46, 5364–5366 RSC.
- F. Zhao, Y. Gao, J. Shi, H. M. Browdy and B. Xu, Langmuir, 2011, 27, 1510–1512 CrossRef CAS.
- Y. Zhang, N. Li, J. Delgado, Y. Gao, Y. Kuang, S. Fraden, I. R. Epstein and B. Xu, Langmuir, 2012, 28, 3063–3066 CrossRef CAS.
- C. Wang, R. J. Stewart and J. Kopeček, Nature, 1999, 397, 417–420 CrossRef CAS.
- R. N. Zuckermann and T. Kodadek, Curr. Opin. Mol. Ther., 2009, 11, 299–307 CAS.
- H. Tran, S. L. Gael, M. D. Connolly and R. N. Zuckermann, J. Visualized Exp., 2011, e3373 CAS.
- M. J. Mahoney and K. S. Anseth, Biomaterials, 2006, 27, 2265–2274 CrossRef CAS.
- O. Jeon, D. S. Alt, S. M. Ahmed and E. Alsberg, Biomaterials, 2012, 33, 3503–3514 CrossRef CAS.
- K. B. Bjugstad, K. Lampe, D. S. Kern and M. Mahoney, J. Biomed. Mater. Res., Part A, 2010, 95, 79–91 CrossRef CAS.
- T. Freier, H. S. Koh, K. Kazazian and M. S. Shoichet, Biomaterials, 2005, 26, 5872–5878 CrossRef CAS.
- W. Wang, S. Itoh, A. Matsuda, S. Ichinose, K. Shinomiya, Y. Hata and J. Tanaka, J. Biomed. Mater. Res., Part A, 2008, 84, 557–566 Search PubMed.
- H. J. Kong, D. Kaigler, K. Kim and D. J. Mooney, Biomacromolecules, 2004, 5, 1720–1727 CrossRef CAS.
- S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmuller, R. Muller, F. E. Weber and J. A. Hubbell, Biomacromolecules, 2006, 7, 3019–3029 CrossRef CAS.
- S. Sokic and G. Papavasiliou, Acta Biomater., 2012, 8, 2213–2222 CrossRef CAS.
- Y. Chau, Y. Luo, A. C. Cheung, Y. Nagai, S. Zhang, J. B. Kobler, S. M. Zeitels and R. Langer, Biomaterials, 2008, 29, 1713–1719 CrossRef.
- H. W. Jun, V. Yuwono, S. E. Paramonov and J. D. Hartgerink, Adv. Mater., 2005, 17, 2612–2617 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CAS.
- L. A. Bagatolli, J. H. Ipsen, A. C. Simonsen and O. G. Mouritsen, Prog. Lipid Res., 2010, 49, 378–389 CrossRef CAS.
- E. Stulz, G. Clever, M. Shionoya and C. D. Mao, Chem. Soc. Rev., 2011, 40, 5633–5635 RSC.
- M. Calamai, J. R. Kumita, J. Mifsud, C. Parrini, M. Ramazzotti, G. Ramponi, N. Taddei, F. Chiti and C. M. Dobson, Biochemistry, 2006, 45, 12806–12815 CrossRef CAS.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. Blake, J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS.
- J. C. Rochet and P. T. Lansbury, Jr., Curr. Opin. Struct. Biol., 2000, 10, 60–68 CrossRef CAS.
- D. Thirumalai, D. K. Klimov and R. I. Dima, Curr. Opin. Struct. Biol., 2003, 13, 146–159 CrossRef CAS.
- C. M. Dobson and M. Karplus, Curr. Opin. Struct. Biol., 1999, 9, 92–101 CrossRef CAS.
- M. W. West, W. Wang, J. Patterson, J. D. Mancias, J. R. Beasley and M. H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11211–11216 CrossRef CAS.
- A. I. Bartlett and S. E. Radford, Nat. Struct. Mol. Biol., 2009, 16, 582–588 CAS.
- S. R. Collins, A. Douglass, R. D. Vale and J. S. Weissman, PLoS Biol., 2004, 2, e321 Search PubMed.
- M. M. Pallitto and R. M. Murphy, Biophys. J., 2001, 81, 1805–1822 CrossRef CAS.
- N. Metropolis and S. Ulam, J. Am. Stat. Assoc., 1949, 44, 335–341 CrossRef CAS.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth and A. H. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS.
- B. J. Alder and T. E. Wainwright, J. Chem. Phys., 1959, 31, 459–466 CrossRef CAS.
- Y. Lu, P. Derreumaux, Z. Guo, N. Mousseau and G. Wei, Proteins: Struct., Funct., Bioinf., 2009, 75, 954–963 CrossRef CAS.
- L. Negureanu and A. Baumketner, J. Mol. Biol., 2009, 389, 921–937 CrossRef CAS.
- H. B. Nam, M. Kouza, H. Zung and M. S. Li, J. Chem. Phys., 2010, 132, 165104 CrossRef.
- Y. Sugita and Y. Okamoto, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- A. R. Leach, Molecular modelling: principles and applications, Prentice Hall, 2001 Search PubMed.
- A. Laio and F. L. Gervasio, Rep. Prog. Phys., 2008, 71, 126601 CrossRef.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS.
- V. Tozzini, Curr. Opin. Struct. Biol., 2005, 15, 144–150 CrossRef CAS.
- J. Zhang and M. Muthukumar, J. Chem. Phys., 2009, 130, 035102 CrossRef.
- M. S. Li, D. K. Klimov, J. E. Straub and D. Thirumalai, J. Chem. Phys., 2008, 129, 175101 CrossRef.
- R. Pellarin, E. Guarnera and A. Caflisch, J. Mol. Biol., 2007, 374, 917–924 CrossRef CAS.
- G. Bellesia and J. E. Shea, J. Chem. Phys., 2009, 130, 145103 CrossRef.
- M. Cheon, I. Chang and C. K. Hall, Proteins: Struct., Funct., Bioinf., 2010, 78, 2950–2960 CrossRef CAS.
- T. P. Stedall, M. F. Butler, D. N. Woolfson and S. Hanna, Eur. Phys. J. E: Soft Matter Biol. Phys., 2011, 34, 1–16 CrossRef CAS.
- D. N. Woolfson, A. M. Smith, S. F. A. Acquah, N. Bone, H. W. Kroto, M. G. Ryadnov, M. S. P. Stevens and D. R. M. Walton, Angew. Chem., Int. Ed., 2005, 44, 325–328 CrossRef.
- N. Mousseau and P. Derreumaux, Front. Biosci., Landmark Ed., 2008, 13, 4495–4516 CAS.
- S. Auer, F. Meersman, C. M. Dobson and M. Vendruscolo, PLoS Comput. Biol., 2008, 4, e1000222 Search PubMed.
- P. J. Hoogerbrugge and J. M. V. A. Koelman, Europhys. Lett., 1992, 19, 155–160 CrossRef.
- H. Tang, M. J. Buehler and B. Moran, Ann. Biomed. Eng., 2009, 37, 1117–1130 CrossRef.
- F. Chiti, M. Stefani, N. Taddei, G. Ramponi and C. M. Dobson, Nature, 2003, 424, 805–808 CrossRef CAS.
- A. Caflisch, Curr. Opin. Chem. Biol., 2006, 10, 437–444 CrossRef CAS.
- G. G. Tartaglia, A. P. Pawar, S. Campioni, C. M. Dobson, F. Chiti and M. Vendruscolo, J. Mol. Biol., 2008, 380, 425–436 CrossRef CAS.
- A. M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS.
- E. I. Lin and M. S. Shell, J. Phys. Chem. B, 2010, 114, 11899–11908 CrossRef CAS.
- I. Daidone, F. Simona, D. Roccatano, R. A. Broglia, G. Tiana, G. Colombo and A. Di Nola, Proteins: Struct., Funct., Bioinf., 2004, 57, 198–204 CrossRef CAS.
- G. A. Saracino, A. Villa, G. Moro, U. Cosentino and M. Salmona, Proteins: Struct., Funct., Bioinf., 2009, 75, 964–976 CrossRef CAS.
- F. Massi, D. Klimov, D. Thirumalai and J. E. Straub, Protein Sci., 2002, 11, 1639–1647 CrossRef CAS.
- A. Baumketner and J. E. Shea, J. Mol. Biol., 2006, 362, 567–579 CrossRef CAS.
- M. M. Murray, M. G. Krone, S. L. Bernstein, A. Baumketner, M. M. Condron, N. D. Lazo, D. B. Teplow, T. Wyttenbach, J. E. Shea and M. T. Bowers, J. Phys. Chem. B, 2009, 113, 6041–6046 CrossRef CAS.
- W. Hwang, S. Zhang, R. D. Kamm and M. Karplus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12916–12921 CrossRef CAS.
- B. Tarus, J. E. Straub and D. Thirumalai, J. Mol. Biol., 2005, 345, 1141–1156 CrossRef CAS.
- A. Baumketner and J. E. Shea, Biophys. J., 2005, 89, 1493–1503 CrossRef CAS.
- G. Favrin, A. Irback and S. Mohanty, Biophys. J., 2004, 87, 3657–3664 CrossRef CAS.
- H. D. Nguyen and C. K. Hall, J. Am. Chem. Soc., 2006, 128, 1890–1901 CrossRef CAS.
- F. Ding, J. J. LaRocque and N. V. Dokholyan, J. Biol. Chem., 2005, 280, 40235–40240 CrossRef CAS.
- G. Singh, I. Brovchenko, A. Oleinikova and R. Winter, Biophys. J., 2008, 95, 3208–3221 CrossRef CAS.
- G. Bellesia and J. E. Shea, Biophys. J., 2009, 96, 875–886 CrossRef CAS.
- M. Lopez de la Paz, G. M. de Mori, L. Serrano and G. Colombo, J. Mol. Biol., 2005, 349, 583–596 CrossRef CAS.
- A. De Simone and P. Derreumaux, J. Chem. Phys., 2010, 132, 165103 CrossRef.
- J. Nasica-Labouze, M. Meli, P. Derreumaux, G. Colombo and N. Mousseau, PLoS Comput. Biol., 2011, 7, e1002051 CAS.
- S. Auer, C. M. Dobson, M. Vendruscolo and A. Maritan, Phys. Rev. Lett., 2008, 101, 258101 CrossRef.
- I. Kheterpal, M. Chen, K. D. Cook and R. Wetzel, J. Mol. Biol., 2006, 361, 785–795 CrossRef CAS.
- M. R. Nichols, M. A. Moss, D. K. Reed, W. L. Lin, R. Mukhopadhyay, J. H. Hoh and T. L. Rosenberry, Biochemistry, 2002, 41, 6115–6127 CrossRef CAS.
- J. E. Straub and D. Thirumalai, Annu. Rev. Phys. Chem., 2011, 62, 437–463 CrossRef CAS.
- G. Reddy, J. E. Straub and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 11948–11953 CrossRef CAS.
- S. Santini, G. Wei, N. Mousseau and P. Derreumaux, Structure, 2004, 12, 1245–1255 CrossRef CAS.
- P. Derreumaux and N. Mousseau, J. Chem. Phys., 2007, 126, 025101 CrossRef.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- L. Esposito, C. Pedone and L. Vitagliano, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11533–11538 CrossRef CAS.
- L. Tjernberg, W. Hosia, N. Bark, J. Thyberg and J. Johansson, J. Biol. Chem., 2002, 277, 43243–43246 CrossRef CAS.
- R. Paparcone, S. Keten and M. J. Buehler, J. Biomech., 2010, 43, 1196–1201 CrossRef.
- J. Park, B. Kahng, R. D. Kamm and W. Hwang, Biophys. J., 2006, 90, 2510–2524 CrossRef CAS.
- M. Schor and P. G. Bolhuis, Phys. Chem. Chem. Phys., 2011, 13, 10457–10467 RSC.
- M. J. Buehler and S. Keten, Appl. Phys. Lett., 2010, 96, 153701 CrossRef.
- A. Nova, S. Keten, N. M. Pugno, A. Redaelli and M. J. Buehler, Nano Lett., 2010, 10, 2626–2634 CrossRef CAS.
- A. Gautieri, S. Vesentini, A. Redaelli and M. J. Buehler, Nano Lett., 2011, 11, 757–766 CrossRef CAS.
- S. Skovstrup, S. G. Hansen, T. Skrydstrup and B. Schiott, Biomacromolecules, 2010, 3196–3207 CrossRef CAS.
- S. Nikolov, M. Petrov, L. Lymperakis, M. Friak, C. Sachs, H. O. Fabritius, D. Raabe and J. Neugebauer, Adv. Mater., 2010, 22, 519–526 CrossRef CAS.
- P.-G. D. Gennes, Scaling concepts in polymer physics, Cornell University Press, Ithaca [N.Y.], London, 1979 Search PubMed.
- P. Flory, Principle of polymer chemistry, Ithaca, New York, 1953 Search PubMed.
- A. V. Dobrynin, Curr. Opin. Colloid Interface Sci., 2008, 13, 376–388 CrossRef CAS.
- A. V. Dobrynin and M. Rubinstein, Prog. Polym. Sci., 2005, 30, 1049–1118 CrossRef CAS.
- G. S. Longo, M. O. de la Cruz and I. Szleifer, Soft Matter, 2012, 8, 1344–1354 RSC.
- J. Walter, V. Ermatchkov, J. Vrabec and H. Hasse, Fluid Phase Equilib., 2010, 296, 164–172 CrossRef CAS.
- Z. Y. Lu and R. Hentschke, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 67, 061807 CrossRef.
- B. A. Mann, C. Holm and K. Kremer, J. Chem. Phys., 2005, 122, 154903 CrossRef.
- I. Carmesin and K. Kremer, Macromolecules, 1988, 21, 2819–2823 CrossRef CAS.
- R. L. Wu, T. Li and E. Nies, Macromol. Theory Simul., 2012, 21, 250–265 CrossRef CAS.
- F. A. Escobedo and J. J. De Pablo, Phys. Rep., 1999, 318, 85–112 CrossRef CAS.
- S. G. Lee, G. F. Brunello, S. S. Jang and D. G. Bucknall, Biomaterials, 2009, 30, 6130–6141 CrossRef CAS.
- E. Chiessi, A. Lonardi and G. Paradossi, J. Phys. Chem. B, 2010, 114, 8301–8312 CrossRef CAS.
- G. Paradossi, I. Finelli, F. Natali, M. T. F. Telling and E. Chiessi, Polymers, 2011, 3, 1805–1832 CrossRef CAS.
- H. Lee, A. H. de Vries, S. J. Marrink and R. W. Pastor, J. Phys. Chem. B, 2009, 113, 13186–13194 CrossRef CAS.
- S.-l. Lin, X.-f. Wen, Z.-q. Cai, P.-h. Pi, D.-f. Zheng, J. Cheng, L.-j. Zhang, Y. Qian and Z.-r. Yang, Phys. Chem. Chem. Phys., 2011, 13, 17323–17332 RSC.
- S. Edgecombe and P. Linse, Polymer, 2008, 49, 1981–1992 CrossRef CAS.
- G. S. Longo, M. O. de la Cruz and I. Szleifer, Macromolecules, 2011, 44, 147–158 CrossRef CAS.
- J. M. Y. Carrillo and A. V. Dobrynin, Macromolecules, 2011, 44, 5798–5816 CrossRef CAS.
- T. Wallmersperger, K. Keller, B. Kroplin, M. Gunther and G. Gerlach, Colloid Polym. Sci., 2011, 289, 535–544 CAS.
- K. S. Straley, C. W. Foo and S. C. Heilshorn, J. Neurotrauma, 2010, 27, 1–19 CrossRef.
- J. Doshi and D. H. Reneker, J. Electrost., 1995, 35, 151–160 CrossRef CAS.
- R. Kessick, J. Fenn and G. Tepper, Polymer, 2004, 45, 2981–2984 CrossRef CAS.
- D. Li, Y. L. Wang and Y. N. Xia, Nano Lett., 2003, 3, 1167–1171 CrossRef CAS.
- D. H. Reneker, A. L. Yarin, H. Fong and S. Koombhongse, J. Appl. Phys., 2000, 87, 4531–4547 CrossRef CAS.
- T. J. Sill and H. A. von Recum, Biomaterials, 2008, 29, 1989–2006 CrossRef CAS.
- S. Ramakrishna, An introduction to electrospinning and nanofibers, World Scientific, Hackensack, NJ, London, 2005 Search PubMed.
- J. Lowery, S. Panseri, C. Cunha and F. Gelain, in Electrospun Nanofibers Research: Recent Developments, NOVA Science Publishers, Hauppauge, NY, 2009, pp. 1–18 Search PubMed.
- B. S. Hsiao, D. Liang and B. Chu, Adv. Drug Delivery Rev., 2007, 59, 1392–1412 CrossRef.
- E. Ruel-Gariepy and J. C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409–426 CrossRef CAS.
- A. Jejurikar, G. Lawrie, D. Martin and L. Grondahl, Biomed. Mater., 2011, 6, 025010 CrossRef.
- R. E. Eliaz and J. Kost, J. Biomed. Mater. Res., 2000, 50, 388–396 CrossRef CAS.
- M. Gonen-Wadmany, L. Oss-Ronen and D. Seliktar, Biomaterials, 2007, 28, 3876–3886 CrossRef CAS.
- C. S. Bahney, C. W. Hsu, M. Bottlang, J. L. West and B. Johnstone, Eur. Cells Mater., 2011, 22, 43–55 CAS.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS.
- H. Susi and D. M. Byler, Methods Enzymol., 1986, 130, 290–311 CrossRef CAS.
- A. M. Mathur and A. B. Scranton, Biomaterials, 1996, 17, 547–557 CrossRef CAS.
- R. Tycko, Annu. Rev. Phys. Chem., 2011, 62, 279–299 CrossRef CAS.
- S. Sharpe, K. Simonetti, J. Yau and P. Walsh, Biomacromolecules, 2011, 12, 1546–1555 CrossRef CAS.
- M. Sunde, L. C. Serpell, M. Bartlam, P. E. Fraser, M. B. Pepys and C. C. F. Blake, J. Mol. Biol., 1997, 273, 729–739 CrossRef CAS.
- M. F. Perutz, J. T. Finch, J. Berriman and A. Lesk, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5591–5595 CrossRef CAS.
- S. K. Burley and G. A. Petsko, Science, 1985, 229, 23–28 CAS.
- D. P. Allison, N. P. Mortensen, C. J. Sullivan and M. J. Doktycz, WIREs Nanomed Nanobiotechnol., 2010, 2, 618–634 CrossRef.
- R. Barattin and N. Voyer, Chem. Commun., 2008, 1513–1532 RSC.
- M. R. Vanlandingham, S. H. McKnight, G. R. Palmese, R. F. Eduljee, J. W. Gillespie and R. L. McCulough, J. Mater. Sci. Lett., 1997, 16, 117–119 CrossRef CAS.
- T. J. Young, M. A. Monclus, T. L. Burnett, W. R. Broughton, S. L. Ogin and P. A. Smith, Meas. Sci. Technol., 2011, 22, 125703 CrossRef.
- S. Zhong and D. J. Pochan, Polym. Rev., 2010, 50, 287–320 CrossRef CAS.
- P. A. Fang, H. C. Margolis, J. F. Conway, J. P. Simmer, G. H. Dickinson and E. Beniash, Cells Tissues Organs, 2011 Search PubMed.
- H. Friedrich, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Angew. Chem., Int. Ed., 2010, 49, 7850–7858 CrossRef CAS.
- H. Cui, T. K. Hodgdon, E. W. Kaler, L. Abezgauz, D. Danino, M. Lubovsky, Y. Talmon and D. J. Pochan, Soft Matter, 2007, 3, 945–955 RSC.
- J. B. Guilbaud and A. Saiani, Chem. Soc. Rev., 2011, 40, 1200–1210 RSC.
- T. Yucel, C. M. Micklitsch, J. P. Schneider and D. J. Pochan, Macromolecules, 2008, 41, 5763–5772 CrossRef CAS.
- L. L. Hyland, M. B. Taraban, Y. Feng, B. Hammouda and Y. B. Yu, Biopolymers, 2012, 97, 177–188 CrossRef CAS.
- S. M. Lanasa, I. T. Hoffecker and S. J. Bryant, J. Biomed. Mater. Res., Part B, 2011, 96, 294–302 CrossRef.
- Q. Tang, X. Sun, Q. Li, J. Wu and J. Lin, J. Colloid Interface Sci., 2009, 339, 45–52 CrossRef CAS.
- J. P. Chen and Y. S. Chang, Colloids Surf., B, 2011, 86, 169–175 CrossRef CAS.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- E. Genove, C. Shen, S. Zhang and C. E. Semino, Biomaterials, 2005, 26, 3341–3351 CrossRef CAS.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS.
- A. V. Shenoy, D. R. Saini and V. M. Nadkarni, Rheol. Acta, 1983, 22, 209–222 CrossRef CAS.
- K. Saha, A. J. Keung, E. F. Irwin, Y. Li, L. Little, D. V. Schaffer and K. E. Healy, Biophys. J., 2008, 95, 4426–4438 CrossRef CAS.
- H. J. Snippert and H. Clevers, EMBO Rep., 2011, 12, 113–122 CrossRef CAS.
- D. E. Cohen and D. Melton, Nat. Rev. Genet., 2011, 12, 243–252 CrossRef CAS.
- G. Giannone and M. P. Sheetz, Trends Cell Biol., 2006, 16, 213–223 CrossRef CAS.
- I. Ahmed, A. S. Ponery, E. K. A. Nur, J. Kamal, A. S. Meshel, M. P. Sheetz, M. Schindler and S. Meiners, Mol. Cell. Biochem., 2007, 301, 241–249 CrossRef CAS.
- Y. J. Li, E. H. Chung, R. T. Rodriguez, M. T. Firpo and K. E. Healy, J. Biomed. Mater. Res., Part A, 2006, 79, 1–5 CrossRef.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS.
- H. Ozawa, T. Matsumoto, T. Ohashi, M. Sato and S. Kokubun, J. Neurosurg., 2001, 95, 221–224 CAS.
- P. C. Georges, W. J. Miller, D. F. Meaney, E. S. Sawyer and P. A. Janmey, Biophys. J., 2006, 90, 3012–3018 CrossRef CAS.
- A. P. Balgude, X. Yu, A. Szymanski and R. V. Bellamkonda, Biomaterials, 2001, 22, 1077–1084 CrossRef CAS.
- J. W. Gunn, S. D. Turner and B. K. Mann, J. Biomed. Mater. Res., Part A, 2005, 72, 91–97 Search PubMed.
- S. Li, J. Lao, B. P. Chen, Y. S. Li, Y. Zhao, J. Chu, K. D. Chen, T. C. Tsou, K. Peck and S. Chien, FASEB J., 2003, 17, 97–99 CrossRef CAS.
- H. J. Evans, J. K. Sweet, R. L. Price, M. Yost and R. L. Goodwin, Am. J. Physiol., 2003, 285, H570–H578 CAS.
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang and A. J. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS.
- L. Richert, D. Binda, G. Hamilton, C. Viollon-Abadie, E. Alexandre, D. Bigot-Lasserre, R. Bars, P. Coassolo and E. LeCluyse, Toxicol. In Vitro, 2002, 16, 89–99 CrossRef CAS.
- E. K. A. Nur, I. Ahmed, J. Kamal, M. Schindler and S. Meiners, Stem Cells, 2006, 24, 426–433 CrossRef.
- E. Garreta, E. Genove, S. Borros and C. E. Semino, Tissue Eng., 2006, 12, 2215–2227 CrossRef CAS.
- C. E. Semino, J. Kasahara, Y. Hayashi and S. Zhang, Tissue Eng., 2004, 10, 643–655 CrossRef CAS.
- M. J. Phillips and D. C. Otteson, Invest. Ophthalmol. Visual Sci., 2011, 52, 1439–1449 CAS.
- C. Cunha, S. Panseri, O. Villa, D. Silva and F. Gelain, Int. J. Nanomed., 2011, 6, 943–955 CrossRef CAS.
- L. D. Nagaprashantha, R. Vatsyayan, P. C. Lelsani, S. Awasthi and S. S. Singhal, Int. J. Cancer, 2011, 128, 743–752 CrossRef CAS.
- G. Haipeng, Z. Yinghui, L. Jianchun, G. Yandao, Z. Nanming and Z. Xiufang, J. Biomed. Mater. Res., 2000, 52, 285–295 CrossRef CAS.
- E. Yavin and Z. Yavin, J. Cell Biol., 1974, 62, 540–546 CrossRef CAS.
- L. N. Novikova, A. Mosahebi, M. Wiberg, G. Terenghi, J. O. Kellerth and L. N. Novikov, J. Biomed. Mater. Res., Part A, 2006, 77, 242–252 CrossRef.
- H. K. Kleinman and G. R. Martin, Semin. Cancer Biol., 2005, 15, 378–386 CrossRef CAS.
- K. A. Venstrom and L. F. Reichardt, FASEB J., 1993, 7, 996–1003 CAS.
- M. Suzuki, S. Itoh, I. Yamaguchi, K. Takakuda, H. Kobayashi, K. Shinomiya and J. Tanaka, J. Neurosci. Res., 2003, 72, 646–659 CrossRef CAS.
- G. Xu, D. Y. Nie, W. Z. Wang, P. H. Zhang, J. Shen, B. T. Ang, G. H. Liu, X. G. Luo, N. L. Chen and Z. C. Xiao, Neuroreport, 2004, 15, 2167–2172 CrossRef CAS.
- K. M. Yamada, J. Biol. Chem., 1991, 266, 12809–12812 CAS.
- P. Liesi, A. Narvanen, J. Soos, H. Sariola and G. Snounou, FEBS Lett., 1989, 244, 141–148 CrossRef CAS.
- A. P. Skubitz, J. B. McCarthy, Q. Zhao, X. Y. Yi and L. T. Furcht, Cancer Res., 1990, 50, 7612–7622 CAS.
- M. Barczyk, S. Carracedo and D. Gullberg, Cell Tissue Res., 2010, 339, 269–280 CrossRef CAS.
- N. K. Loh, S. Woerly, S. M. Bunt, S. D. Wilton and A. R. Harvey, Exp. Neurol., 2001, 170, 72–84 CrossRef CAS.
- F. Z. Cui, W. M. Tian, S. P. Hou, Q. Y. Xu and I. S. Lee, J. Mater. Sci.: Mater. Med., 2006, 17, 1393–1401 CrossRef CAS.
- J. C. Schense, J. Bloch, P. Aebischer and J. A. Hubbell, Nat. Biotechnol., 2000, 18, 415–419 CrossRef CAS.
- T. T. Yu and M. S. Shoichet, Biomaterials, 2005, 26, 1507–1514 CrossRef CAS.
- J. Xie, M. R. MacEwan, A. G. Schwartz and Y. Xia, Nanoscale, 2010, 2, 35–44 RSC.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- T. C. Holmes, S. de Lacalle, X. Su, G. Liu, A. Rich and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- H. Hosseinkhani, M. Hosseinkhani and H. Kobayashi, Biomed. Mater., 2006, 1, 8–15 CrossRef CAS.
- K. Saha, E. F. Irwin, J. Kozhukh, D. V. Schaffer and K. E. Healy, J. Biomed. Mater. Res., Part A, 2007, 81, 240–249 CrossRef.
- H. R. Hoogenboom, Trends Biotechnol., 1997, 15, 62–70 CrossRef CAS.
- K. Manoutcharian, G. Gevorkian, A. Cano and J. C. Almagro, Curr. Pharm. Biotechnol., 2001, 2, 217–223 CAS.
- R. H. Hoess, Chem. Rev., 2001, 101, 3205–3218 CrossRef CAS.
- S. M. Willerth, K. J. Arendas, D. I. Gottlieb and S. E. Sakiyama-Elbert, Biomaterials, 2006, 27, 5990–6003 CrossRef CAS.
- S. I. Stupp, J. J. J. M. Donners, G. A. Silva, H. A. Behanna and S. G. Anthony, Self-assembling peptide amphiphiles and related methods for growth factor delivery, US 7544661 B2, June 9, 2009 Search PubMed.
- S. Brown, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8651–8655 CrossRef CAS.
- S. R. Whaley, D. S. English, E. L. Hu, P. F. Barbara and A. M. Belcher, Nature, 2000, 405, 665–668 CrossRef CAS.
- M. G. Kolonin, L. Bover, J. Sun, A. J. Zurita, K. A. Do, J. Lahdenranta, M. Cardo-Vila, R. J. Giordano, D. E. Jaalouk, M. G. Ozawa, C. A. Moya, G. R. Souza, F. I. Staquicini, A. Kunyiasu, D. A. Scudiero, S. L. Holbeck, E. A. Sausville, W. Arap and R. Pasqualini, Cancer Res., 2006, 66, 34–40 CrossRef CAS.
- S. Zitzmann, W. Mier, A. Schad, R. Kinscherf, V. Askoxylakis, S. Kramer, A. Altmann, M. Eisenhut and U. Haberkorn, Clin. Cancer Res., 2005, 11, 139–146 CAS.
- M. L. Balestrieri and C. Napoli, Eur. J. Cancer, 2007, 43, 1242–1250 CrossRef CAS.
- J. K. Liu, Q. Teng, M. Garrity-Moses, T. Federici, D. Tanase, M. J. Imperiale and N. M. Boulis, Neurobiol. Dis., 2005, 19, 407–418 CrossRef CAS.
- S. T. Hou, M. Dove, E. Anderson, J. Zhang and C. R. MacKenzie, J. Neurosci. Methods, 2004, 138, 39–44 CrossRef CAS.
- W. Zhao, H. Yuan, X. Xu and L. Ma, J. Biomol. Screening, 2010, 15, 687–694 CrossRef CAS.
- A. Merzlyak, S. Indrakanti and S. W. Lee, Nano Lett., 2009, 9, 846–852 CrossRef CAS.
- F. I. Staquicini, E. Dias-Neto, J. Li, E. Y. Snyder, R. L. Sidman, R. Pasqualini and W. Arap, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2903–2908 CrossRef CAS.
- M. Trepel, W. Arap and R. Pasqualini, Curr. Opin. Chem. Biol., 2002, 6, 399–404 CrossRef CAS.
- S. Kanki, D. E. Jaalouk, S. Lee, A. Y. Yu, J. Gannon and R. T. Lee, J. Mol. Cell. Cardiol., 2011, 50, 841–848 CrossRef CAS.
- M. Zahid, B. E. Phillips, S. M. Albers, N. Giannoukakis, S. C. Watkins and P. D. Robbins, PLoS One, 2010, 5, e12252 Search PubMed.
- H. Y. Hong, J. S. Choi, Y. J. Kim, H. Y. Lee, W. Kwak, J. Yoo, J. T. Lee, T. H. Kwon, I. S. Kim, H. S. Han and B. H. Lee, J. Controlled Release, 2008, 131, 167–172 CrossRef CAS.
- S. Ueberberg and S. Schneider, Regul. Pept., 2010, 160, 1–8 CrossRef CAS.
- W. Arap, M. G. Kolonin, M. Trepel, J. Lahdenranta, M. Cardo-Vila, R. J. Giordano, P. J. Mintz, P. U. Ardelt, V. J. Yao, C. I. Vidal, L. Chen, A. Flamm, H. Valtanen, L. M. Weavind, M. E. Hicks, R. E. Pollock, G. H. Botz, C. D. Bucana, E. Koivunen, D. Cahill, P. Troncoso, K. A. Baggerly, R. D. Pentz, K. A. Do, C. J. Logothetis and R. Pasqualini, Nat. Med., 2002, 8, 121–127 CrossRef CAS.
- J. Ito, A. Ghosh, L. A. Moreira, E. A. Wimmer and M. Jacobs-Lorena, Nature, 2002, 417, 452–455 CrossRef CAS.
- X. Jiang, S. H. Lim, H. Q. Mao and S. Y. Chew, Exp. Neurol., 2010, 223, 86–101 CrossRef.
- E. C. Tsai, P. D. Dalton, M. S. Shoichet and C. H. Tator, J. Neurotrauma, 2004, 21, 789–804 CrossRef.
- F. M. Bareyre, N. Garzorz, C. Lang, T. Misgeld, H. Buning and M. Kerschensteiner, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6282–6287 CrossRef CAS.
- X. Gu, F. Ding, Y. Yang and J. Liu, Prog. Neurobiol., 2011, 93, 204–230 CrossRef CAS.
- L. R. Williams, F. M. Longo, H. C. Powell, G. Lundborg and S. Varon, J. Comp. Neurol., 1983, 218, 460–470 CrossRef CAS.
- J. S. Taras, V. Nanavati and P. Steelman, J. Hand Ther., 2005, 18, 191–197 CrossRef.
- W. Wang, S. Itoh, A. Matsuda, T. Aizawa, M. Demura, S. Ichinose, K. Shinomiya and J. Tanaka, J. Biomed. Mater. Res., Part A, 2008, 85, 919–928 CrossRef.
- S. Y. Chew, R. Mi, A. Hoke and K. W. Leong, Adv. Funct. Mater., 2007, 17, 1288–1296 CrossRef CAS.
- W. Wang, S. Itoh, K. Konno, T. Kikkawa, S. Ichinose, K. Sakai, T. Ohkuma and K. Watabe, J. Biomed. Mater. Res., Part A, 2009, 91, 994–1005 CrossRef.
- Y. T. Kim, V. K. Haftel, S. Kumar and R. V. Bellamkonda, Biomaterials, 2008, 29, 3117–3127 CrossRef CAS.
- M. S. Beattie, J. C. Bresnahan, J. Komon, C. A. Tovar, M. Van Meter, D. K. Anderson, A. I. Faden, C. Y. Hsu, L. J. Noble, S. Salzman and W. Young, Exp. Neurol., 1997, 148, 453–463 CrossRef CAS.
- C. Watson, The spinal cord: a Christopher and Dana Reeve Foundation text and atlas, Academic, London, 2009 Search PubMed.
- E. A. Joosten, P. R. Bar and W. H. Gispen, J. Neurosci. Res., 1995, 41, 481–490 CrossRef CAS.
- A. Iwata, K. D. Browne, B. J. Pfister, J. A. Gruner and D. H. Smith, Tissue Eng., 2006, 12, 101–110 CrossRef CAS.
- M. S. Shoichet, C. H. Tator, P. Poon, C. Kang and M. D. Baumann, Prog. Brain Res., 2007, 161, 385–392 CrossRef.
- P. J. Johnson, S. R. Parker and S. E. Sakiyama-Elbert, J. Biomed. Mater. Res., Part A, 2010, 92, 152–163 CrossRef.
- P. J. Johnson, A. Tatara, A. Shiu and S. E. Sakiyama-Elbert, Cell Transplant., 2010, 19, 89–101 CrossRef.
- V. R. King, D. Hewazy, A. Alovskaya, J. B. Phillips, R. A. Brown and J. V. Priestley, Neuroscience, 2010, 168, 523–530 CrossRef CAS.
- P. Prang, R. Muller, A. Eljaouhari, K. Heckmann, W. Kunz, T. Weber, C. Faber, M. Vroemen, U. Bogdahn and N. Weidner, Biomaterials, 2006, 27, 3560–3569 CAS.
- Y. Cho, R. Shi and R. B. Borgens, J. Exp. Biol., 2010, 213, 1513–1520 CrossRef CAS.
- A. Jain, Y. T. Kim, R. J. McKeon and R. V. Bellamkonda, Biomaterials, 2006, 27, 497–504 CrossRef CAS.
- T. Kamada, M. Koda, M. Dezawa, R. Anahara, Y. Toyama, K. Yoshinaga, M. Hashimoto, S. Koshizuka, Y. Nishio, C. Mannoji, A. Okawa and M. Yamazaki, Neuropathology, 2011, 31, 48–58 CrossRef.
- S. H. Lee, Y. N. Chung, Y. H. Kim, Y. J. Kim, J. P. Park, D. K. Kwon, O. S. Kwon, J. H. Heo, S. Ryu, H. J. Kang, S. H. Paek, K. C. Wang, S. U. Kim and B. W. Yoon, Neurol. Res., 2009, 31, 996–1002 CrossRef.
- J. Piantino, J. A. Burdick, D. Goldberg, R. Langer and L. I. Benowitz, Exp. Neurol., 2006, 201, 359–367 CrossRef CAS.
- A. Hejcl, L. Urdzikova, J. Sedy, P. Lesny, M. Pradny, J. Michalek, M. Burian, M. Hajek, J. Zamecnik, P. Jendelova and E. Sykova, J. Neurosurg., 2008, 8, 67–73 Search PubMed.
- S. Woerly, Neurosurg. Rev., 2000, 23, 59–77 CAS ; discussion 78–79.
- S. Woerly, E. Pinet, L. de Robertis, D. Van Diep and M. Bousmina, Biomaterials, 2001, 22, 1095–1111 CrossRef CAS.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS.
- V. M. Tysseling, V. Sahni, E. T. Pashuck, D. Birch, A. Hebert, C. Czeisler, S. I. Stupp and J. A. Kessler, J. Neurosci. Res., 2010, 88, 3161–3170 CrossRef CAS.
- J. Guo, H. Su, Y. Zeng, Y. X. Liang, W. M. Wong, R. G. Ellis-Behnke, K. F. So and W. Wu, Nanomedicine, 2007, 3, 311–321 CrossRef CAS.
- D. Cigognini, A. Satta, B. Colleoni, D. Silva, M. Donega, S. Antonini and F. Gelain, PLoS One, 2011, 6, e19782 CAS.
- E. C. Tsai, P. D. Dalton, M. S. Shoichet and C. H. Tator, Biomaterials, 2006, 27, 519–533 CrossRef CAS.
- F. Gelain, S. Panseri, S. Antonini, C. Cunha, M. Donega, J. Lowery, F. Taraballi, G. Cerri, M. Montagna, F. Baldissera and A. Vescovi, ACS Nano, 2011, 5, 227–236 CrossRef CAS.
- S. Meiners, I. Ahmed, A. S. Ponery, N. Amor, S. L. Harris, V. Ayres, Y. Fan, Q. Chen, R. Delgado-Rivera and A. N. Babu, Polym. Int., 2007, 56, 1340–1348 CrossRef CAS.
- A. Durukan and T. Tatlisumak, Pharmacol., Biochem. Behav., 2007, 87, 179–197 CrossRef CAS.
- P. A. Walker, K. R. Aroom, F. Jimenez, S. K. Shah, M. T. Harting, B. S. Gill and C. S. Cox, Jr., Stem Cell Rev., 2009, 5, 283–300 CrossRef CAS.
- S. Hou, Q. Xu, W. Tian, F. Cui, Q. Cai, J. Ma and I. S. Lee, J. Neurosci. Methods, 2005, 148, 60–70 CrossRef CAS.
- Y. T. Wei, W. M. Tian, X. Yu, F. Z. Cui, S. P. Hou, Q. Y. Xu and I. S. Lee, Biomed. Mater., 2007, 2, S142–S146 CrossRef CAS.
- J. Ma, W. M. Tian, S. P. Hou, Q. Y. Xu, M. Spector and F. Z. Cui, Biomed. Mater., 2007, 2, 233–240 CrossRef CAS.
- M. C. Tate, D. A. Shear, S. W. Hoffman, D. G. Stein and M. C. LaPlaca, Biomaterials, 2001, 22, 1113–1123 CrossRef CAS.
- Y. Xiong, C. Qu, A. Mahmood, Z. Liu, R. Ning, Y. Li, D. L. Kaplan, T. Schallert and M. Chopp, Brain Res., 2009, 1263, 183–191 CrossRef CAS.
- M. C. Tate, D. A. Shear, S. W. Hoffman, D. G. Stein, D. R. Archer and M. C. LaPlaca, Cell Transplant., 2002, 11, 283–295 Search PubMed.
- H. Yasuda, S. Kuroda, H. Shichinohe, S. Kamei, R. Kawamura and Y. Iwasaki, J. Neurosurg., 2010, 112, 336–344 CrossRef CAS.
- K. I. Park, Y. D. Teng and E. Y. Snyder, Nat. Biotechnol., 2002, 20, 1111–1117 CrossRef CAS.
- R. G. Ellis-Behnke, Y. X. Liang, S. W. You, D. K. Tay, S. Zhang, K. F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054–5059 CrossRef CAS.
- J. Guo, K. K. Leung, H. Su, Q. Yuan, L. Wang, T. H. Chu, W. Zhang, J. K. Pu, G. K. Ng, W. M. Wong, X. Dai and W. Wu, Nanomedicine, 2009, 5, 345–351 CrossRef CAS.
- D. Y. Wong, P. H. Krebsbach and S. J. Hollister, J. Neurosurg., 2008, 109, 715–722 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |