“Clicking” on/with polymers: a rapidly expanding field for the straightforward preparation of novel macromolecular architectures
Kristian
Kempe†
ab,
Andreas
Krieg†
ab,
C. Remzi
Becer
*ac and
Ulrich S.
Schubert
*abc
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich-Schiller-University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de; C.R.Becer@warwick.ac.uk; Fax: +49 3641 948 202
bJena Center for Soft Matter (JCSM), Friedrich-Schiller-University Jena, Humboldtstr. 10, 07743 Jena, Germany
cDutch Polymer Institute (DPI), John F. Kennedylaan 2, 5612 AB Eindhoven, The Netherlands
First published on 1st July 2011
Abstract
The combination of controlled polymerization techniques and “click” reactions form an efficient platform for the preparation of polymers in various architectures. In this critical review, an update of our 2007 review in Chem. Soc. Rev., we focus on the “click” reactions that have been used widely in the last four years to create new polymer architectures. Not only block copolymers and star-shaped polymers but also cyclic and dendritic macromolecules could be synthesized using these robust “click” reactions (205 references).
 Kristian Kempe | Kristian Kempe was born in 1983 in Borna (Germany) and studied chemistry at the Friedrich-Schiller-University Jena (Germany) and the Eindhoven University of Technology (The Netherlands). In 2007 he obtained his MSc degree in Chemistry. Since 2007 he is performing his PhD studies in Jena under the supervision of Prof. Ulrich S. Schubert. In 2010 he spent five months at the University of California, Santa Barbara (UCSB) where he worked under the supervision of Prof. Craig J. Hawker. His research focuses on the combination of the microwave-assisted preparation of novel (co-)poly(2-oxazoline)s and their post-modification using efficient (“click”) reactions. |
 Andreas Krieg | Andreas Krieg was born in 1981 in Eisenach, Germany. He studied chemistry at the Friedrich-Schiller-University Jena, Germany and received his MSc in Chemistry in 2008. Since 2008 he is a PhD student in the group of Prof. Ulrich S. Schubert in Jena focussing amongst others on the controlled radical polymerization of well-defined homo and copolymers. |
 C. Remzi Becer | C. Remzi Becer was born in 1980 in Izmir, Turkey. He received his BSc degree in 2003 at the Chemistry Department of the Istanbul Technical University (ITU). In 2005, he received his MSc degree in Polymer Science and Technology at the ITU. He completed his PhD study titled as “Controlling Polymer Architectures” in 2009 under the supervision of Prof. Ulrich S. Schubert at the Eindhoven University of Technology (Netherlands) and the Friedrich-Schiller-University Jena (Germany). Since late 2009, he has been a Marie Curie Research Fellow at the University of Warwick (United Kingdom). Recently, he received a Science City Independent Research Fellowship to conduct research in the area of magnetic nanomedicine at the University of Warwick. |
 Ulrich S. Schubert | Ulrich S. Schubert was born in Tübingen (Germany) in 1969. He studied chemistry in Frankfurt and Bayreuth (both Germany) and the Virginia Commonwealth University, Richmond (USA). His PhD studies were performed at the Universities of Bayreuth and South Florida/Tampa. After postdoctoral training with J.-M. Lehn at the University in Strasbourg (France), he moved to the TU München (Germany) and obtained his Habilitation in 1999. 1999–2000 he was professor at the Center for NanoScience, University of Munich (Germany), and 2000–2007 Full-Professor at the Eindhoven University of Technology. Currently he holds a chair at the Friedrich-Schiller-University Jena (Germany). His awards include the Bayerischen Habilitations-Förderpreis, the Habilitandenpreis of the GDCh (Makromolekulare Chemie), the Heisenberg-Stipendium of the DFG, the Dozenten-Stipendium of the Fonds der Chemischen Industrie the VICI award of the NWO, the Piet-Lemstra innovation award of the DPI and the BPG biannual international award. |
1. Introduction
Scientists and engineers have always been interested in constructing difficult or even impossible structures. Pyramids as one of the oldest and the most stable structures on earth were built more than 5000 years ago and they can still survive desert conditions owing to their wide base and solid structure formed of thousands of cubic blocks. Material scientists are always in need of combining various simple pieces to form complex and highly functional systems. The journey of polymer chemists started a century ago in 1907 with the invention of the first synthetic polymer by Leo Baekeland.1 It took five decades to gain a good control over the chain growth, which was achieved by the living anionic polymerization invented by Szwarc in 1956.2,3 In the following four decades radical polymerization advanced very rapidly and several controlled or “living” radical polymerization techniques were developed.These techniques are based on the activation and deactivation of a reactive chain end via either chemical or physical stimuli. For instance, atom transfer radical polymerization (ATRP),4–6 single electron transfer living radical polymerization,7–10 and chain transfer polymerization techniques11–14 rely on chemical stimuli whereas the nitroxide mediated polymerization (NMP)15 requires relatively higher temperatures for activation/deactivation. A clever selection of the catalytic system or of the reaction conditions allows the preparation of polymers with well-defined chain length and end group functionalities.14,16,17 Thousands of scientific contributions have been published on these techniques using various (functional) monomers, initiators, catalysts, and solvents to demonstrate the power of controlled/“living” polymerization techniques (CLP). Furthermore, the exploitation of multifunctional initiators, monomers and inimers enabled the synthesis of various architectures with CLP techniques.18–24
Moreover, another giant step was taken a decade ago when chemists were reminded on the opportunities to use highly efficient organic reactions to modify complex polymer chains.25–29 Sharpless and coworkers introduced the “click chemistry” concept that defines any reaction which can be applied for a broad range of reactions, gives very high yields, is stereospecific, generates only inoffensive byproducts that can be removed by non-chromatographic methods, uses readily available starting materials and reagents, preferably using no solvent, and allows for an easy isolation of the product.30 Moreover, very recently a group of leading scientists in the polymer chemistry field discussed the advantages and disadvantages of utilizing “click” reactions in polymer science. They have defined that the list of “click” reactions criteria for polymer chemistry should be slightly modified and equimolarity, large scale purification, fast timescale, high yields, stable compounds, modular, wide in scope, chemoselective and single reaction trajectory requirements should also be taken into account carefully.31
Initially, the majority of researchers focused on the use of the copper catalyzed azide–alkyne cycloaddition (CuAAC) reaction.32–34 Indeed, polymers with azide end groups could be easily prepared by transforming the bromine end groups to azide obtained by using ATRP procedures.35 Not only small compounds such as dyes or carbohydrates were “clicked” to the chain end of these macromolecules but also larger systems such as synthetic polymers or proteins were conjugated using CuAAC “click” reactions.36–38 As a consequence, a variety of polymer architectures were accessible as critically reviewed by Schubert and coworkers in 2007.39 However, the necessity of the copper catalyst and its sensitivity to air limits its use under physiological conditions and in living organisms.40 Therefore, several other organic reactions were re-examined for their successful application in polymer chemistry.41,42 Most of these reactions were conducted metal-free and showed great success under certain conditions.26,43,44
In particular, thiol–ene (TE) reactions (Fig. 1) attracted attention due to the wide range of commercially available thiols and alkenes.45 Additionally, TE reactions can proceed via two routes, anti-Markovnikov radical addition or base catalyzed Michael-addition (MA).46–49 Hawker and coworkers demonstrated the use of TE “click” reactions for the formation of monodisperse dendrimers.50 Schlaad and coworkers successfully utilized this chemistry for the functionalization of 1,2-polybutadiene side groups,51 as well as poly(2-oxazoline)s bearing pendant alkene moieties.52 Other thiol based “click” reactions have been developed and can be listed as thiol–maleimide addition, thiol–isocyanate addition, pyridyl–disulfide exchange, and thiol–para fluoro.53–58 Thiol terminated polymers can be easily obtained by reversible addition–fragmentation chain transfer (RAFT) polymerization of a wide range of monomers and a subsequent cleavage of the chain transfer agent.28 Alternatively, disulfide containing bifunctional ATRP initiators can be used for the preparation of thiol functionalized polymers.59,60
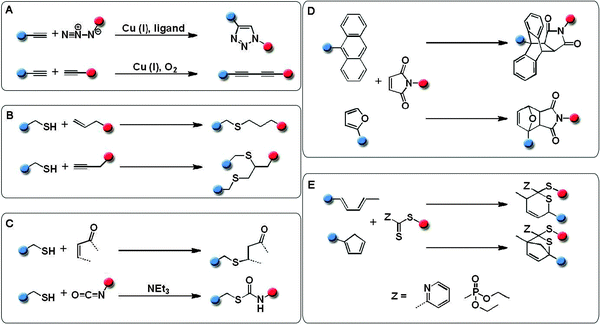 | ||
| Fig. 1 Schematic representation of the “click” reactions exploited for the preparation of macromolecular architectures. | ||
Diels–Alder (DA) cycloaddition reactions were first reported in 1928 and they are amongst the most fascinating organic reactions that involve the formation of carbon–carbon bonds. In combination with the hetero Diels–Alder (HDA) approach these reactions cover a wide range of possible starting materials and reaction conditions, e.g. enabling reactions to be performed at high or even below room temperature depending on the choice of the system.61–70 Furthermore, DA reactions feature a reversible character (retro-DA), which can be exploited for the preparation of, e.g., self-healing materials.71 Similarly, strain promoted azide–alkyne cycloaddition (SPAAC) reactions can be performed at low temperatures and in the absence of a copper catalyst. The driving force in these reactions is the ring strain of the cyclooctyne. The robustness as well as orthogonality of this reaction was demonstrated by labeling living cells and even animals.40,41,72,73
In this review, we discuss efficient reactions covered under the term “click chemistry” as a tool for the preparation of block copolymers, cyclic polymers, side chain functionalization of the polymers, dendritic and hyperbranched polymers, star-shaped polymers, cross-linked polymers and their use in particle stabilization and surface modification (Fig. 2). Special attention is drawn to lately recognized “click”-type reactions, such as TE and DA reactions. Representative examples are given to demonstrate the success of these “click” reactions in combination with controlled living polymerizations to prepare different polymer architectures.
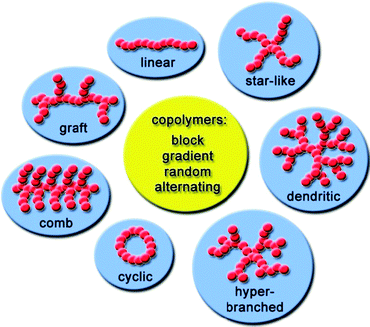 | ||
| Fig. 2 Overview of the discussed architectures. | ||
2. “Click chemistry” in polymer science
2.1 Linear (co-)polymers
“Click” reactions has enabled access to a multitude of complex and sophisticated polymeric architectures since this straightforward concept has been transferred from organic chemistry to polymer science.69,74,75 The great success of this approach is owed to the availability of different living/controlled polymerization techniques which allow the introduction and transformation of specific end groups. Thus, over the last years several nearly perfect combinations of polymerization techniques and “clickable” functionalities evolved providing high degrees of α- and/or ω-functionalization of the polymer backbone ideally suited for efficient and controlled coupling reactions. The halogen end groups resulting from the ATRP mechanism are typically converted into azide functionalities using sodium azide, directly applicable to the CuAAC.76 The necessary alkyne compounds are usually obtained via usage of functional initiators bearing protected or unprotected terminal alkynes.77,78 The reaction conditions of the ATRP polymerization and the CuAAC reaction are remarkable similar in terms of the used reagents, like catalysts and ligands, enabling the successful development of one pot approaches.79,80 All these advantages made the combination of ATRP and CuAAC a highly versatile synthetic strategy to obtain complex polymeric architectures. The use of functionalized initiators is of course also applicable to other polymerization techniques as well, such as RAFT polymerization,81 NMP,82 or cationic ring-opening polymerization (CROP).83,84For the goal of combining as many macromolecular building blocks with as few as possible involved reaction steps, orthogonal coupling mechanisms are required. A technique which goes along with the conditions of the CuAAC without losing selectivity is the TE coupling reaction which can be counted towards the “click-like” reactions due to its selectivity and efficiency.85 Since RAFT polymers naturally bear sulfur containing end groups, the cleavage of the chain transfer agent (CTA) via aminolysis became a widespread method to generate the thiol compound necessary for TE and thiol–yne (TY) coupling reactions.86,87 An issue correlated with the cleavage of RAFT CTAs is the possible S–S coupling of two polymer chains. This side reaction can be suppressed employing hydrazine hydrate as cleaving agent as presented by Shen et al.86 Furthermore, Spruell et al. used functionalized acrylates acting as Michael acceptors to transform the CTA end group of RAFT polymers after reduction with NaBH4.88 Sumerlin and coworkers demonstrated the preparation of maleimido-terminated poly(N-isopropylacrylamide) (PNIPAm) via reaction of 1,8-bismaleimidodiethyleneglycol with the thiol functionality obtained after aminolysis of the trithiocarbonate CTA.57 The obtained polymers were exploited for the performance of MA as well as DA coupling reactions. Near quantitative conversions were achieved using small molecules, e.g. 4-methoxybenzyl mercaptan, 1-dodecanethiol and 9-anthracenemethanol. Furthermore, a thiol-terminated polystyrene (PS) was coupled to the maleimido-terminated PNIPAM yielding well-defined PNIPAM-b-PS (Scheme 1).
![Schematic representation of the RAFT end group modification with bismaleimides towards the use in MA and DA reactions [adopted from ref. 57].](/image/article/2012/CS/c1cs15107j/c1cs15107j-s1.gif) | ||
| Scheme 1 Schematic representation of the RAFT end group modification with bismaleimides towards the use in MA and DA reactions [adopted from ref. 57]. | ||
In particular, the latter can be added towards the “click-like” reactions which are increasingly used in the construction of polymeric architectures due to its orthogonality to the other mentioned coupling techniques. The incorporation of dienes and dienophiles can be performed straightforward using the correspondingly functionalized initiators.82,89 Rivaling the established “click” procedures in terms of speed and efficiency is an elegant approach which was presented by Barner-Kowollik and coworkers who reported a new “click” concept involving a hetero DA mechanism with the sulfur–carbon double bond of RAFT polymers serving as dienophile.90 The necessary diene compounds were reported to be easily accessible by quantitative functionalization of polymers bearing electrophilic end groups (e.g. polymers bearing a terminal bromide moiety obtained by ATRP) with sodium cyclopentadienide.
Another technique to couple polymers in a rather efficient way and thereby deserving the title efficient (“click”) reaction is the Glaser-type C–C bond formation via reaction of two alkynes. While being a possible side reaction during CuAAC it can be turned into an efficient and quantitative coupling reaction depending on the right conditions as shown by Duxbury et al. (Scheme 2).91
![Schematic representation of the Glaser coupling of alkyne-terminated PS and the subsequent symmetrical chain extension reaction [adopted from ref. 91].](/image/article/2012/CS/c1cs15107j/c1cs15107j-s2.gif) | ||
| Scheme 2 Schematic representation of the Glaser coupling of alkyne-terminated PS and the subsequent symmetrical chain extension reaction [adopted from ref. 91]. | ||
Although seemingly the most simple structure of the advanced macromolecular architectures, linear block copolymers are of permanent high interest in polymer research due to their potential to form phase-separated structures in bulk as well as in solution.92,93 With classical polymer chemistry methods the preparation of well-defined block copolymer is still a demanding task, whereas “click” chemistry offers a straightforward access to generate the desired macromolecules.26,39 The orthogonality of certain “click” mechanisms enables the controlled one-pot combination of multiple macromolecules of otherwise nearly incompatible types to prepare the corresponding block copolymers.94–96 An example for the simultaneous usage of orthogonal “click” reactions is the preparation of a H-shaped quintopolymer in a one-pot approach reported by Tunca and coworkers.94 Using an anthracene functionalized ATRP initiator for the polymerization of poly(t-butyl acrylate) (PtBA) followed by the conversion of the terminal bromine into an azide led to the telechelic homopolymer. Two block copolymers with corresponding alkyne and dienophile groups at the center were prepared by using an alkyne functionalized bifunctional initiator able to polymerize PS via NMP as well as polycaprolactone (PCL) via ring-opening polymerization (ROP) for the first block copolymer. A maleimide functionalized polyethylene glycol (PEG)-macroinitiator for the ATRP of methyl methacrylate (MMA) resulted in the second block copolymer. In this respect, another reaction emerged to be of interest for the coupling of polymer chains in combination with other “click” reactions. The atom transfer nitroxide radical coupling (ATNRC), which covers reactions of halide (obtained by ATRP) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) end-functionalized polymers was applied for the one-pot synthesis of linear triblock terpolymers,97 as well as of tetrablock quarterpolymers.98
2.2 Cyclic polymers
Since Laurent and Scott demonstrated the feasibility of CuAAC for the synthesis of cyclic PS polymers a variety of new cyclic polymers using this “click” approach were developed.99 Besides other cyclic homopolymers,100–104 block copolymers,105 as well as crown ether based macrocycles,106 more sophisticated cyclic polymer structures, such as tadpole-shaped copolymers,107–110 were prepared using similar conditions. A very recent example for the preparation of cyclic polymers was demonstrated by Lutz and coworkers where the authors used a combination of ATRP and CuAAC reactions for the synthesis.111 The idea behind the preparation of cyclic polymers was to mimic the folding process of sequence-defined biopolymers. To achieve this purpose, the authors conducted ATRP of styrene; at a certain time protected maleimide monomers were injected to the reaction medium. Due to the higher reactivity of maleimide, it is rapidly consumed and styrene kept polymerizing. Thus, functional monomers could be inserted at any part of the polymer chain by simply following the chain growth of polystyrene. This approach allows the controlled preparation of various cyclic polymers encompassing tadpole (P-shaped), pseudocyclic (Q-shaped), bicyclic (8-shaped) and knotted (alpha-shaped) architectures. Thus, in the last years the CuAAC emerged to be the method of choice for this kind of structures. Nevertheless, various examples have highlighted the usage of alternatively efficient coupling methods. A very elegant approach was described by Stanford et al. who were able to cyclisize maleimide end functionalized poly(lactide)s under TE “click” conditions.112 The MA was performed by a slow addition of 1,2-ethanedithiol as co-reagent and the corresponding difunctional polylactic acid (PLA) to a solution of triethylamine in dichloromethane (Fig. 3). The high efficiency of these reactions was illustrated by MALDI-TOF MS and SEC.![Schematic representation of the synthesis of a stereoregular cyclic poly(lactide) using dithiolethane as cyclization agent in a MA approach (left) and SEC traces of (a) linear and (b) cyclic PLA (right) [adopted from ref. 112].](/image/article/2012/CS/c1cs15107j/c1cs15107j-f3.gif) | ||
| Fig. 3 Schematic representation of the synthesis of a stereoregular cyclic poly(lactide) using dithiolethane as cyclization agent in a MA approach (left) and SEC traces of (a) linear and (b) cyclic PLA (right) [adopted from ref. 112]. | ||
Furthermore, Anseth and coworkers reported the on-resin cyclization of polypeptides exploiting amino acid cysteine as thiol source in a TE approach.113,114 In order to incorporate alkene functionalities into the peptide sequence allyloxycarbonyl as well as norbonene functional groups were used in the solid phase peptide synthesis. The cyclic products were obtained in either case in higher yields relative to other on-resin reactions. Furthermore, TE reactions applying norbonene as alkene substituent were performed with accelerated reaction kinetics compared to the reaction with the allyl compound due to the higher reactivity of the strained bicyclic alkene. More recently DA reactions were applied for the synthesis of cyclic homo as well as block copolymers by Durmaz et al.115 The end functionalization of PS, PtBA, PS-b-PtBA and PS-b-PCL with groups known to undergo an efficient DA reaction at high temperatures (anthracene, maleimide) yielded telechelic precursors for cyclic polymers. As reported for the CuAAC reactions mentioned before highly diluted conditions were necessary in order to prevent intermolecular coupling reactions which could be considered as a prerequisite for all cyclization reactions performed in solution.
2.3 Side-chain functionalization
The design of polymer properties in a controlled and predictable way is one of the foremost goals in polymer chemistry. The polymerization mechanisms often limit the potential use of functional monomers making post-modifications inevitable. Hindering sterical effects and potential sensibilities of the polymer backbone demand mild and high efficient coupling techniques. The preparation of branched polymers via the graft to approach also requires highly efficient coupling reactions.To permit subsequent modification of polymers via CuAAC, in principle two different approaches are possible and have been explored. Either the azido or the acetylene compound has to be incorporated into the polymer by means of functional monomers.116–118 Hereby azido functionalized polymers are mostly obtained by an indirect approach utilizing alkyl halogenide bearing monomers as precursors119 due to the thermal lability of the azide group although the direct polymerization of azido functionalized monomers is also possible.120,121 The incorporation of the alkyne compound is easily possible using monomers bearing terminal acetylene functionalities, e.g. propargyl methacrylate.122 However, in particular the use of controlled radical polymerization (CRP) methods requires the use of protecting groups like TMS to prevent radical side reactions leading to ill defined polymers.123
In order to allow TE coupling reactions polymers have to contain either thiol functionalities, which can be introduced via protected monomers as reported by Bulmus and coworkers124 or alkene groups. The ease of incorporating pendant alkene functionalities and its application for TE modifications was demonstrated by Schlaad and coworkers (Scheme 3).51,52 1,2-Polybutadiene as well as poly(2-(3-butenyl)-2-oxazoline) bearing pendant alkene functionalities, obtained by the living CROP, were modified by the photo-addition of various thiols onto the double bonds using a mercury UV lamp and sunlight for the generation of thiyl radicals, respectively. Thus, this mild procedure allows a tuning of the polymer properties, e.g. of the hydrophilic/hydrophobic character and enable the access to biohybrid polymers when using bio-active thiols, e.g. N-Boc-L-cysteine, 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranose and thiocholesterol. Recently, this approach was extended to the functionalization of polypeptides.125 A similar approach was used by Kempe et al. for the synthesis of diverse glycopolymers based on (co)polymers consisting of 2-ethyl-2-oxazoline and 2-dec(-9-enyl)-2-oxazoline.126–128 While ionic polymerization mechanisms permit the unproblematic use of alkene functionalized monomers CRP-mechanisms again proof to be more demanding.129
 | ||
| Scheme 3 Schematic representation of the side-chain functionalization of alkene bearing polymers by TE reactions.51,52,125 | ||
However, Hawker and coworkers demonstrated the copolymerization of monomers bearing terminal double bonds based on styrene, MMA and caprolactone (CL) by RAFT and ROP without losing the controlled character of the polymerizations.130 The polymers obtained were applied for a detailed study of the efficiency and orthogonality of the thermal- and photo-initiated TE reaction. The latter was found to show a higher efficiency, proceed faster and with a higher tolerance to various backbones and functional groups. In contrast, the thermal TE reaction showed a complete orthogonality with the CuAAC.
Besides alkenes and alkyne functionalities thiol bearing molecules can also be coupled to pentafluorostyrene moieties substituting the para-positioned fluorine.131,132 Schubert and coworkers demonstrated the copolymerization of pentafluorostyrene (PFS) with styrene via NMP followed by subsequent coupling of sugar units as proven by SEC and MALDI-TOF MS (Scheme 4).55 Furthermore classical DA reactions as well as HDA are exploited to functionalize polymers introducing an additional orthogonal coupling possibility. Polymerization of monomers permitting subsequent functionalizations by DA reaction has been described, e.g. for RAFT,133 and ATRP.134 Barner-Kowollik and coworkers reported the synthesis of graft copolymers via the HDA-coupling of poly(n-butyl acrylate) (PnBA) prepared by RAFT with the CTA acting as dienophile onto statistical copolymers of styrene and trans, trans-hexa-2,4-dienylacrylate.133 Again, due to the radical polymerization mechanism, the degree of substitution is limited to avoid possible side reactions caused by the double bonds of the diene functionalized monomer.
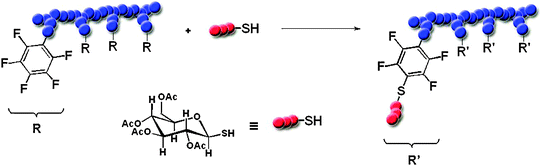 | ||
| Scheme 4 Schematic representation of the thio-para fluoro “click” reaction for the preparation of side-chain glycosylated poly(pentafluorostyrene).55 | ||
Furthermore, ATNRC chemistry was applied for the preparation of heterograft copolymers.135 Fu et al. described the synthesis of poly(ethylene oxide) (PEO)-based copolymers with TEMPO side groups, which were exploited for the coupling of PS and PtBA obtained by ATRP.135 The reactions were accomplished in the presence of CuBr and N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) at 90 °C. Under these conditions the secondary carbon radicals of PS and PtBA are trapped by the nitroxide radicals resulting in the formation of the corresponding graft copolymers. In a similar approach the same group reported the conversion of polymer backbones bearing secondary bromine functionalities with TEMPO end functionalized polymers.136
Apart from the main-stream “clickable” functionalities, functional groups rather uncommon in polymer chemistry like ketenes can be incorporated in polymers to enable subsequent, high efficient coupling reactions. Yagci and coworkers synthesized graft copolymers based on the photoinduced reaction of benzodioxinone, which forms intermediate ketenes suitable for the conversion with hydroxyl functions.137 Thus, benzodioxinone-functionalized PS was prepared and coupled under UV light to a copolymer consisting of MMA and 2-hydroxyethyl methacrylate (HEMA). Hawker and coworkers reported the preparation of well-defined polymers bearing Meldrum's acid moieties via ATRP (styrene based) and ROMP (norbornene based).138 Thermal treatment led to the formation of ketene functionalities which could be efficiently attacked by nucleophiles like amino functionalized dyes (Scheme 5). A competing reaction that has to be considered is the dimerization of ketene leading to crosslinked networks in the described example. However, the balance between both reactions can be influenced by the amount of added nucleophile as well as the utilized thermal treatment conditions.
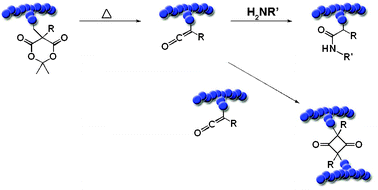 | ||
| Scheme 5 Schematic representation of the modification of Meldrum's acid containing copolymers obtained by ROMP and ATRP.138 | ||
2.4 Dendritic and hyperbranched structures
Dendrimers are well-defined macromolecular structures being of great interest in material science and medicine, which is owed to their unique properties, such as high functional group density, high solubility and low viscosity combined with a superb control over size and polydispersity. Albeit typically an enormous synthetic effort is necessary, numerous reports were published in the last years using different techniques for the preparation of these systems. In general, dendritic structures are the result of successive reactions of organic compounds performed in a convergent or divergent manner. The requirement of coupling reactions with high yields, an ease in the isolation of the product and a tolerance toward diverse functional groups has established the CuAAC as perfectly suited reaction for the preparation of dendrimers. However, the usage of other “click” coupling reactions such as TE/MA and DA cycloaddition reactions has emerged to be an additional strategy exhibiting all advantages of the CuAAC without the drawback of a heavy metal catalyst, which was recently summarized in an excellent review from Franc and Kakkar.139Up to now the DA cycloaddition is only little explored regarding dendrimer synthesis. However, due to its reversible character, known as retro-DA, it allows for the fabrication of responsive dendrimers, which has been exploited by the groups of McElhanon and Sanyal.62,140 Both groups report the synthesis of dendrimers using peripheral functionalized dendrons which were either “clicked” with a functional linker exhibiting the complementary “clickable” group or by a direct conjugation of two differently functionalized dendrons. In either case the assembly/reassembly potential of the DA reaction of furan and maleimide was studied as could be proven by SEC and 1H NMR spectroscopy. Furthermore, Sanyal and coworkers used (furan protected) maleimide functionalized dendrons for the functionalization of linear copolymers in a DA reaction and as Michael acceptor in a MA reaction for the conjugation with a tripeptide.141,142 More recently, Vieyres et al. demonstrated for the first time the utilization of the DA reaction in combination with CuAAC for the synthesis of dendrimers in a divergent approach.143 The authors synthesized two different building blocks, a maleimide functionalized with two acetylenes and a compound consisting of an azide and two furan moieties, which were “clicked” sequentially under microwave irradiation yielding G1 to G3 dendrimers (Scheme 6).
![Synthesis of second generation dendrimers exploiting the DA reaction of furan and maleimide [adopted from ref. 143].](/image/article/2012/CS/c1cs15107j/c1cs15107j-s6.gif) | ||
| Scheme 6 Synthesis of second generation dendrimers exploiting the DA reaction of furan and maleimide [adopted from ref. 143]. | ||
TE, TY and MA reactions, however, are other favorable methods for the synthesis of dendrimers. The divergent approach, commonly used for the synthesis of dendrimers, prefers the coupling with small molecules and, thus, the thiol-based reactions are highly suited since they are known to work excellent for small molecules compared to polymer–polymer conjugation. Hawker, Malkoch and coworkers explored the divergent synthesis of dendrimers using TE reactions. In a couple of impressive publications the authors describe the combination of TE reactions and esterification reactions for a series of different generations of dendrimers and the constant improvement of their synthesis.50,144–146 Using commercially available compounds, namely thioglycerol and 4-pentenoic anhydride the authors succeeded in the preparation of a G4 dendrimer in 8 steps via alternating TE and esterification reactions (Fig. 4). All TE reactions were prepared in bulk in the presence of an UV initiator, namely, 2,2-dimethoxy-2-phenylacetophenone (DMPA) at 365 nm light within 30 minutes. The synthesis of more complex coupling compounds yielded dendrimers with the same functional group density in just half of the number of steps. Furthermore, the utilization of the TE chemistry for the functionalization of the periphery with diverse thio-compounds as well as for the fabrication of hydrogels with HS–PEG–SH was demonstrated. In an alternative approach Hawker and coworkers combined TE with efficient epoxy amine reactions which resulted in the formation of dendrimers bearing hydroxyl groups at each dendritic layer able to be functionalized with various acids. More recently, the same group demonstrated the synthesis of a G6 dendrimer in one day by sequential TE as well as CuAAC reactions. Using TY and esterification reactions Stenzel and coworkers prepared G3 dendrimers with 192 hydroxy periphery groups in 5 steps which were tested as conjugation partner for cis-platin.147 Furthermore, Shen and coworkers introduced an asymmetric monomer bearing two different alkene functionalities, namely a methacrylate as well as an acrylate residue which exhibit different reactivities.148 Whereas the acrylate group reacts selectively with amines the methacrylate moieties is prone to undergo a reaction with thiol groups. By means of these reactions the authors describe the synthesis of G4 dendrimers as well as dendronized polyethylene imine (PEI). Furthermore, the synthesis of mechanistically asymmetric functionalized monomers and their employment for the preparation of a G4 dendrimer is described. Another “click”-type approach was reported by Percec and coworkers, who prepared dendrimers exploiting the base-mediated thioetherification of α-bromoesters with thiol compounds.53,149 In a divergent manner sequential thio–bromo and esterification reactions yielded G1–G4 dendrimers, which were used as macroinitiators for methyl acrylate under the formation of dendritic macromolecules with PMA chains in between the different junctions.
![Synthesis of G4 dendrimers exploiting a TE/esterification approach [adopted from ref. 50].](/image/article/2012/CS/c1cs15107j/c1cs15107j-f4.gif) | ||
| Fig. 4 Synthesis of G4 dendrimers exploiting a TE/esterification approach [adopted from ref. 50]. | ||
Similar reactions involving diverse thiol groups were additionally used in the last years for the synthesis of the less defined counterparts of the dendrimers, the hyperbranched polymers. The control over size and polydispersity index is limited for these systems, but at the same time the synthesis is comparatively easy. Lowe, Davis and co-workers reported the synthesis of hyperbranched systems combing RAFT polymerization and subsequent thiol–bromo reactions.150 Well-defined PMA was prepared with RAFT agents bearing two α-bromoester functional groups which coupled in situ to thiol groups generated by the aminolysis of the RAFT agent. The power of TY reactions for the synthesis of hyperbranched polymers was explored by Perrier and co-workers.151 Different compounds exhibiting a terminal alkene and thiol group were exposed to UV light under DMPA catalysis in DMF and hence hyperbranched systems were obtained in only one simple step.
2.5 Star-shaped (co-)polymers
Star-shaped (co-)polymers can either be considered as branched polymers with just one junction or as dendritic-type systems exhibiting a lower functional group density. Thus, their synthesis using “click chemistry” encompasses similar methods as applied for the preparation of the mentioned structures. As named by the geometrical shape of a star these structures feature a core as interior with a number of arms which form the exterior. Depending on the structural composition of the arms, star (co-)polymers and miktoarm star (co-)polymers can be distinguished. The former are characterized by uniform arms which consist of a defined block sequence, whereas the latter possess unequal arms. In view of the application of “click chemistry” for the synthesis of star-shaped polymers in either case convergent methods (“arm-first”) as well as divergent methods (“core-first”) can be exploited. Thus, the conversion of α- and/or ω-functionalized (co-)polymers with a multifunctional core as well as “click” modifications of star-shaped polymers obtained by multifunctional initiators are perfectly suited for the preparation of various star-shaped architectures. Foremost method for the conjugation is the CuAAC due to the ease in transforming or introducing the complementary functional groups. The work of Gao and Matyjaszewski, who demonstrated the conversion of the bromide end group of an ATRP initiator to an azide, is just one example,152 which was used in the following years for a variety of star and mikto-arm block copolymers.153–155 Further successes in the synthesis of star-shaped copolymers were achieved using multifunctional initiators bearing initiating groups for different living and/or controlled polymerization techniques in combination with azide or acetylene functionalities.156–160In the last years DA reactions have emerged to act as an appropriate alternative/partner for the conjugation. The groups of Tunca and Hizal have focused on the reaction of maleimide and anthracene as synthetic tool for the formation of diverse star-shaped block copolymers. In an “arm-first” approach polymeric precursors were prepared using α-functionalized ATRP initiators and esterification reactions in order to prepare maleimide and anthracene end functionalized polymers, respectively. The DA reaction was performed in toluene under reflux for 48 h. In order to prepare star (block) (co-)polymers tri-anthracene functional linking agents and cross-linked PS-cores with an anthracene periphery were used.66,161
The same groups demonstrated the feasibility of these approaches for one-pot and sequential double “click” reactions with CuAAC, respectively.64,162,163 Thus, the orthogonality of either reactions enable the synthesis of multiarm star triblock copolymers as well as multi-miktoarm star block copolymers using α,ω-functionalized polymers and cross-linked PS-cores possessing anthracene and/or alkyne exteriors (Fig. 5). Another DA approach for the synthesis of star-shaped polymers was reported by Barner-Kowollik and coworkers.164–166 While the reaction of anthracene and maleimide involves exclusively carbon–carbon double bonds, hetero atom containing double bonds can undergo a DA cycloaddition reaction, known as HDA, as well. The authors reported the synthesis of building blocks containing electron-deficient dithioester end groups derived from an accordingly functionalized RAFT agent (diethoxyphosphoryldithioformate or pyridine-2-yldithioformate) which can be applied in HDA reactions with diene functionalized compounds. For the synthesis of 2-, 3- and 4-star polymers di-, tri- and tetrafunctional diene cores were converted with both types of the PS-RAFT precursors.164 Depending on the choice of the RAFT agent and the number of arms yields between 65 and 91% could be achieved, in which the pyridine-2-yldithioformate PS showed the higher yields in any case. In a further study the same group presented the combination of the HDA and the CuAAC for the preparation of PCL-b-PS star copolymers.165 In order to prepare telechelic PCL, a diene terminus containing PCL was converted in an esterification reaction with 4-pentynoic acid. Thus, the polymer obtained is capable to undergo a reaction with an electron-deficient dithioester and an azide compound as well. For the synthesis of a star block copolymer a trifunctional azide compound and a PS containing a dithioester end group was used. Best results in view of conversion and molar mass distribution were obtained for the “arm-first” method performing the HDA reaction (block copolymer formation) prior to the CuAAC. The “core first” method resulted in the formation of a triazole ring which might cause competitive reactions with the TFA, which is necessary to activate the RAFT-PS for the HDA reaction (Fig. 6). Thus, this approach is limited to sequential reactions since in a one-pot reaction the acidic conditions for the HDA would be in contrast to the basic conditions for the CuAAC.
![Preparation of star block copolymers via CuAAC and DA reactions on a cross-linked PS core [adopted from ref. 162].](/image/article/2012/CS/c1cs15107j/c1cs15107j-f5.gif) | ||
| Fig. 5 Preparation of star block copolymers via CuAAC and DA reactions on a cross-linked PS core [adopted from ref. 162]. | ||
![Synthesis of PCL-b-PS star block copolymers via an arm-first or core-first approach using an α-diene ω-alkyne functional PCL, a trifunctional azide compound and a PS with an electron deficient dithioester end group [adopted from ref. 165].](/image/article/2012/CS/c1cs15107j/c1cs15107j-f6.gif) | ||
| Fig. 6 Synthesis of PCL-b-PS star block copolymers via an arm-first or core-first approach using an α-diene ω-alkyne functional PCL, a trifunctional azide compound and a PS with an electron deficient dithioester end group [adopted from ref. 165]. | ||
While TE and TY chemistry as well as MA reactions are perfectly suited for the construction of dendritic and hyperbranched systems using small organic components, their application in view of star-shaped polymers is limited. However, Hoyle and Lowe were able to synthesize 3-arm star poly(N,N-diethylacrylamide) by (thiol-)MA.167 In an one pot reaction a RAFT polymer with a thiocarbonylthio end group reacted with a trifunctionalized ene coupling agent under phoshine catalysis within 5 min. The reduction of the RAFT end group was managed simultaneously by a primary amine. Also in this case an excess of polymer was used most likely to guaranty a full conversion of the double bonds as proven by 1H NMR and IR spectroscopy as well as MALDI TOF MS. Pounder et al. used the (thiol-)MA reaction for the synthesis of 3-arm star poly(lactide).56 A trifunctional thiol compound was conjugated to a maleimide functionalized PLA, the Michael acceptor, in 2 h under NEt3 catalysis and with a slight excess of the thiol.
Moreover, “click chemistry” can be used to functionalize star-shaped scaffolds in a post-modification step to prepare more advanced structures. Recently, Stenzel and coworkers described the synthesis of a 4-arm star polymer via RAFT polymerization of vinylbenzyl chloride, just to highlight one.168 In an efficient “click-type” reaction the corresponding star glycopolymer was prepared by the nucleophilic substitution of the chlorine with the sodium salt of 1-thio-β-D-glucose at 40 °C. Despite the fact that this reaction is slow the main benefit of this reaction is the absence of a hazardous side product or any other “special” reaction conditions, such as a UV source.
Apart from this kind of reactions another “click-type” reaction, the Glaser coupling was reported more recently in the context of star-shaped (co)polymers. As a first example Gungor et al. described the preparation of A2B2 type miktoarm star copolymers, (PS)2–(PMMA)2 and (PCL)2–(PS)2 exploiting the Glaser coupling as efficient coupling reaction.169 This approach was subsequently further extended for the synthesis of mikto star-shaped copolymers containing PS, poly(isoprene) (PI), PtBA, PCL and poly(acrylic acid) (PAA) block sequences, respectively.170,171
2.6 Cross-linked networks
In order to prepare cross-linked materials with high densities and functionalities, reactions occurring under mild conditions combined with a high functional group tolerance are desirable. Since “click” reactions, in particular CuAAC, provide these properties they were applied in the past for the preparation of cross-linked networks,172,173 with the potential to be used as drug delivery systems, cell-encapsulating materials and tissue engineering.Even if TE chemistry is a relatively new member of the “click-family” it has been already broadly used in the field of hydrogels in the last years. TE systems form structurally homogenous networks via a step-growth free-radical process, in contrast to the well-known acrylate photopolymerizations which follow a chain-growth polymerization mechanism. In the last years, in particular the effect of the thiol-addition to a multifunctional (meth-)acrylate systems was investigated.28 The combination of the homopolymerization of acrylates and the thiol–acrylate reaction leads to more uniform cross-linked networks, with a lower shrinkage and a reduced oxygen inhibition.174 In a similar manner several PEG based hydrogel systems were prepared using thiol-functionalized PEG as well as (multi-)functional PEG with various Michael-acceptors, such as vinyl sulfones.175,176 Advantageous of the TE chemistry is its compatibility to cells which enables the performance of these reactions in living systems. Thus, it is possible to encapsulate cells directly during the hydrogel process as well as to functionalize existing hydrogels with a range of thiol-compounds as illustrated by Anseth and coworkers.177–179 In an orthogonal approach the same group investigated the formation of 3D gels starting from a macromolecular precursor exhibiting cyclooctyne moieties suitable for (copper-free) SPAAC reactions, which allow for a direct encapsulation of cells within the hydrogels (Fig. 7).180 In order to achieve a further functionalization of the hydrogels biomolecules, e.g. a cysteine-containing fluorescently labeled RGD sequence, were attached to the hydrogel by the bio-orthogonal TE photoaddition. Furthermore, the exploitation of PEG based TE reactions for the fabrication of hydrogel-supported microarrays was reported by Hawker and coworkers.181 Substrates exhibiting thiol or alkene functionalities were printed on glass prior to an embedding under a TE prepolymer solution. The curing of this solution yielded hydrogel embedded microarrays. For the immobilization of bioactive and diagnostic molecules different strategies were described encompassing the direct incorporation of the active compound as well as the post-modification of orthogonal functional groups during the TE process. The synthesis of degradable networks composed of anhydrides using the TE reaction was recently described by Shipp and coworkers.182,183 Commercially available tetrathiols and diene anhydrides were exposed to UV light yielding elastomeric polyanhydrides which were characterized with regard to thermomechanical properties. The occurrence of possible side reactions was studied in detail revealing the formation of thioesters during the photoaddition, however, with a much slower rate. Recently, Nordberg et al. reported the employment of TE photoaddition reactions of triazine-based compounds for the fabrication of fiber-reinforced adhesive patches.184 To enhance the adhesion of the hydrophobic TE matrix to moist bones they were pre-treated with phenolic dopamine as well as poly(p-hydroxystyrene) primers. Furthermore, this bone fracture treatment benefits from excellent shear rates and low cytotoxicities making it an alternative to the commercially available cyanoacrylate systems.
![Synthesis of a peptide–PEG based hydrogel containing pendant alkene functionalities by the copper-free strain-promoted alkyne azide cycloaddition [adopted from ref. 180].](/image/article/2012/CS/c1cs15107j/c1cs15107j-f7.gif) | ||
| Fig. 7 Synthesis of a peptide–PEG based hydrogel containing pendant alkene functionalities by the copper-free strain-promoted alkyne azide cycloaddition [adopted from ref. 180]. | ||
Recently, the TY reaction emerged to be of special interest for this kind of structures, as well. This reaction seems to be very well suited to be used in network formation reactions. The double reaction of an alkyne functionality with two thiols exhibits a perfect basis for a cross-linking junction since networks with higher cross-linking density can be obtained. Chan et al. exploited this approach to prepare a series of networks with different cross-link densities.185 The reaction proceeded under UV irradiation and was supported by the addition of 1 wt% photoinitiator. In order to study the effect of cross-link density, starting materials with nearly the same chemical composition but different number of functional groups (thiol, yne) were used resulting in networks in high yields already after a few minutes. The polymerization rate was found to be dependent on the monomer functionality showing a decrease in the rate for higher functionalized TY compounds. More recently, Cameron and coworkers described the synthesis of porous polymer networks mediated by TE as well as TY photoreactions using an emulsion templating approach (Fig. 8). The materials obtained showed defined morphologies as well as a mechanical property dependency on the cross-linking system (TE or TY).186
![Preparation of porous polymer networks applying TE as well as TY chemistry [adopted from ref. 186].](/image/article/2012/CS/c1cs15107j/c1cs15107j-f8.gif) | ||
| Fig. 8 Preparation of porous polymer networks applying TE as well as TY chemistry [adopted from ref. 186]. | ||
Another “click-type” approach for the preparation of three-dimensional crosslinked networks was described by Nishide and coworkers. Michael (poly-)additions of a TEMPO-substituted acetoacetate monomer and tri- as well as tetraacrylate systems were applied to form poly(β-ketoester)s.187 For the reaction all components were mixed and cured at room temperature in the presence of 1 wt% 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to form films for the fabrication of polymer-electrodes. This was possible since the nitroxide moieties stayed active upon the thermal-curing process and the half-life of the radicals was found to be more than one year in the film state. A similar strategy was already applied in 2007 by Long and coworkers, who demonstrated the formation of networks based on the MA of poly(propylene glycol) bis(acetoacetate) with diacrylate compounds under DBU catalysis.188 The incorporation of urethane functionalities in the acrylate compound revealed networks with improved mechanical properties which refer to their hydrogen bonding ability.
Intermediately formed ketenes can undergo an addition reaction in the presence of alcohols as it was already demonstrated for the preparation of graft copolymers. Talinli, Yagci and coworkers used the potential of benzodioxinones to form ketenes under UV irradiation for the preparation of crosslinked networks.189 For this purpose, bisbenzodioxinones were prepared and converted with polymers containing pendant hydroxy groups under UV light. Furthermore, radicals released during the UV induced formation of ketenes were shown to act as initiators for the radical polymerization of HEMA.190 As a result, this approach allows polymerization and cross-linking in one step.
2.7 Particle stabilization and surface modification
The formation of cross-linked self-assembled structures as well as the surface modification of microspheres and (nano)particles represent another emerging field for “click chemistry”. In the following a few recent examples are highlighted further demonstrating the extended scope of the “click” reactions as an efficient coupling strategy.The self-assembly of (block) copolymers in (aqueous) solution may result in the formation of, for instance, particles or micelles. In general, the incorporation of “clickable” groups in a polymer allows its cross-linking, which can result, in terms of block copolymers, in the stabilization of various cores, shells or layers, depending on the self-assembled structure. The ease of incorporation of alkyne and azide groups in copolymers using functionalized comonomers resulted in the preparation of plenty of materials suitable for the cross-linking applying CuAAC chemistry. This approach resulted in the synthesis of, e.g. capsules,191,192 core cross-linked micelles,193,194 and cross-linked nanoparticles.195 In a similar manner TE as well as TY reactions were exploited for the direct synthesis of nanoparticles,196 nonporous functional polymer beads,197 and nanocapsules.198 Sung, Kim and coworkers reported the direct synthesis of nanocapsules based on TE photoaddition reactions of cucurbit[6]uril (CB[6]) exhibiting 12 allyloxy groups at the periphery and various dithiols. In order to gain a deeper understanding of the capsule formation the reaction was monitored by dynamic light scattering (DLS), IR spectroscopy and elemental analysis. As a result, intermediate 2D oligomeric patches were observed resulting in the formation of hollow spheres. By performing the nanocapsule creation in the presence of guest molecules an entrapment was achieved. Furthermore, the surface composed of CB[6] units allow for the formation of stable noncovalent host–guest systems with polyamines, as demonstrated by a fluorescein isothiocyanate-tagged polyamine (Fig. 9).
![Entrapment of dye molecules during the synthesis of polymer nanocapsules exploiting the TE photoaddition and noncovalent surface modification of the nanocapsules using a FITC-tagged polyamine [adopted from ref. 198].](/image/article/2012/CS/c1cs15107j/c1cs15107j-f9.gif) | ||
| Fig. 9 Entrapment of dye molecules during the synthesis of polymer nanocapsules exploiting the TE photoaddition and noncovalent surface modification of the nanocapsules using a FITC-tagged polyamine [adopted from ref. 198]. | ||
Functional surfaces demonstrate an additional platform for the application of “click” reactions. In the last years various approaches for the preparation of functional particles were developed encompassing the modification of, e.g. poly(divinylbenzene) microspheres,199,200 silica–polymer hybrid nanoparticles,201–204 as well as 1,2-polybutadiene particles.205 In this regard, CuAAC as well as TE reactions are mainly reported. Goldmann et al. reported the modification of poly(divinylbenzene) (PDVB) microspheres by TE reactions of the residual double bonds on the sphere surfaces with thiol end functionalized PNIPAm in an one pot procedure.199 Furthermore, the double bonds were converted to azide end groups using 1-azido-undecane-11-thiol followed by subsequent CuAAC with an alkyne endfunctionalized pHEMA. The preparation of PDVB microspheres containing surface expressed dithioester groups via a precipitation RAFT polymerization was described by Barner-Kowollik, Barner and coworkers.200 The available dithioester end groups of the RAFT agent enabled grafting of diene-functionalized PCL onto the microsphere surface by a HDA conjugation.
3. Summary and outlook
Telechelic or side chain functional polymers have been prepared successfully using controlled polymerization techniques that have been developed in the last two decades. The preparation of various macromolecular architectures has been demonstrated using these well-defined and functional macromolecules as building blocks. In the first half of the last decade, polymer chemists investigated the use of copper catalyzed [3+2] cycloaddition reactions of azide–alkyne to prepare new macromolecular architectures. However, as we have noted in our previous review that “future research should focus on the development of true “click” reactions that do not require a (toxic) metal catalysts”. That is exactly how it happened and in the second half of the last decade, the main focus was on the development of “click” reactions that can be conducted in the absence of metal catalysts and under ambient conditions.Several polymer research groups introduced new and improved metal free “click” reactions. All these techniques can be categorized into three groups: (i) metal free [3+2] cycloaddition reaction of azide and activated alkynes, (ii) [4+2] Diels–Alder reactions, and (iii) thiol based “click” reactions. The latter attracted enormous attention in polymer science due to the wide range of available thiols and corresponding reagents that can readily react with thiols. These reagents can be listed as alkene, alkyne, para-fluoro phenyl, bromo, chloro, sulfo, and isocyanate groups containing compounds. As discussed in this review, thiol based “click” reactions can proceed via radical or base catalyzed mechanism and both routes are very robust and efficient. Various architectures such as block copolymers, dendrimers, hyperbranched systems, highly crosslinked networks, graft copolymers and telechelic polymers could be synthesized using thiol-“click” reactions. However, significant disadvantages of the thiol-“click” reactions should be also pointed out as the prevalent thiol odor, particularly for low molar mass materials, as well as the lack of shelf stability for many formulations.
In the light of the rapid developments in the last decade in polymer chemistry, it is evident that the ultimate goal will be to gain an absolute control over each repeating unit of the polymer chain. Apparently, preparation of the sequence controlled macromolecular structures will be the main challenge of the next decade. We believe that this will be achievable by combining controlled and living polymerization procedures with “click” reactions.
Acknowledgements
The authors thank the Dutch Polymer Institute (DPI, Technology area HTE) and the Thuringian Ministry for Education, Science and Culture (grant #B514-09051, NanoConSens). K.K. is grateful to the Landesgraduiertenfoerderung Thueringen for financial support.References
- J. Gillis and R. E. Oesper, J. Chem. Educ., 1964, 41, 224 CrossRef.
- M. Szwarc, M. Levy and R. Milkovich, J. Am. Chem. Soc., 1956, 78, 2656 CrossRef CAS.
- M. Szwarc, Nature, 1956, 178, 1168 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- V. Percec, A. V. Popov, E. Ramirez-Castillo and O. Weichold, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6364 CrossRef CAS.
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156 CrossRef CAS.
- M. E. Levere, I. Willoughby, S. O'Donohue, A. de Cuendias, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1086 RSC.
- M. E. Levere, I. Willoughby, S. O'Donohue, P. M. Wright, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1753 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- G. Moad and S. H. Thang, Aust. J. Chem., 2009, 62, 1379 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- P. L. Golas, N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2006, 39, 6451 CrossRef CAS.
- W. Tang and K. Matyjaszewski, Macromolecules, 2006, 39, 4953 CrossRef CAS.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133 CrossRef CAS.
- C. R. Becer, R. M. Paulus, S. Hoppener, R. Hoogenboom, C. A. Fustin, J.-F. Gohy and U. S. Schubert, Macromolecules, 2008, 41, 5210 CrossRef CAS.
- C. R. Becer, C. Haensch, S. Hoeppener and U. S. Schubert, Small, 2007, 3, 220 CrossRef CAS.
- C. R. Becer, R. Hoogenboom, D. Fournier and U. S. Schubert, Macromol. Rapid Commun., 2007, 28, 1161 CrossRef CAS.
- R. M. Paulus, C. R. Becer, R. Hoogenboom and U. S. Schubert, Aust. J. Chem., 2009, 62, 254 CrossRef CAS.
- R. M. Paulus, C. R. Becer, R. Hoogenboorn and U. S. Schubert, Macromol. Chem. Phys., 2008, 209, 794 CrossRef CAS.
- A. H. Soeriyadi, C. Boyer, J. Burns, C. R. Becer, M. R. Whittaker, D. M. Haddleton and T. P. Davis, Chem. Commun., 2010, 46, 6338 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- U. Mansfeld, C. Pietsch, R. Hoogenboom, C. R. Becer and U. S. Schubert, Polym. Chem., 2010, 1, 1560 RSC.
- W. D. Sharpless, P. Wu, T. V. Hansen and J. G. Lindberg, J. Chem. Educ., 2005, 82, 1833 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60 CrossRef CAS.
- D. D. Diaz, S. Punna, P. Holzer, A. K. Mcpherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392 CrossRef CAS.
- P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Frechet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928 CrossRef CAS.
- M. Malkoch, K. Schleicher, E. Drockenmuller, C. J. Hawker, T. P. Russell, P. Wu and V. V. Fokin, Macromolecules, 2005, 38, 3663 CrossRef CAS.
- P. L. Golas and K. Matyjaszewski, QSAR Comb. Sci., 2007, 26, 1116 CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727 RSC.
- D. M. Haddleton, G. Mantovani and V. Ladmiral, Abstr. Pap., Jt. Conf. –Chem. Inst. Can. Am. Chem. Soc., 2006, 231, 123 Search PubMed.
- J. Geng, G. Mantovani, L. Tao, J. Nicolas, G. Chen, R. Wallis, D. A. Mitchell, B. R. G. Johnson, S. D. Evans and D. M. Haddleton, J. Am. Chem. Soc., 2007, 129, 15156 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369 RSC.
- P. V. Chang, J. A. Prescher, E. M. Sletten, J. M. Baskin, I. A. Miller, N. J. Agard, A. Lo and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1821 CrossRef CAS.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS.
- J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211 CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2008, 47, 2182 CrossRef CAS.
- S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699 CrossRef CAS.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010, 20, 4745 RSC.
- G. Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
- A. H. Soeriyadi, G. Z. Li, S. Slavin, M. W. Jones, C. M. Amos, C. R. Becer, M. R. Whittaker, D. M. Haddleton, C. Boyer and T. P. Davis, Polym. Chem., 2011, 2, 815 RSC.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062 CrossRef CAS.
- N. ten Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946 CrossRef CAS.
- A. Gress, A. Volkel and H. Schlaad, Macromolecules, 2007, 40, 7928 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3931 CrossRef CAS.
- C. R. Becer, K. Kokado, C. Weber, A. Can, Y. Chujo and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1278 CrossRef CAS.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387 CrossRef CAS.
- R. J. Pounder, M. J. Stanford, P. Brooks, S. P. Richards and A. P. Dove, Chem. Commun., 2008, 5158 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093 CrossRef CAS.
- H. B. Li, B. Yu, H. Matsushima, C. E. Hoyle and A. B. Lowe, Macromolecules, 2009, 42, 6537 CrossRef CAS.
- A. Klaikherd, S. Ghosh and S. Thayumanavan, Macromolecules, 2007, 40, 8518 CrossRef CAS.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978 RSC.
- T. Dispinar, R. Sanyal and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4545 CrossRef CAS.
- M. L. Szalai, D. V. McGrath, D. R. Wheeler, T. Zifer and J. R. McElhanon, Macromolecules, 2007, 40, 818 CrossRef CAS.
- B. Voit, S. Fleischmann, H. Komber, A. Scheel and K. Stumpe, Macromol. Symp., 2007, 254, 16 CAS.
- H. Durmaz, A. Dag, A. Hizal, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7091 CrossRef CAS.
- B. Gacal, H. Akat, D. K. Balta, N. Arsu and Y. Yagci, Macromolecules, 2008, 41, 2401 CrossRef CAS.
- A. Dag, H. Durmaz, U. Tunca and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 178 CrossRef CAS.
- A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2009, 30, 1792 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630 CrossRef CAS.
- B. S. Sumerlin and A. P. Vogt, Macromolecules, 2010, 43, 1 CrossRef CAS.
- J. A. Syrett, G. Mantovani, W. R. S. Barton, D. Price and D. M. Haddleton, Polym. Chem., 2010, 1, 102 RSC.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176 CrossRef CAS.
- S. T. Laughlin and C. R. Bertozzi, ACS Chem. Biol., 2009, 4, 1068 CrossRef CAS.
- P. V. Chang, X. Chen, C. Smyrniotis, A. Xenakis, T. S. Hu, C. R. Bertozzi and P. Wu, Angew. Chem., Int. Ed., 2009, 48, 4030 CrossRef CAS.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1052 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57 RSC.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558 CrossRef CAS.
- J. A. Opsteen and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2913 CrossRef CAS.
- G. Mantovani, V. Ladmiral, L. Tao and D. M. Haddleton, Chem. Commun., 2005, 2089 RSC.
- R. K. Jing, G. W. Wang, Y. N. Zhang and J. L. Huang, Macromolecules, 2011, 44, 805 CrossRef CAS.
- K. T. Wiss and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4758 CrossRef CAS.
- S. Fleischmann, H. Kornber, D. Appelhans and B. I. Voit, Macromol. Chem. Phys., 2007, 208, 1050 CrossRef CAS.
- M. W. M. Fijten, C. Haensch, B. M. van Lankvelt, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2008, 209, 1887 CrossRef CAS.
- G. Volet, T. X. Lav, J. Babinot and C. Amiel, Macromol. Chem. Phys., 2011, 212, 118 CrossRef CAS.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301 CrossRef CAS.
- W. Q. Shen, Q. A. Qiu, Y. Wang, M. A. Miao, B. S. Li, T. S. Zhang, A. N. Cao and Z. S. An, Macromol. Rapid Commun., 2010, 31, 1444 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773 CrossRef CAS.
- J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, J. F. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346 CrossRef CAS.
- H. Durmaz, A. Dag, O. Altintas, T. Erdogan, G. Hizal and U. Tunca, Macromolecules, 2007, 40, 191 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, M. H. Stenzel and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2009, 48, 2411 CAS.
- C. J. Duxbury, D. Cummins and A. Heise, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3795 CrossRef CAS.
- E. L. Thomas, D. M. Anderson, C. S. Henkee and D. Hoffman, Nature, 1988, 334, 598 CrossRef CAS.
- T. P. Lodge, Macromol. Chem. Phys., 2003, 204, 265 CrossRef CAS.
- E. Gungor, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3409 CrossRef CAS.
- S. Hilf, N. Hanik and A. F. M. Kilbinger, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2913 CrossRef CAS.
- P. Lundberg, C. J. Hawker, A. Hult and M. Malkoch, Macromol. Rapid Commun., 2008, 29, 998 CrossRef CAS.
- W. Lin, Q. Fu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 4127 CrossRef CAS.
- H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1962 CrossRef CAS.
- B. A. Laurent and S. M. Grayson, J. Am. Chem. Soc., 2006, 128, 4238 CrossRef CAS.
- J. N. Hoskins and S. M. Grayson, Macromolecules, 2009, 42, 6406 CrossRef CAS.
- X.-P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069 CrossRef CAS.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103 CrossRef CAS.
- D. Han, X. Tong, Y. Zhao, T. Galstian and Y. Zhao, Macromolecules, 2010, 43, 3664 CrossRef CAS.
- H. Misaka, R. Kakuchi, C. Zhang, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 5091 CrossRef CAS.
- D. M. Eugene and S. M. Grayson, Macromolecules, 2008, 41, 5082 CrossRef CAS.
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654 CrossRef CAS.
- G. Y. Shi, X. Z. Tang and C. Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2390 CrossRef CAS.
- L. Y. Li, W. D. He, J. Li, S. C. Han, X. L. Sun and B. Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 7066 CrossRef CAS.
- H. Y. Li, R. Riva, R. Jerome and P. Lecomte, Macromolecules, 2007, 40, 824 CrossRef CAS.
- Y.-Q. Dong, Y.-Y. Tong, B.-T. Dong, F.-S. Du and Z.-C. Li, Macromolecules, 2009, 42, 2940 CrossRef CAS.
- B. V. K. J. Schmidt, N. Fechler, J. Falkenhagen and J.-F. Lutz, Nat. Chem., 2011, 3, 236 CrossRef.
- M. J. Stanford, R. L. Pflughaupt and A. P. Dove, Macromolecules, 2010, 43, 6538 CrossRef CAS.
- A. A. Aimetti, R. K. Shoemaker, C. C. Lin and K. S. Anseth, Chem. Commun., 2010, 46, 4061 RSC.
- A. A. Aimetti, K. R. Feaver and K. S. Anseth, Chem. Commun., 2010, 46, 5781 RSC.
- H. Durmaz, A. Dag, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5083 CrossRef CAS.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130 CrossRef CAS.
- N. Vinson, Y. Z. Gou, C. R. Becer, D. M. Haddleton and M. I. Gibson, Polym. Chem., 2011, 2, 107 RSC.
- A. Krieg, C. R. Becer, R. Hoogenboom and U. S. Schubert, Macromol. Symp., 2009, 275–276, 73 CrossRef CAS.
- D. Wu, X. Song, T. Tang and H. Zhao, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 443 CrossRef CAS.
- Y. Li, J. W. Yang and B. C. Benicewicz, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4300 CrossRef CAS.
- B. S. Sumerlin, N. V. Tsarevsky, G. Louche, R. Y. Lee and K. Matyjaszewski, Macromolecules, 2005, 38, 7540 CrossRef CAS.
- P. D. Topham, N. Sandon, E. S. Read, J. Madsen, A. J. Ryan and S. P. Armes, Macromolecules, 2008, 41, 9542 CrossRef CAS.
- D. Quemener, M. Le Hellaye, C. Bissett, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 155 CrossRef CAS.
- L. J. Wong, C. Boyer, Z. F. Jia, H. M. Zareie, T. P. Davis and V. Bulmus, Biomacromolecules, 2008, 9, 1934 CrossRef CAS.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445 CrossRef CAS.
- K. Kempe, C. Weber, K. Babiuch, M. Gottschadt, R. Hoogenboom and U. S. Schubert, Biomacromolecules, 2011 DOI:10.1021/bm2003847.
- K. Kempe, T. Neuwirth, J. Czaplewska, M. Gottschadt, R. Hoogenboom and U. S. Schubert, Polym. Chem., 2011 10.1039/c1py00099c.
- K. Kempe, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2011 DOI:10.1002/marc.201100271.
- Z. F. Jia, J. Q. Liu, T. P. Davis and V. Bulmus, Polymer, 2009, 50, 5928 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063 CrossRef CAS.
- K. Babiuch, C. R. Becer, M. Gottschaldt, J. T. Delaney, J. R. Weisser, B. Beer, R. Wyrwa, M. Schnabelrauch and U. S. Schubert, Macromol. Biosci., 2011, 11, 535 CrossRef CAS.
- K. Babiuch, R. Wyrwa, K. Wagner, T. Seemann, S. Hoeppener, C. R. Becer, R. Linke, M. Gottschaldt, J. Weisser, M. Schnabelrauch and U. S. Schubert, Biomacromolecules, 2011, 12, 681 CrossRef CAS.
- A. Bousquet, C. Barner-Kowollik and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1773 CrossRef CAS.
- A. A. Kavitha and N. K. Singha, Macromol. Chem. Phys., 2007, 208, 2569 CrossRef CAS.
- Q. Fu, C. Liu, W. Lin and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6770 CrossRef CAS.
- G. Wang, Y. Zhang and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1633 CrossRef CAS.
- Y. Y. Durmaz, V. Kumbaraci, A. L. Demirel, N. Talinli and Y. Yagci, Macromolecules, 2009, 42, 3743 CrossRef CAS.
- F. A. Leibfarth, M. Kang, M. Ham, J. Kim, L. M. Campos, N. Gupta, B. Moon and C. J. Hawker, Nat. Chem., 2010, 2, 207 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536 RSC.
- M. M. Kose, G. Yesilbag and A. Sanyal, Org. Lett., 2008, 10, 2353 CrossRef CAS.
- M. Tonga, N. Cengiz, M. M. Kose, T. Dede and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 410 CrossRef CAS.
- O. Gok, H. Durmaz, E. S. Ozdes, G. Hizal, U. Tunca and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2546 CrossRef CAS.
- A. Vieyres, T. Lam, R. Gillet, G. Franc, A. Castonguay and A. Kakkar, Chem. Commun., 2010, 46, 1875 RSC.
- M. I. Montanez, L. M. Campos, P. Antoni, Y. Hed, M. V. Walter, B. T. Krull, A. Khan, A. Hult, C. J. Hawker and M. Malkoch, Macromolecules, 2010, 43, 6004 CrossRef CAS.
- T. Kang, R. J. Amir, A. Khan, K. Ohshimizu, J. N. Hunt, K. Sivanandan, M. I. Montanez, M. Malkoch, M. Ueda and C. J. Hawker, Chem. Commun., 2010, 46, 1556 RSC.
- P. Antoni, M. J. Robb, L. Campos, M. Montanez, A. Hult, E. Malmstrom, M. Malkoch and C. J. Hawker, Macromolecules, 2010, 43, 6625 CrossRef CAS.
- G. J. Chen, J. Kumar, A. Gregory and M. H. Stenzel, Chem. Commun., 2009, 6291 RSC.
- X. P. Ma, J. B. Tang, Y. Q. Shen, M. H. Fan, H. D. Tang and M. Radosz, J. Am. Chem. Soc., 2009, 131, 14795 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3940 CrossRef CAS.
- J. T. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2010, 43, 20 CrossRef CAS.
- D. Konkolewicz, A. Gray-Weale and S. Perrier, J. Am. Chem. Soc., 2009, 131, 18075 CrossRef CAS.
- H. F. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960 CrossRef CAS.
- M. Lammens, D. Fournier, M. W. M. Fijten, R. Hoogenboom and F. Du Prez, Macromol. Rapid Commun., 2009, 30, 2049 CrossRef CAS.
- Z. S. Ge, D. Wang, Y. M. Zhou, H. W. Liu and S. Y. Liu, Macromolecules, 2009, 42, 2903 CrossRef CAS.
- L. P. Yang, X. H. Dong and C. Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7757 CrossRef CAS.
- O. Altintas, B. Yankul, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3588 CrossRef CAS.
- Q. Fu, G. W. Wang, W. C. Lin and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 986 CrossRef CAS.
- G. W. Wang, X. L. Luo, C. Liu and J. L. Huang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2154 CrossRef CAS.
- L. P. Yang, H. X. Zhou, G. Y. Shi, Y. Wang and C. Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6641 CrossRef CAS.
- E. Gungor, G. Cote, T. Erdogan, H. Durmaz, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1055 CrossRef CAS.
- A. Dag, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 302 CrossRef CAS.
- A. Dag, H. Durmaz, V. Kirmizi, G. Hizal and U. Tunca, Polym. Chem., 2010, 1, 621 RSC.
- H. Durmaz, A. Dag, D. Gursoy, A. L. Demirel, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1557 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120 CrossRef CAS.
- S. Sinnwell, A. J. Inglis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2008, 29, 1090 CrossRef CAS.
- S. Sinnwell, M. Lammens, M. H. Stenzel, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2207 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959 RSC.
- Y. Chen, G. J. Chen and M. H. Stenzel, Macromolecules, 2010, 43, 8109 CrossRef CAS.
- E. Gungor, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6703 CrossRef CAS.
- G. W. Wang, B. Hu and J. L. Huang, Macromolecules, 2010, 43, 6939 CrossRef CAS.
- G. Wang, X. Fan and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5313 CrossRef CAS.
- S. Q. Liu, P. L. R. Ee, C. Y. Ke, J. L. Hedrick and Y. Y. Yang, Biomaterials, 2009, 30, 1453 CrossRef CAS.
- L. Mespouille, O. Coulembier, D. Paneva, P. Degee, I. Rashkov and P. Dubois, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4997 CrossRef CAS.
- A. F. Senyurt, H. Y. Wei, C. E. Hoyle, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 4901 CrossRef CAS.
- C. Hiemstra, L. J. van der Aa, Z. Zhong, P. J. Dijkstra and J. Feijen, Macromolecules, 2007, 40, 1165 CrossRef CAS.
- C. Hiemstra, L. J. van der Aa, Z. Zhong, P. J. Dijkstra and J. Feijen, Biomacromolecules, 2007, 8, 1548 CrossRef CAS.
- M. C. Cushing and K. S. Anseth, Science, 2007, 316, 1133 CrossRef CAS.
- C. N. Salinas, B. B. Cole, A. M. Kasko and K. S. Anseth, Tissue Eng., 2007, 13, 1025 CrossRef CAS.
- C. A. DeForest, E. A. Sims and K. S. Anseth, Chem. Mater., 2010, 22, 4783 CrossRef CAS.
- C. A. DeForest, B. D. Polizzotti and K. S. Anseth, Nat. Mater., 2009, 8, 659 CrossRef CAS.
- N. Gupta, B. F. Lin, L. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138 CrossRef CAS.
- D. A. Shipp, C. W. McQuinn, B. G. Rutherglen and R. A. McBath, Chem. Commun., 2009, 6415 RSC.
- B. G. Rutherglen, R. A. McBath, Y. L. Huang and D. A. Shipp, Macromolecules, 2010, 43, 10297 CrossRef CAS.
- A. Nordberg, P. Antoni, M. I. Montanìfez, A. Hult, H. Von Holst and M. Malkoch, ACS Appl. Mater. Interfaces, 2010, 2, 654 CAS.
- J. W. Chan, J. Shin, C. E. Hoyle, C. N. Bowman and A. B. Lowe, Macromolecules, 2010, 43, 4937 CrossRef CAS.
- E. Lovelady, S. D. Kimmins, J. Wu and N. R. Cameron, Polym. Chem., 2011, 2, 559 RSC.
- T. Ibe, R. B. Frings, A. Lachowicz, S. Kyo and H. Nishide, Chem. Commun., 2010, 46, 3475 RSC.
- S. R. Williams, B. D. Mather, K. M. Miller and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4118 CrossRef CAS.
- V. Kumbaraci, N. Talinli and Y. Yagci, Macromol. Rapid Commun., 2007, 28, 72 CrossRef CAS.
- M. A. Tasdelen, V. Kumbaraci, N. Talinli and Y. Yagci, Macromolecules, 2007, 40, 4406 CrossRef CAS.
- G. K. Such, E. Tjipto, A. Postma, A. P. R. Johnston and F. Caruso, Nano Lett., 2007, 7, 1706 CrossRef CAS.
- C. J. Ochs, G. K. Such, Y. Yan, M. P. van Koeverden and F. Caruso, ACS Nano, 2010, 4, 1653 CrossRef CAS.
- X. Z. Jiang, J. Y. Zhang, Y. M. Zhou, J. Xu and S. Y. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 860 CrossRef CAS.
- J. Zhang, Y. Zhou, Z. Zhu, Z. Ge and S. Liu, Macromolecules, 2008, 41, 1444 CrossRef CAS.
- L. Q. Xu, F. Yao, G.-D. Fu and L. Shen, Macromolecules, 2009, 42, 6385 CrossRef CAS.
- A. E. van der Ende, J. Harrell, V. Sathiyakumar, M. Meschievitz, J. Katz, K. Adcock and E. Harth, Macromolecules, 2010, 43, 5665 CrossRef CAS.
- R. A. Prasath, M. T. Gokmen, P. Espeel and F. E. Du Prez, Polym. Chem., 2010, 1, 685 RSC.
- D. Kim, E. Kim, J. Lee, S. Hong, W. Sung, N. Lim, C. G. Park and K. Kim, J. Am. Chem. Soc., 2010, 132, 9908 CrossRef CAS.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Mueller, Macromolecules, 2009, 42, 3707 CrossRef CAS.
- L. Nebhani, S. Sinnwell, A. J. Inglis, M. H. Stenzel, C. Barner-Kowollik and L. Barner, Macromol. Rapid Commun., 2008, 29, 1431 CrossRef CAS.
- G. L. Li, D. Wan, K. G. Neoh and E. T. Kang, Macromolecules, 2010, 43, 10275 CrossRef CAS.
- G. L. Li, L. Q. Xu, X. Tang, K. G. Neoh and E. T. Kang, Macromolecules, 2010, 43, 5797 CrossRef CAS.
- R. Ranjan and W. J. Brittain, Macromolecules, 2007, 40, 6217 CrossRef CAS.
- Y. Li and B. C. Benicewicz, Macromolecules, 2008, 41, 7986 CrossRef CAS.
- B. Korthals, M. C. Morant-Minana, M. Schmid and S. Mecking, Macromolecules, 2010, 43, 8071 CrossRef CAS.
Footnote |
| † Both authors contributed equally to this review. |
| This journal is © The Royal Society of Chemistry 2012 |