Traps and cages for universal SERS detection
Ramon A.
Alvarez-Puebla
* and
Luis M.
Liz-Marzán
*
Departamento de Química Física and Unidad Asociada CSIC-Universidade de Vigo, 36310, Vigo, Spain. E-mail: ramon.alvarez@uvigo.es; lmarzan@uvigo.es; Fax: +34 9868 12556; Tel: +34 9868 12298
First published on 4th August 2011
Abstract
In this tutorial review, we provide an overview of the recent research toward surface functionalization of plasmonic nanoparticles for the generation of advanced optical sensors that make possible the analysis of various moieties by means of surface enhanced Raman scattering (SERS). Such moieties include atomic ions, low affinity target molecules, inorganic anions, biometabolites, pathogen markers and/or other analytes of interest even under very demanding circumstances such as those related to real life samples. We expect this review to be of interest to researchers in a broad diversity of fields that can take advantage of the unprecedented sensitivity of this type of molecular spectroscopy, in a wide variety of analytical and bioanalytical problems.
 Ramon A. Alvarez-Puebla | Dr Ramon Alvarez-Puebla is currently a Research Scientist at the Department of Physical Chemistry, University of Vigo. He worked as a postdoc in the Department of Chemistry and Biochemistry of the University of Windsor (Canada) with Prof. Ricardo Aroca and he was appointed as a Research Officer at the National Institute for Nanotechnology of the National Research Council of Canada. He has co-authored over 80 articles and holds 2 patents. His current interests involve electronic and vibrational spectroscopy, surface enhanced spectroscopy and their application for sensor fabrication. |
 Luis M. Liz-Marzán | Luis M. Liz-Marzán is a PhD from the University of Santiago de Compostela (1992) and has been a postdoc at Utrecht University and (more recently) a visiting professor at Tohoku University, Michigan, Melbourne and Hamburg. He holds a chair in Physical Chemistry at the University of Vigo, where he is head of the Colloid Chemistry Group. He is a co-author of over 200 publications and 5 patents, has received several research awards, and is an editor and editorial advisory board member of several chemistry, nanotechnology and materials science journals. His current interests include nanoparticle synthesis and assembly, nanoplasmonics, and development of nanoparticle-based sensing and diagnostic tools. |
1. Introduction
There is currently a high demand for detection techniques that fulfil requirements relevant to extremely important social problems such as early stage diagnosis, environmental pollution, or terrorist threats, to name a few. These problems not only require extremely high sensitivity that allows detection of minute amounts of the relevant analyte, but also easy-to-handle devices and reduced (or ideally eliminated) sample preparation. Naturally, a universal detection technique would be highly preferred, so that various tests can be simultaneously run. Nanotechnology has been proposed as the perfect framework in which such techniques should be developed, and a number of nanostructured materials and devices have indeed been developed, including quantum dots, magnetic nanoparticles and plasmonic nanostructures, among others. In the context of nanoplasmonics (manipulation of light by metals with sub-wavelength dimensions), surface-enhanced Raman scattering (SERS) spectroscopy1 has been established as a true ultrasensitive, ultra-rapid and universal analytical technique, which can provide detection limits even down to the single molecule limit. A number of direct and label-free applications have been recently developed in fields as diverse as biomedicine, multiplex high-throughput screening, pollutant monitoring or molecular and materials characterization.2,3 The high sensitivity of SERS is however compromised by optimization of the enhanced electromagnetic near field at the surface of metal nanostructures when localized surface plasmon modes are excited, thus requiring the analyte to be in close contact with the metal surface. Therefore, until recently the applications of SERS were restricted to molecular analytes with chemical affinity for the common plasmonic substrates under the specific analysis conditions (i.e. surface composition and solution pH, ionic strength and/or composition). The rational design of efficient and flexible nanostructured substrates for SERS detection thus requires controlling the surface chemistry of the metallic nanostructures, so that a wider variety of chemical moieties can be analysed. This is thus one of the key challenges that needs to be achieved prior to universal application of the technique.As a summary, the surface of metallic nanostructures can be modified to improve the contact between the analyte and the plasmonic material by avoiding the presence of undesired surfactant/polymer species that may hinder this contact, either during the synthesis or by designing protocols to remove them, since such capping agents are often necessary for the fabrication of particles with a given shape and size.4 An alternative route consists of promoting adsorption of the analyte (i.e. increasing the partition coefficient between the surface and the bulk solution) through the increase of attractive electrostatic/hydrophobic interactions.5,6 In fact, an additional step may comprise forcing the adsorption of chemical species with very limited affinity for the metallic surfaces by using, in conjunction with the former, completely unspecific interactions such as mechanical forces.7,8 On the other hand, surface functionalization involving selected molecular systems can be used as well for the ultrasensitive analysis of species that give rise to very weak or negligible SERS signals by generating specific complexes between the analyte and the surface molecule, which can be more easily identified.9 Thus, the ligand usually comprises a molecule with a high SERS cross-section, which changes its electronic distribution, molecular geometry or both, upon complexation with the target analyte. This approach has recently become very popular in particular for the analysis of species such as atomic cations and anions, oxoanions or other molecular species with remarkably low cross-sections in SERS,10 and thus very difficult to detect. In relation with this, the occurrence of highly specific binding events between the surface molecule and the target analyte is also often exploited. Such events may include antibody–antigen recognition,11,12 as well as nucleic acid interactions and peptidic or nucleic acid aptamers.13 These biomolecules offer a suitable alternative for the determination of minute amounts of the analyte in highly complex media such as biological fluids or natural and waste waters directly, i.e. with no need for previous separation/purification, and thus improving the applicability and speed of the SERS analysis.
The purpose of this tutorial review is to summarize and discuss the rational design of plasmonic materials, with particular attention to the control of the environment close to the metallic surface, for efficient trapping of atomic species, low affinity target molecules, inorganic anions and/or other analytes of interest, even under highly demanding circumstances such as those of real life samples. We describe a variety of methods, including self-assembly, polymer grafting, layer-by-layer assembly, and bioconjugation, among others, which have been applied for the generation of advanced optical sensors allowing for optimized SERS analysis.
2. Capping agents that are undesired for SERS
The SERS effect is known to be a first layer effect. In fact, and contrary to what is often believed (and reported), the Raman signal is only amplified at distances not larger than 5 nm from the metallic surface, and the signal intensity exponentially decreases with the separation distance.1,14 Additionally, while gap distances within hot spots are known to extend over several nanometres (ca. 5 nm),15–17 recent reports indicate that extraordinary enhancements can occur at interparticle distances smaller than 2 nm due to the generation of non-local effects and quantum confinement.18,19 It is thus clear that the closer the analyte is to the surface, the higher SERS intensity will be registered. Bearing this in mind it becomes clear that, as a rule of thumb, clean plasmonic surfaces will have a better probability to adsorb the analyte and therefore give rise to a higher SERS signal. Unfortunately, this is impossible in normal practice, as even if the plasmonic nanostructures have been prepared by ultra-clean methods (i.e.physical vapour deposition, PVD) the operation of the sensor in contact with the atmosphere or with solutions carrying the analyte will be enough for the surface to retain undesired species such as O2, CO2, H2O, etc. This is not a big issue for common analytes containing functional groups such as thiols, amines, cyanides, carboxylic acids and others, which can often easily replace the undesired ligands because of their higher affinities for binding silver or gold,20 the most common plasmonic surfaces. However, it has been demonstrated that even in these cases the SERS signals augment when the O2 and other atmospheric gases are removed from the solution,21 and PVD systems are anyway rather expensive and restricted to a limited number of on-film geometries and sizes. Thus, colloidal plasmonic particles are commonly utilized, since they can be obtained in a rich variety of particle geometries and sizes and they can be either used directly in solution or even comprise the building blocks that give rise to other more refined plasmonic materials. Unfortunately, the fabrication of wet chemistry nanostructures typically requires the use of chemical reducing agents and surfactants to control the particle size, shape and colloidal stability and these chemical species may weakly interact with gold surfaces. For example, one of the most extended colloidal nanostructures exploited as SERS platforms comprises the use of citrate anions as reducing agents. In the case of gold, colloidal particles prepared in this way exclusively yield spherical geometries within a limited size range (below 30 nm). Interestingly, when the same method is used for silver, heterogeneous samples are typically obtained containing various geometries such as spheres, rods and plates of different sizes ranging from 10 to 80 nm. Although citrate has a well-defined (while weak) SERS signature,22 these colloids have been shown to perform extraordinarily well for many applications. However, when a better degree of size and shape control is required, different capping agents must be used.In general, all surfactants or solvents containing thiols, amines, or ionic halides should be avoided if the particles are to be used as SERS platforms. However, the cationic surfactant cetyl trimethylammonium bromide (CTAB) is still widely used as one of the most common shape-directing agents. This surfactant binds strongly to gold surfaces through the quaternary ammonium group and forms a bilayer structure around the particles, thus hindering the retention of most analytes. Because several of the most popular and interesting nanostructures (i.e.Au nanorods) require the use of excess CTAB for their efficient fabrication, the only solution to obtain a suitable SERS signal from these particles is post-synthesis cleaning and removal of the surfactant. As a first alternative, careful centrifugation has been employed, with the result of removing most of the excess of CTAB from the colloidal dispersion, but a single bilayer seems to consistently remain on the nanorod surface. Even in the presence of this protective bilayer, these structures have been demonstrated to yield higher SERS signals than those from spheres; however, the signal can be maximized if CTAB is efficiently removed, for example using an oxygen plasma, as recently demonstrated for nanorod supercrystals (Fig. 1A and B).4 Regarding polymers, other common shape-directing agents, significant effects have been found especially when non-porous macromolecules were used. Typical examples of this are polyvinylpyrrolidone (PVP) or poly(2-vinyl pyridine) and poly(4-vinyl pyridine) (P2VP and P4VP, respectively). The former can be almost completely removed after several centrifugation/redispersion cycles, yielding a progressively increased SERS intensity (Fig. 1C and D).23 However, the only way to remove the latter is, again, by application of an oxygen plasma treatment.
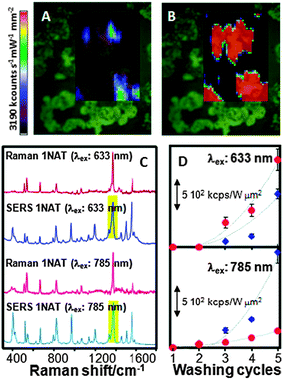 | ||
| Fig. 1 (A, B) Effect of plasma cleaning on the SERS signal of benzenethiol adsorbed onto Au nanorod supercrystals before (A) and after (B) plasma treatment. A substantial increase in SERS intensity was obtained after cleaning because of the removal of all the capping molecules present on the gold particles. (C, D) Raman, and SERS spectra (C) of 1-naphthalenethiol (1NAT) and variation of the SERS intensity as a function of the number of washing cycles and nanoparticle shape (spheres, red; stars, blue) (D) for two laser lines. The bands highlighted in (C) were used for intensity integration. Reproduced with permission from ref. 4 and 23. Copyright 2011, Proceedings of the National Academy of Sciences of the United States of America, and 2010, American Chemical Society, respectively. | ||
3. The simplest approach: electrostatic and hydrophobic interactions
In the previous section we have learnt that the intensity of the SERS signal gets improved as the analyte has a higher affinity toward the surface. The interactions of analytes with plasmonic surfaces are equivalent to those classically studied for heterogeneous catalysis, including transport of molecules to the interface, diffusion through the pores (if present) and adsorption on the solid surface. Regarding molecular transport to the surface, the involved driving forces are essentially of electrostatic or hydrophobic nature, depending on the specific nature of the analyte. The hydrophobic character of metallic surfaces is questionable when dealing with colloidal metals in solution, which has been demonstrated to be solvated and surrounded by an electrostatic double layer. This concept is very important because it means that before interacting with the surface, the analyte needs to be attracted by the electrostatic (or hydrophobic) nature of the capping counterions. In the case of the most common colloidal systems for SERS (i.e.citrate or borohydride reduced gold or silver particles) the capping agent is mainly formed by citrate ions and thus negatively charged. Bearing in mind that most of the conventional charged organic molecules carry negative charges, the interaction between the double layer and the analyte gets hindered. Historically, this problem has been partially resolved by using the so called “colloidal activation” which comprises the addition of an electrolyte solution such as nitrates, sulfates, perchlorates, halides or others to increase the ionic strength of the system, with a corresponding reduction of surface charge. This leads to a smaller electrostatic repulsion between the surface and the analyte, as well as an increase in analyte retention. The method compromises however the colloidal stability of the system while it only acts on the sols, not affecting the analytes. A step further involves the use of mineral acids (i.e.HNO3, H2SO4 or HClO4) to decrease the solution pH. The addition of acidic solution to the colloidal dispersion increases the ionic strength but it also increases the proton concentration, thereby decreasing the negative charge of carboxylate groups more efficiently (Fig. 2A).24 Additionally, these agents may also interact with analytes making the organic acids less negative, or even neutral, whereas amines are made more positive, thus improving their electrostatic interaction with surfaces and resulting in a consistent increase of the SERS signal at low pH (Fig. 2). Still, by using acids, the surface charge remains negative. Therefore, to reverse the surface charge from negative to positive and further improve the SERS signal, a rational choice of capping agents should be made. One of the best alternatives for citrate includes the aliphatic amino acids, which are characterized by their zwitterionic nature with a remarkably low cross-section for SERS. As a function of the amino acid nature (number of amino groups) and the pH of the medium, the surface charge on the particles can be tuned from +30 to −50 mV.25 It is however important to remark that not all amino acids can be used, since those containing thiol groups (Cys and Met) effectively passivate gold or silver surfaces, thereby inhibiting the retention of the molecules under study and thus SERS detection.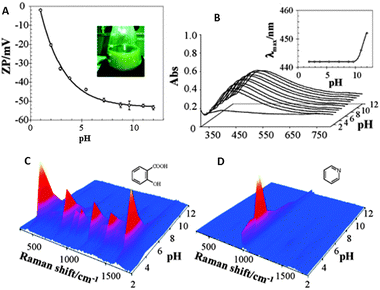 | ||
| Fig. 2 Variation of the ζ potential (A) and localized surface plasmon resonance (B) of citrate reduced silver sols as a function of solution pH. Insets in the surface plasmon spectra show the evolution of the absorption maximum as a function of pH. SERS spectra of salicylic acid (C) and pyridine (D) as a function of pH. Adapted with permission from ref. 24. Copyright 2005 the American Chemical society. | ||
All the described methods are efficient for polar molecules containing functional groups with affinity for gold and silver. Notwithstanding, there are many important analytes with no affinity for gold or silver and which are highly hydrophobic. One of the classical examples is the case of the polycyclic aromatic hydrocarbons (PAHs), hazardous atmospheric pollutants, that consist of fused aromatic rings and do not contain heteroatoms or carry any substituents. PAHs cannot be analysed using conventional SERS substrates as they are not retained at metallic surfaces. Hence, new approaches have been developed to increase the surface:solution partition coefficient. All such strategies rely on the functionalization of the surface with hydrophobic molecular monolayers. Perhaps the simplest but one of the most powerful techniques comprises the generation of a self-assembled monolayer of amino- or thio-aliphatic chains. Since these capping agents are not soluble in water, nanostructured solid thin films are preferred in this case, using any type of nanostructured plasmonic films.26 The films can be subsequently functionalized with the desired capping ligands (Fig. 3A). For example, PAHs5 and polychlorinated biphenyls27 have been detected by using alkyl monolayers. This technique, however, is very versatile and the chemical nature of the coating monolayer can be selected to accumulate other analytes with low affinity near the plasmonic surface. In another example, it has been possible to detect glucose in serum or Alzheimer markers by using (1-mercaptoundeca-11-yl)tri(ethylene glycol)6 (Fig. 3B) or 11-mercaptoundecanoic acid/1-octanethiol28 as capping monolayers, respectively.
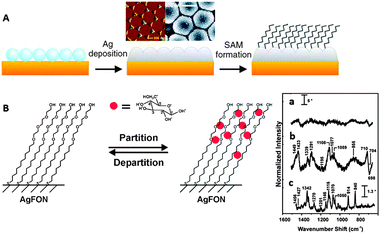 | ||
| Fig. 3 (A) Diagram demonstrating the fabrication process of the partition-layer modified Ag film over nanospheres. Silica spheres self-assemble on clean copper disks, then 200 nm of Ag is deposited onto the nanosphere mask, and the monolayer is formed during exposure of the substrate to an ethanolic alkanethiol solution. (B) Schematic representation of a hypothetical glucose concentration gradient created by a (1-mercaptoundeca-11-yl)tri(ethylene glycol) partition layer. SERS spectra of serum albumin (a) and serum albumin with glucose (b), both of them after subtraction of the monolayer signals. Raman spectrum of glucose (c). Adapted with permission from ref. 6. Copyright 2004 the American Chemical society. | ||
The advances in the understanding of supramolecular chemistry, and host–guest interactions in particular, have also been applied in this context. This approach is, in essence, very similar to the surface functionalization with aliphatic monolayers, but in this case it is carried out by means of molecules (host) showing specific supramolecular interactions––electrostatic, hydrophobic or both––with a particular family of target analytes (guest). This, a priori, slight difference is of key importance because such supramolecular interactions impart on the molecules preferential orientations with respect to the surface, contrary to the random orientation obtained by exploiting the partition coefficients in alkyl chains. Therefore, by using host molecules, the analyte is retained in a different geometry as compared to an open surface (for example, it is clear in Fig. 4B that the aromatic rings of the R,R isomer are more perpendicular to the surface than those of the S,S isomer, yielding an increase in the relative intensity of the in-plane C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretchings for the former) and thus surface selection rules29 can be applied, thereby increasing the level of selectivity of the technique. In fact, this approach has been recently applied for the direct recognition and quantification of the R,R- and S,S-hydrobenzoin enantiomers using thiolated cyclodextrins (CDs) as host molecules (Fig. 4).30 CDs have also been applied to the ultrasensitive detection of explosives,31 PAHs,32 and other pollutants such as carbendazim33 or parathion.34 As most sugars, CDs are characterized by extremely low SERS cross-sections, which results perfect for their trapping task while avoiding the presence of spurious signals in the spectra. Nevertheless, other hosts of different nature were successfully used for the detection of organic pollutants including calixarenes,35,36 viologen37 or even natural humic substances.38
C stretchings for the former) and thus surface selection rules29 can be applied, thereby increasing the level of selectivity of the technique. In fact, this approach has been recently applied for the direct recognition and quantification of the R,R- and S,S-hydrobenzoin enantiomers using thiolated cyclodextrins (CDs) as host molecules (Fig. 4).30 CDs have also been applied to the ultrasensitive detection of explosives,31 PAHs,32 and other pollutants such as carbendazim33 or parathion.34 As most sugars, CDs are characterized by extremely low SERS cross-sections, which results perfect for their trapping task while avoiding the presence of spurious signals in the spectra. Nevertheless, other hosts of different nature were successfully used for the detection of organic pollutants including calixarenes,35,36 viologen37 or even natural humic substances.38
![(A) Formation of silver nanoparticle-coated microspheres functionalized with thiolated cyclodextrin (Tβ-CD). Step (1): micron-sized polystyrene beads (PS) are wrapped with polyelectrolytes by sequential layer-by-layer (LbL) assembly of poly(diallyldimethylammonium chloride) [PDDA], polystyrene sulfonate (PSS), and poly(allylamine hydrochloride) [PAH]. Functionalized beads are then coated with densely packed silver nanoparticles (PS@AgNPs) (2), followed by silver surface functionalization with Tb-CDs (PS@AgNPs@Tb-CD) (3). Finally, the addition of hydrobenzoin enantiomers leads to the formation of supramolecular complexes, increasing the relative concentration of probes close to the active SERS surface (4). (B) DFT molecular models for the most probable conformation for each enantiomer within the cyclodextrin. (C) SERS spectra of PS@AgNP@Tb-CD and its supramolecular complexes with R,R- and S,S-hydrobenzoin enantiomers. Adapted with permission from ref. 30. Copyright 2011 Wiley-VCH.](/image/article/2012/CS/c1cs15155j/c1cs15155j-f4.gif) | ||
| Fig. 4 (A) Formation of silver nanoparticle-coated microspheres functionalized with thiolated cyclodextrin (Tβ-CD). Step (1): micron-sized polystyrene beads (PS) are wrapped with polyelectrolytes by sequential layer-by-layer (LbL) assembly of poly(diallyldimethylammonium chloride) [PDDA], polystyrene sulfonate (PSS), and poly(allylamine hydrochloride) [PAH]. Functionalized beads are then coated with densely packed silver nanoparticles (PS@AgNPs) (2), followed by silver surface functionalization with Tb-CDs (PS@AgNPs@Tb-CD) (3). Finally, the addition of hydrobenzoin enantiomers leads to the formation of supramolecular complexes, increasing the relative concentration of probes close to the active SERS surface (4). (B) DFT molecular models for the most probable conformation for each enantiomer within the cyclodextrin. (C) SERS spectra of PS@AgNP@Tb-CD and its supramolecular complexes with R,R- and S,S-hydrobenzoin enantiomers. Adapted with permission from ref. 30. Copyright 2011 Wiley-VCH. | ||
4. Mechanical trapping
The idea behind mechanical trapping comprises surrounding the active plasmonic surface with a responsive matrix that is capable of trapping and retaining the analyte of interest. Upon trapping, the matrix is retracted by applying a chemical or physical stimulus, thereby dragging all the trapped molecules toward the metal surface. The reversibility of this effect is mainly determined by the analyte–surface interaction. Analytes carrying functional groups with high affinity for gold and silver can readily passivate the surface, thereby not allowing reutilization of the sensor. Nevertheless, when the analytes are characterised by weak interactions with gold or silver surfaces, reversible and reusable sensors can be obtained.8,39 However, the fabrication of inorganic nanoparticle–polymer composites for SERS application has remained elusive for a long time because the loading of particles into the polymer matrix was typically too low. Recently, approaches such as grafting polymers directly on the particles, layer-by-layer deposition, molecular imprinting of polymers or in situreduction of metal ions within the polymer have made possible to increase the metal concentration and therefore to obtain composite materials with synergic properties that can be applied for optical sensing.The most commonly used stimuli are pH and temperature, but others such as ionic strength or dehydration can also be found in the literature. Regarding temperature-responsive polymers, poly-N-isopropylacrylamide (pNIPAM) has been the most popular choice. This material is characterized by a lower critical solution temperature (LCST) at around 32 °C, displaying an expanded porous network at lower temperatures and collapsing at higher temperatures. Demonstration of the use of pNIPAM to trap analytes for SERS detection has been recently reported using core–shell colloids comprising gold nanoparticle cores homogeneously coated with a microgel shell (Fig. 5A).7 Reversible trapping of small molecules with low affinity toward gold (1-naphthol) was demonstrated when the pNIPAM shell was collapsed at high temperature, so that they were concentrated close to the gold surface, yielding a high SERS signal. Unfortunately, the pNIPAM shell prevents close contact between metal particles and the corresponding electromagnetic coupling necessary to form hot spots, thus limiting the enhancement factors. Therefore, alternative composite materials were devised comprising multiple plasmonic40,41 particles, or even bifunctional plasmonic/magnetic42 particles inside each sub-micron sized pNIPAM bead (Fig. 5B). In these materials, the collapse of the gel at high temperature makes the nanoparticles approach each other and generate highly active hot spots. Additionally, the trapping properties of this material are not only restricted to their mechanical behaviour but electrostatic interactions can also be tunable through the use of pNIPAM block-copolymers with, for example, methacrylic acid40 or N-vinyl-2-pyrrolidone.41 On the other hand, it is important to note that the trapping efficiency of pNIPAM is not restricted to colloidal cross-linked particles but has also been demonstrated with polymer brushes in solid thin films, where two approaches are possible. The first one consists of growing the polymer fibres directly on the nanostructured plasmonic surface;8 while the second is based on the growth of the fibres onto a smooth gold surface followed by functionalization of their terminal groups with nanoparticles.43 For SERS applications, the second alternative has been shown to yield higher intensity (Fig. 5C). The SERS intensity of the analyte under study, trapped inside the fibres while the film is expanded, notably increases when it collapses because of both the optical coupling between nanoparticles and the non-local effects provided by coupling between the nanoparticles and the smooth gold surface.44
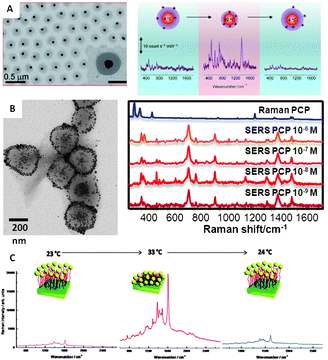 | ||
| Fig. 5 (A) Spherical gold nanoparticles coated with pNIPAM and variation of the SERS (λex = 785 nm) intensity of 1-naphthol as a function of solution temperature (from 4 to 60 °C and back to 4 °C). (B) Magnetite–Ag@pNIPAM composite microgels and their application to the SERS ultradetection of pentachlorophenol after concentration of the material in a spot using a permanent magnet (λex = 785 nm). (C) SERS spectra of methylene blue adsorbed on Au–PNIPAM–NPs substrates at various temperatures: from 23 to 33 °C and back to 24 °C (λex = 633 nm). Adapted with permission from ref. 7, 8, and 42. Copyright 2009 Wiley-VCH; and 2011 the American Chemical Society. | ||
Regarding materials that are responsive towards pH, the driving force to collapse or expand the polymer fibres or matrix essentially comprises the electrostatic forces generated at the functional groups (usually carboxylic or amino groups) as a function of proton concentration. This response can be tuned as a function of the nature of the analyte. For example, if cationic analytes (amines) are to be detected, the most widely used polymers are polyacrylic acid45,46 and its block-copolymers with polyethylene glycol, for example.47 At high pH, these polymers are expanded because of the electrostatic repulsion between the differently ionized carboxylate groups. When pH is decreased carboxylates are converted into carboxylic acids and the polymer collapses due to formation of intramolecular H-bonds. In the case of anionic analytes, polycarboxylic polymers are replaced by amino polymers such as polylysine.48 The behaviour of these materials is the contrary, they expand at low pH and collapse when pH increases. Notably, polymer engineering allows also for the design of à la carte responses by using the appropriate block-copolymers. Thus, materials such as nanoparticles coated with poly(4-vinylphenylboronic acid-co-2-(dimethylamino)ethyl acrylate) have been designed to expand only at physiological pH, allowing the in vivodetection of glucose or monitoring of insulin.49
Another alternative method for selective trapping is the direct imprinting of polymers with the shape of the molecular analyte (molecularly imprinted polymers, MIP) on the nanostructured plasmonic surface.50 Briefly, this method comprises the deposition of a thin polymer layer, with the analyte embedded within the polymer matrix. The analyte is subsequently removed by soft methods generating a void that perfectly replicates the desired molecule. MIP benefits from high selectivity since it is based on the key and lock concept, where the key would be the analyte and the lock the imprinted polymer. This has been demonstrated to work for medium-large sized analytes such as propranolol51 or TNT.52 Still, the separation between the analyte and the plasmonic surface, imparted by the polymer matrix, results in modest enhancement factors.
Another major group of responsive materials includes those with a remarkable volume change as a function of the internal hydration. Here, the idea relies on the free diffusion of the solution containing the analytes through the swollen polymer–nanoparticle composite. Once the substrate is dehydrated, the material collapses trapping the analytes within the hot spots generated by the close proximity of the plasmonic nanoparticles. These materials are usually prepared as thin films or bulk materials. Composite thin films have been prepared using polymer brushes of α-hydroxy-ω-carboxy-terminated polystyrene and polyglycidyl methacrylate capped with nanoparticles.53 However, the most powerful alternative still makes use of the layer-by-layer (LbL) protocol.54 This method, based on the successive deposition of layers of oppositely charged components, leads to materials with a high nanoparticle![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶polymer ratio, providing easy-to-handle platforms. For example, LbL films have been prepared using a variety of polyelectrolytes, dendrimers and biopolymers.54 Notably, a novel type of LbL films, known as exponentially grown layer-by-layer (eLbL) films, has emerged as an efficient SERS platform because they can be densely loaded with nanoparticles. Additionally, their volume changes upon dehydration and hydrophobic/hydrophilic character can be tuned as a function of the composition, allowing for the detection of organic pollutants such as dioxins.55 Finally, these materials can also be produced in bulk form, with nanoparticles embedded within the polymer matrix by a variety of techniques including microfluidics,56,57 and suspension58 or bulk polymerization.59 Advantages of these materials are their extraordinary decrease of volume upon dehydration (usually more than one order of magnitude) and their micro/macroscopic size, which permits direct integration into sensing devices. Further, as the nanoparticles are trapped inside the polymer networks, the polymer behaves as a dialyzing membrane, allowing reusability upon application of a washing flow.39
∶polymer ratio, providing easy-to-handle platforms. For example, LbL films have been prepared using a variety of polyelectrolytes, dendrimers and biopolymers.54 Notably, a novel type of LbL films, known as exponentially grown layer-by-layer (eLbL) films, has emerged as an efficient SERS platform because they can be densely loaded with nanoparticles. Additionally, their volume changes upon dehydration and hydrophobic/hydrophilic character can be tuned as a function of the composition, allowing for the detection of organic pollutants such as dioxins.55 Finally, these materials can also be produced in bulk form, with nanoparticles embedded within the polymer matrix by a variety of techniques including microfluidics,56,57 and suspension58 or bulk polymerization.59 Advantages of these materials are their extraordinary decrease of volume upon dehydration (usually more than one order of magnitude) and their micro/macroscopic size, which permits direct integration into sensing devices. Further, as the nanoparticles are trapped inside the polymer networks, the polymer behaves as a dialyzing membrane, allowing reusability upon application of a washing flow.39
5. Detection through the changes induced by the analyte on a selective ligand
We have discussed so far methods that can increase the concentration of chemical species, including those with no chemical affinity, in close proximity to plasmonic surfaces, so that the analytes could be detected through their own characteristic SERS spectra (vibrational fingerprint). In this final section, we describe methods where the analyte is detected taking advantage of geometrical and/or electronic changes that occur in a different molecule, previously attached to the surface of the plasmonic nanostructure. These indirect methods have an essential contrary requirement to those based on trapping the analyte, i.e. the capping agent must have a high SERS cross-section (opposite to cyclodextrins, polycarbohydrates or aliphatic polymers) so that changes are easy to detect. These methods are particularly suitable for the detection of analytes with a low SERS cross-section such as atomic cations, including protons and metals, inorganic oxoanions or organic aliphatic molecules, as well as the direct detection of other species, regardless of their SERS cross-section, that are usually dispersed in very complex matrices such as biological fluids and natural or waste waters.Probably the first report of this kind was based on changes in the SERS spectrum of the carboxylic group of the 4-mercapto benzoic acid (MBA) upon ionization/deionization, as a function of the medium pH (Fig. 6A).10 This molecule was able to recognise minute changes in pH in the range between 2–9, becoming the first “all optical” pH-meter. These sensors are suited for remote pH determination and thus MBA60,61 has been used to measure pH at different regions inside living cells (Fig. 6B).60,62 The importance of these reports not only relies on the pH sensing itself, but they have fuelled research toward novel optical and remote methods for the determination and quantification of other atomic species, which was previously restricted to atomic spectroscopy or chromatography. For example Zn(II) has been analysed9 using 4-(N-piperazinyl)terpyridine-dithiocarbamate as a capture agent, whereas the simultaneous detection of Hg(II) and Pb(II) was possible by using MBA in a chromatographic column.63 Contrary to atomic cations, oxoanions are molecular species and thus can be analysed by direct SERS,64 but their SERS cross-sections are still low, resulting in very modest detection limits. The use of selective ligands to dramatically increase the SERS signal of chromate was demonstrated by using nanostructured surfaces capped with 4-(2-mercaptoethyl)pyridinium.65 Although a modest activity has been identified in this field so far, we anticipate an increase in the interest of the SERS community, specially taking advantage of the fully developed colorimetric and fluorescence assays.66
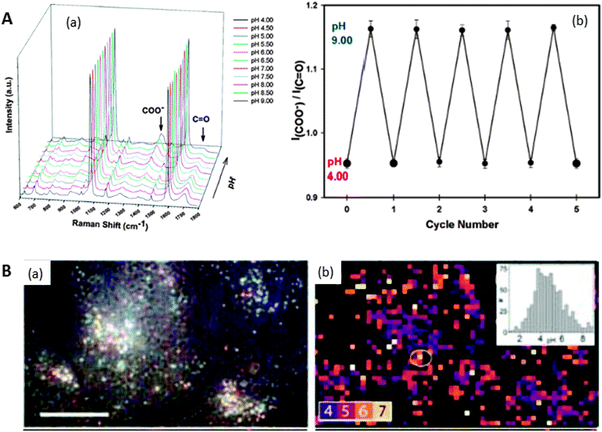 | ||
| Fig. 6 (A) SERS spectra of MBA on nanoshells at various pH values ranging from pH 4.0 to pH 9.0 in steps of 0.50 pH units (a). Repetitive cycling of the Raman pH nanosensor measured by monitoring the change in the I(COO−)/I(CO) (1702 cm−1/1393 cm−1) Stokes modes (b). Error bars represent the standard deviation for the measurements. (B) Reflected white-light image of encapsulated Ag clusters within HeLa cells. Scale bar 20 μm (a). SERS map coloured for pH value determination from ratios of pH-sensitive bands. Regions of low SERS are masked black (b). Inset is the histogram of pH values. Adapted with permission from ref. 10 and 62. Copyright 2010 the American Chemical Society and 2010 Wiley-VCH. | ||
Selective ligands can also be used for the detection of organic molecules and this is also of extreme interest. While common organic ligands such as boronic acids have been used for the detection of organic species such as sugars (glucose),49 the most active current direction is the use of biosensors. A biosensor can be defined as a sensor where the sensing element is of biological nature. These platforms are thus not restricted to biological samples but can be utilized in all analytical problems. For sure, the use of biological interfaces results expensive as compared with conventional ligands but their use is justified by advantages such as the extraordinary chemical selectivity and often their biocompatibility. Regarding the chemical selectivity, for comparison, boronic acid reacts with any kind of sugar present in the sample, but an antibody designed against glucose will only recognise this particular chemical species. The design of optical biosensors, however, is not trivial. Antibodies (Ab) and nucleic acids require very well defined conditions of medium composition (i.e. pH and ionic strength) and sometimes even temperature, to ensure optimal operation. Therefore, conventional colloidal suspensions cannot be used as the SERS platform (small particle size and ionic strength of the solution compromise colloidal stability). The fabrication of optical biosensors is thus mainly carried out on nanostructured surfaces or hybrid colloids comprising a micron sized support where the particles are retained.
The use of antibodies as the sensing element is recent and is based on the structural differences induced by the antigen upon complexation with the antibody (Fig. 7). Notably, this method can be successfully applied to the analysis of minute amounts of the target molecule in very complex solutions with no need for previous separation or purification protocols. For example, it has been used for the determination of exogenous proteins in milk,11 but also for the analysis of small metabolites such as benzoylecgonine, a biomarker of cocaine drug abuse.12 The use of DNA/RNA fragments as the sensitive layer is also extended.
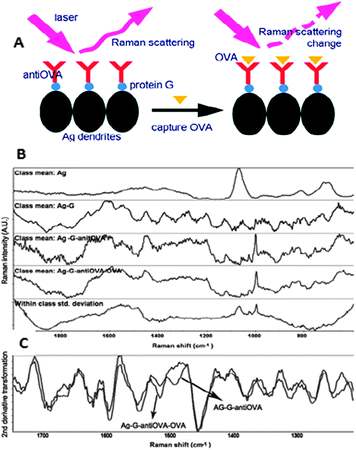 | ||
| Fig. 7 (A) Schematic illustration of the SERS detection of ovalbumin (OVA) in milk based on the antibody modified silver dendrites complex. (B) Raw SERS spectra of Ag, Ag–G, Ag–G–antiOVA, and Ag–G–antiOVA–OVA and (C) the second derivative transformation of spectra of Ag–G–antiOVA, and Ag–G–antiOVA–OVA. Ag, silver; G, protein G; OVA, ovalbumin. Adapted with permission from ref. 11. Copyright 2011 the American Chemical Society. | ||
Rather than the selectivity due to the complexation sites characteristic of antibodies, purine and pyrimidine bases that compose the nucleic acids specifically recognise their corresponding pairs. Thus, a given DNA or RNA sequence will recognise its specific corresponding fragment. Such ultrasensitive tests have extremely important implications for forensic science,67 but can also be applied to the recognition of proteins through specific interactions.68 Notably, in the last two years the use of aptamers is gaining relevance.13Aptamers are peptide or oligonucleotide molecules that bind to a specific target molecule by forming unique secondary and tertiary structures. They are able to detect very small structural changes in their target molecule, such as the presence or absence of certain functional groups. Aptamers can be generated for any protein targets through a systematic evolution of ligands by an exponential enrichment procedure which does not depend on an in vivo system. Aptamers can also be denatured and then renatured for several rounds, which confers them reusability, contrary to what happens with proteins. Further, their small size and smaller requirement for chemical stability allow them to be used with conventional colloids while maximizing the changes induced by complexation of the target molecule.69 As in the case of antibodies, although extensive application is being developed for protein detection,70 they have already been applied to the detection of small molecules71 and even atomic cations.72
6. Conclusions and outlook
Ultrasensitive detection based on SERS has been traditionally restricted to molecules carrying functional groups with high affinity toward gold or silver surfaces. Therefore, the general application of SERS as a highly sensitive detection technique requires the development of materials that are able to capture the analyte in a selective and efficient manner and drag it toward the plasmonic metal surface. This is a topic of current expansion but a number of composite materials are available that can efficiently fulfil this task. Exploiting general forces, such as those resulting from electrostatic or hydrophobic interactions, has allowed the detection of important contaminants such as PAHs, whereas molecular cages such as cyclodextrins make possible the specific detection of certain ions. Analytes can also be trapped within polymeric networks, which constitute real smart materials, even comprising dynamically forming hot spots. Methods have even been developed to analyse the presence of substances with negligible SERS cross-sections, such as pH or to carry out highly selective detection, by monitoring changes in (bio)receptors that are anchored to metallic nanostructures.Therefore, we can undoubtedly state that the recent advances in the chemical design of nanostructured materials have opened the way toward generalized SERS detection, which will ultimately reach the marketplace and compete or even replace current detection methods.
Acknowledgements
R.A.A.-P. acknowledges the RyC Program (Ministerio de Ciencia e Innovación, Spain). This work was funded by the Spanish Ministerio de Ciencia e Innovacion (MAT2010-15374 and MAT2008-05755) and the Xunta de Galicia (08TMT008314PR and 09TMT011314PR). L.M.L.-M. acknowledges funding from the ERC (PLASMAQUO, Advanced Grant 267867).Notes and references
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. R. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS.
- J. Kneipp, H. Kneipp and K. Kneipp, Chem. Soc. Rev., 2008, 37, 1052–1060 RSC.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- R. A. Alvarez-Puebla, A. Agarwal, P. Manna, B. P. Khanal, P. Aldeanueva-Potel, E. Carbo-Argibay, N. Pazos-Perez, L. Vigderman, E. R. Zubarev, N. A. Kotov and L. M. Liz-Marzan, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8157–8161 CrossRef CAS.
- C. L. Jones, K. C. Bantz and C. L. Haynes, Anal. Bioanal. Chem., 2009, 39, 303–311 CrossRef.
- C. R. Yonzon, C. L. Haynes, X. Zhang, J. T. Walsh and R. P. Van Duyne, Anal. Chem., 2004, 76, 78–85 CrossRef CAS.
- R. A. Alvarez-Puebla, R. Contreras-Caceres, I. Pastoriza-Santos, J. Perez-Juste and L. M. Liz-Marzan, Angew. Chem., Int. Ed., 2009, 48, 138–143 CrossRef CAS.
- H. Gehan, C. Mangeney, J. Aubard, G. Lévi, A. Hohenau, J. R. Krenn, E. Lacaze and N. Félidj, J. Phys. Chem. Lett., 2011, 2, 926–931 CrossRef CAS.
- Y. Zhao, J. N. Newton, J. Liu and A. Wei, Langmuir, 2009, 25, 13833–13839 CrossRef CAS.
- S. W. Bishnoi, C. J. Rozell, C. S. Levin, M. K. Gheith, B. R. Johnson, D. H. Johnson and N. J. Halas, Nano Lett., 2006, 6, 1687–1692 CrossRef CAS.
- L. L. He, T. Rodda, C. L. Haynes, T. Deschaines, T. Strother, F. Diez-Gonzalez and T. P. Labuza, Anal. Chem., 2011, 83, 1510–1513 CrossRef CAS.
- M. Sanles-Sobrido, L. Rodríguez-Lorenzo, S. Lorenzo-Abalde, A. González-Fernández, M. A. Correa-Duarte, R. A. Álvarez-Puebla and L. M. Liz-Marzán, Nanoscale, 2009, 1, 153–158 RSC.
- G. Q. Wang, Z. P. Chen and L. X. Chen, Prog. Chem., 2010, 22, 489–499 CAS.
- G. J. Kovacs, R. O. Loutfy, P. S. Vincett, C. Jennings and R. Aroca, Langmuir, 1986, 2, 689–694 CrossRef CAS.
- Y. Fang, N.-H. Seong and D. D. Dlott, Science, 2008, 321, 388–392 CrossRef CAS.
- J. P. Camden, J. A. Dieringer, Y. Wang, D. J. Masiello, L. D. Marks, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2008, 130, 12616–12617 CrossRef CAS.
- S. L. Kleinman, E. Ringe, N. Valley, K. L. Wustholz, E. Phillips, K. A. Scheidt, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2011, 133, 4115–4122 CrossRef CAS.
- F. J. Garcia de Abajo, J. Phys. Chem. C, 2008, 112, 17983–17987 CAS.
- F. J. Garcia de Abajo, Rev. Mod. Phys., 2010, 82, 209 CrossRef CAS.
- S. E. J. Bell and N. M. S. Sirimuthu, J. Phys. Chem. A, 2005, 109, 7405–7410 CrossRef CAS.
- M. Erol, Y. Han, S. K. Stanley, C. M. Stafford, H. Du and S. Sukhishvili, J. Am. Chem. Soc., 2009, 131, 7480–7481 CrossRef CAS.
- C. H. Munro, W. E. Smith, M. Garner, J. Clarkson and P. C. White, Langmuir, 1995, 11, 3712–3720 CrossRef CAS.
- L. Rodriguez-Lorenzo, R. A. Alvarez-Puebla, F. J. G. de Abajo and L. M. Liz-Marzan, J. Phys. Chem. C, 2010, 114, 7336–7340 CAS.
- R. A. Alvarez-Puebla, E. Arceo, P. J. G. Goulet, J. J. Garrido and R. F. Aroca, J. Phys. Chem. B, 2005, 109, 3787–3792 CrossRef CAS.
- R. A. Alvarez-Puebla and R. F. Aroca, Anal. Chem., 2009, 81, 2280–2285 CrossRef CAS.
- J. P. Camden, J. A. Dieringer, J. Zhao and R. P. Van Duyne, Acc. Chem. Res., 2008, 41, 1653–1661 CrossRef CAS.
- K. C. Bantz and C. L. Haynes, Vib. Spectrosc., 2009, 50, 29–35 CrossRef CAS.
- A. J. Haes, L. Chang, W. L. Klein and R. P. Van Duyne, J. Am. Chem. Soc., 2005, 127, 2264–2271 CrossRef CAS.
- M. Moskovits and J. S. Suh, J. Phys. Chem., 1984, 88, 5526–5530 CrossRef CAS.
- S. Abalde-Cela, J. M. Hermida-Ramón, P. Contreras-Carballada, L. De Cola, A. Guerrero-Martínez, R. A. Alvarez-Puebla and L. M. Liz-Marzán, ChemPhysChem, 2011, 12, 1529–1535 CrossRef CAS.
- J. Y. Xu, J. Wang, L. T. Kong, G. C. Zheng, Z. Guo and J. H. Liu, J. Raman Spectrosc., 2011 DOI:10.1002/jrs.2932.
- Y. Xie, X. Wang, X. Han, X. Xue, W. Ji, Z. Qi, J. Liu, B. Zhao and Y. Ozaki, Analyst, 2010, 135, 1389–1394 RSC.
- A. D. Strickland and C. A. Batt, Anal. Chem., 2009, 81, 2895–2903 CrossRef CAS.
- J. Wang, L. Kong, Z. Guo, J. Xu and J. Liu, J. Mater. Chem., 2010, 20, 5271–5279 RSC.
- E. Del Puerto, S. Sánchez-Cortés, J. V. García-Ramos and C. Domingo, Chem. Commun., 2011, 47, 1854–1856 RSC.
- L. Guerrini, J. V. Garcia-Ramos, C. Domingo and S. Sanchez-Cortes, Phys. Chem. Chem. Phys., 2009, 11, 1787–1793 RSC.
- I. López-Tocón, J. C. Otero, J. F. Arenas, J. V. Garcia-Ramos and S. Sanchez-Cortes, Anal. Chem., 2011, 83, 2518–2525 CrossRef.
- R. A. Alvarez-Puebla, D. S. Dos Santos Jr and R. F. Aroca, Analyst, 2007, 132, 1210–1214 RSC.
- P. Aldeanueva-Potel, E. Faoucher, R. A. Alvarez-Puebla, L. M. Liz-Marzan and M. Brust, Anal. Chem., 2009, 81, 9233–9238 CrossRef CAS.
- P.-G. Yin, Y. Chen, L. Jiang, T.-T. You, X.-Y. Lu, L. Guo and S. Yang, Macromol. Rapid Commun., 2011, 13, 1000–1006 CrossRef.
- C. Jiang, Y. Qian, Q. Gao, J. Dong and W. Qian, J. Mater. Chem., 2010, 20, 8711–8716 RSC.
- R. Contreras-Cáceres, S. Abalde-Cela, P. Guardia-Girós, A. Fernández-Barbero, J. Pérez-Juste, R. A. Alvarez-Puebla and L. M. Liz-Marzán, Langmuir, 2011, 27, 4520–4525 CrossRef.
- H. Gehan, L. Fillaud, M. M. Chehimi, J. Aubard, A. Hohenau, N. Felidj and C. Mangeney, ACS Nano, 2010, 4, 6491–6500 CrossRef CAS.
- R. Alvarez-Puebla, L. M. Liz-Marzán and F. J. García De Abajo, J. Phys. Chem. Lett., 2010, 1, 2428–2434 CrossRef CAS.
- M. K. Gupta, S. Chang, S. Singamaneni, L. F. Drummy, R. Gunawidjaja, R. R. Naik and V. V. Tsukruk, Small, 2011, 7, 1192–1198 CrossRef CAS.
- V. Kozlovskaya, E. Kharlampieva, B. P. Khanal, P. Manna, E. R. Zubarev and V. V. Tsukruk, Chem. Mater., 2008, 20, 7474–7485 CrossRef CAS.
- X. Qian, J. Li and S. Nie, J. Am. Chem. Soc., 2009, 131, 7540–7541 CrossRef CAS.
- D.-Y. Wang, T.-S. Teng, Y.-C. Wu, Y.-C. Lee, K.-H. Chen, C.-H. Chen, Y.-C. Chang and C.-C. Chen, J. Phys. Chem. C, 2009, 113, 13498–13504 CAS.
- W. Wu, N. Mitra, E. C. Y. Yan and S. Zhou, ACS Nano, 2010, 4, 4831–4839 CrossRef CAS.
- M. Bompart, Y. De Wilde and K. Haupt, Adv. Mater., 2010, 22, 2343–2348 CrossRef CAS.
- K. Kantarovich, I. Tsarfati, L. A. Gheber, K. Haupt and I. Bar, Anal. Chem., 2009, 81, 5686–5690 CrossRef CAS.
- E. L. Holthoff, D. N. Stratis-Cullum and M. E. Hankus, Sensors, 2011, 11, 2700–2714 CrossRef CAS.
- S. Gupta, M. Agrawal, P. Uhlmann, F. Simon, U. Oertel and M. Stamm, Macromolecules, 2008, 41, 8152–8158 CrossRef CAS.
- C. Jiang and V. V. Tsukruk, Adv. Mater., 2006, 18, 829–840 CrossRef CAS.
- S. Abalde-Cela, S. Ho, B. Rodriguez-Gonzalez, M. A. Correa-Duarte, R. A. Alvarez-Puebla, L. M. Liz-Marzan and N. A. Kotov, Angew. Chem., Int. Ed., 2009, 48, 5326–5329 CrossRef CAS.
- S. Abalde-Cela, B. Auguié, M. Fischlechner, W. T. S. Huck, R. A. Alvarez-Puebla, L. M. Liz-Marzán and C. Abell, Soft Matter, 2011, 7, 1321–1325 RSC.
- R. K. Shah, J.-W. Kim, J. J. Agresti, D. A. Weitz and L.-Y. Chu, Soft Matter, 2008, 4, 2303–2309 RSC.
- J. Zhang, S. Xu and E. Kumacheva, J. Am. Chem. Soc., 2004, 126, 7908–7914 CrossRef CAS.
- E. Faoucher, P. Nativo, K. Black, J. B. Claridge, M. Gass, S. Romani, A. L. Bleloch and M. Brust, Chem. Commun., 2009, 6661–6663 RSC.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, J. Phys. Chem. C, 2010, 114, 7421–7426 CAS.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nano Lett., 2007, 7, 2819–2823 CrossRef CAS.
- A. Pallaoro, G. B. Braun, N. O. Reich and M. Moskovits, Small, 2010, 6, 618–622 CrossRef CAS.
- S. J. Lee and M. Moskovits, Nano Lett., 2011, 11, 145–150 CrossRef CAS.
- L. Rintoul, K. Crawford, H. F. Shurvell and P. M. Fredericks, Vib. Spectrosc., 1997, 15, 171–177 CrossRef CAS.
- P. A. Mosier-Boss and S. H. Lieberman, Langmuir, 2003, 19, 6826–6836 CrossRef CAS.
- D. Liu, Z. Wang and X. Jiang, Nanoscale, 2011, 3, 1421–1433 RSC.
- D. Graham, B. J. Mallinder, D. Whitcombe, N. D. Watson and W. E. Smith, Anal. Chem., 2002, 74, 1069–1074 CrossRef CAS.
- A. J. Bonham, G. Braun, I. Pavel, M. Moskovits and N. O. Reich, J. Am. Chem. Soc., 2007, 129, 14572–14573 CrossRef CAS.
- O. Neumann, D. M. Zhang, F. Tam, S. Lal, P. Wittung-Stafshede and N. J. Halas, Anal. Chem., 2009, 81, 10002–10006 CrossRef CAS.
- L. Fabris, M. Dante, T. Q. Nguyen, J. B. H. Tok and G. C. Bazan, Adv. Funct. Mater., 2008, 18, 2518–2525 CrossRef CAS.
- J. W. Chen, J. H. Jiang, X. Gao, G. K. Liu, G. L. Shen and R. Q. Yu, Chem.–Eur. J., 2008, 14, 8374–8382 CrossRef CAS.
- Y. L. Wang and J. Irudayaraj, Chem. Commun., 2011, 47, 4394–4396 RSC.
| This journal is © The Royal Society of Chemistry 2012 |