Activation of C–Cl by ground-state aluminum atoms: an EPR and DFT investigation
Helen A.
Joly
*,
Trevor
Newton†
and
Maxine
Myre
Department of Chemistry and Biochemistry, Laurentian University, Ramsey Lake Road, Sudbury, ON P3E 2C6, Canada. E-mail: hjoly@laurentian.ca; Fax: 705-675-4844; Tel: 705-675-1151 ext. 2333
First published on 16th November 2011
Abstract
The reaction of ground-state Al atoms with dichloromethane (CH2Cl2) in an adamantane matrix at 77 K yielded two mononuclear Al species. The magnetic parameters, extracted from the axial EPR spectrum of Species A/A′ (g1 = 2.0037, g2 = g3 = 2.0030, aAl,1 = 1307 MHz, aAl,2 = aAl,3 = 1273 MHz, a35Cl = 34 MHz and a37Cl = 28 MHz) were assigned to the Al-atom insertion product, ClCH2AlCl. Density functional theory (DFT) calculations of the values of the Al and Cl hyperfine interaction (hfi) of the Cl1–Cl2gauche conformer were in close agreement with the experimental values of ClCH2AlCl. The second species, B/B′, had identical magnetic parameters to those of ClCH2AlCl with the exception that the Al hfi was 15% smaller. Coordination of a ligand, possessing a lone pair of electrons, to the Al atom of the insertion product, [ClCH2AlCl]:X, could cause the aAl to decrease by 15%. Alternatively, it is possible that the Cl1–Cl2 anti conformer of ClCH2AlCl is also isolated in the matrix. Support for the spectral assignments is given by calculation of the nuclear hfi of [ClCH2AlCl]:H2O and the Cl1–Cl2 anti conformer of ClCH2AlCl using a DFT method. The potential energy hypersurface for an Al atom approaching CH2Cl2, calculated at the B3LYP level, suggests that Al atom abstraction of Cl forming AlCl and CH2Cl is favoured in the gas phase. When produced in a matrix, the close proximity of AlCl and CH2Cl could account for the formation of ClCH2AlCl. EPR evidence was also found for the formation of the CHCl2 radical.
Introduction
A strategy used to destroy man-made environmental pollutants, such as halogenated organic compounds, involves finding ways to activate the C–Cl or C–F bonds.1,2 Ground-state Al (2P1/2) atoms have been shown to activate a number of different types of bonds resulting in the formation of insertion products. Experiments involving NH3,3,4H2O,5 H2S, H2Se,6CH3OCH3,7,8 and CH49 yielded HAlNH2, HAlOH, HAlSH, HAlSeH, CH3AlOCH3 and CH3AlH, respectively, indicating that Al atoms can activate N–H, O–H, S–H, Se–H, C–O and C–H bonds. Al atoms have also been reported to activate the C–C bond of cyclohexanol,101-methylcyclohexanol10 and diethyl ether.8,11 With respect to halogenated compounds, co-condensation of Al atoms with HCl in an Ar matrix resulted in the formation of HAlCl12,13 at low concentration while increasing the HCl concentration to ∼8% yielded AlCl2. Finally, reaction of ground-state Al atoms with bromocyclopropane (CpBr) in adamantane at 77 K gave the two C–Br insertion products CpAl79Br and CpAl81Br as well as the allyl radical.14The above-mentioned Al-centered radicals were characterized by EPR spectroscopy. The magnitude of the isotropic Al hyperfine interactions (hfi) of the radicals is related to the nature of the ligands attached to Al, i.e., the Al hfi increases as the electron-withdrawing ability of the ligands increase.
Matrix-isolation infrared spectroscopy was used to study the reaction of Group 13 metal atoms (M) with halomethanes (CH3X).15,16 In the case of CH3Br, a weak CH3Br-M complex16 formed at low temperatures (<77 K) while higher temperatures favoured the formation of a “Grignard reagent”, CH3MBr, via the oxidative addition15,16 of CH3Br to the metal. Insertion into the C–Br bond is the preferred reaction because of the low first ionization potentials (IP) of the Group 13 metal atoms and the large Group 13 metal–Br bond energies.16 In a recent study17 involving laser–ablated Pd atoms, Cho et al. found that only insertion complexes, HCX2PdX formed in reactions with halomethanes containing H.
In the present study, the EPR investigation of the paramagnetic products resulting from the reaction of dichloromethane (CH2Cl2) with Al atoms, under matrix-isolation conditions, confirmed that C–Cl activation was possible. The spectral features are attributed to the Al-atom C–Cl insertion product, ClCH2AlCl. Annealing the sample to higher temperatures led to the detection of a product with spectral features similar to those of ClCH2AlCl with the exception of the magnitude of the Al hfi. The species is either ClCH2AlCl in another conformation or [ClCH2AlCl]:X where X is H2O. A comparison of the experimental values of the Al and Cl hfi to those calculated for ClCH2AlCl using density functional theory (DFT) supports the spectral assignments. A map of the potential energy hypersurface corresponding to the approach of an Al atom to CH2Cl2 indicated that in the gas phase formation of AlCl is barrierless. The combination of AlCl with CH2Cl gives the observed insertion product. Finally, CHCl2 dominates the center of the spectrum.
Experimental section
Preparation of Al atom–CH2Cl2 reaction mixtures
The CH2Cl2, CD2Cl2 and adamantane were the purest available commercial products (Aldrich) and were used as received after being subjected to a number of freeze-thaw cycles on a vacuum line.The method used to prepare the reaction mixture of ground-state Al atoms with CH2Cl2 or CD2Cl2, has been described in detail elsewhere.18Aluminum wire, (Alpha Products) in a tungsten basket (No. 12070, Ernest F. Fullam, Inc., Schenectady, NY) suspended between the electrodes of a furnace, was resistively heated to produce Al atoms. The Al atoms, CH2Cl2 or CD2Cl2 and adamantane were co-condensed on the surface of the rotating liquid nitrogen-filled drum situated in the metal-atom reactor. During the experiment (ca. 10 to 12 min) the reactor was maintained at <10−5 Torr. A sample of the reaction mixture was scraped into a suprasil quartz tube and sealed under vacuum while maintaining a temperature of 77 K. EPR analysis was carried out between 77 and 298 K on a Varian E line spectrometer operating an X-band. A Varian gaussmeter and a Systron-Donner 6016 frequency counter were used in obtaining calibrated spectra. The magnetic parameters of the paramagnetic products were determined using the computer programs, ESRLSQ,19 EPRNMR20 or ISOPLOT.19
Computational methods
(i) Geometry and nuclear hyperfine interaction (hfi)
The Gaussian 0921 suite of programs was used to calculate the values of the Al, H and 35Cl hfi for ClCH2AlCl. Full geometry optimization of ClCH2AlCl was carried out at the correlated ab initio (QCISD method) and density functional levels of theory (B3LYP and B3P86). In the QCISD method22 the electron correlation was included through configuration interaction. The B3LYP23 functional combines Becke's three parameter exchange functional24 with Lee, Yang and Parr's25 correlation functional which includes both local and non-local terms. B3P86 combines the exchange functional mentioned above with the non-local correlation introduced by Perdew.26 The split valence 6-31G(df,p) basis set was used in the optimization of the insertion radical's geometry. In an exploratory investigation of the ClCH2AlCl radical the 6-31G(df,p) basis set gave values for the Al hfi that agreed more closely with the experimental values than the basis set most often used in these types of calculations, namely the 6-31G(d,p).10,13 The radical was characterized using frequency analysis. The Al, H and 35Cl nuclear hfi values were determined from single point calculations using the following methods: B3LYP/6-311+G(2df,p), BHandHLYP/6-311+G(2df,p), BHandHLYP25,27,28/6-311G(d,p), and mPWP8626,29,30/IGLO-III.31(ii) Born–Oppenheimer potential energy surface
The Born–Oppenheimer potential energy surface for the lowest-energy doublet was calculated at the B3LYP level with a 6-31G(df,p) basis set by optimizing the geometry of the radical with fixed C–Cl1 and C–Al distances. We chose to investigate the energy cross-section over the (C–Al, C–Cl1) plane using geometries in which the dihedral angle defined by Cl2, C, Al, and Cl1 was approximately 90°. In calculations where no constraint was placed on the structures, the dihedral angle defined by Cl2, C, Al, and Cl1 for the most stable geometry was 91.1°. The B3LYP functional was used so that a comparison could be made with results previously reported for a similar system, namely, the reaction of Al atoms with CpBr.14Results
EPR analysis of the products resulting from the reaction of Al atoms with CH2Cl2 revealed two overlapping sextets, labeled 35A/37A and 35B/37B (Fig. 1), in addition to a strong central feature (C). Five of the six transitions, characteristic of mononuclear Al (I = 5/2) radicals, are indicated for both 35A/37A and 35B/37B on the EPR spectrum recorded at 145 K. Close inspection of the Al transitions for each of 35A/37A and 35B/37B revealed superhfi consistent with the unpaired electron interacting with a nucleus with I = 3/2. Chlorine has two EPR active isotopes 35Cl (I = 3/2, μ = +0.82181βn, natural abundance = 75.53%)32 and 37Cl (I = 3/2, μ = +0.68407 βn, natural abundance = 24.47%).32 The high and low field features were simulated by assuming a mixture of 35A/37A, with the magnetic parameters g1 = 2.0037 ± 0.0003, g2 = g3 = 2.0030 ± 0.0003, a1(Al) = 1307 ± 1 MHz, a2(Al) = a3 (Al) = 1273 ± 1 MHz, a35Cl = 34 ± 1 MHz and a37Cl = 28 ± 1 MHz and 35B/37B with the magnetic parameters g1 = g2 = g3 = 2.0030 ± 0.0003, a1 (Al) = a2(Al) = a3 (Al) = 1086 ± 1 MHz, a35Cl = 34 ± 1 MHz and a37Cl = 28 ± 1 MHz. The a35Cl/a37Cl = μ35Cl/μ37Cl = 1.21 and the value of the 35A/37A and 35B/37B ratios used in the simulation was 3.09, i.e., the ratio of the natural abundance of 35Cl to 37Cl. The best simulation was obtained using a ratio of 35A to 35B of 13 to 1.![(a) Five of the six Al EPR transitions for the paramagnetic species (35A/37A and 35B/37B) formed in the reaction of Al atoms with CH2Cl2 in an adamantane matrix. The low- and high-field regions of the EPR spectrum were recorded at 145 K (9120 MHz, microwave power [m.p. = 2 mW]); (b) Simulation of the low- and high-field regions of the EPR spectrum assuming the magnetic parameters for Species 35A/37A and for Species 35B/37B found in the text.](/image/article/2012/CP/c1cp22398d/c1cp22398d-f1.gif) | ||
| Fig. 1 (a) Five of the six Al EPR transitions for the paramagnetic species (35A/37A and 35B/37B) formed in the reaction of Al atoms with CH2Cl2 in an adamantane matrix. The low- and high-field regions of the EPR spectrum were recorded at 145 K (9120 MHz, microwave power [m.p. = 2 mW]); (b) Simulation of the low- and high-field regions of the EPR spectrum assuming the magnetic parameters for Species 35A/37A and for Species 35B/37B found in the text. | ||
The EPR spectrum of the major product, Species C, recorded at 195 K, shown in Fig. 2, was simulated using the magnetic parameters g = 2.011, aH(1) = 47.6 MHz and aCl(2) = 11.2 MHz.
 | ||
| Fig. 2 (a) The central region of the EPR spectrum of the Al–CH2Cl2 reaction mixture recorded at 195 K (9120 MHz, m.p. = 20 mW). (b) Simulation of the central feature assuming g = 2.011, aH(1) = 47.6 MHz, and aCl(2) = 11.2 MHz. | ||
Mononuclear Al radicals with magnetic parameters similar to those of Species 35A/37A and 35B/37B were found for the reaction of Al atoms with CD2Cl2. A comparison of the Al transitions of 35A/37A, Fig. 3a, and 35B/37B, Fig. 3c, with those obtained in the Al atom–CD2Cl2 reaction (Fig. 3b and d) did not show noticeable narrowing indicating that the unpaired electron on Al does not interact with H atoms in the CH2Cl2. The EPR spectrum of the central feature of the Al atom–CD2Cl2 reaction was simulated with the magnetic parameters g = 2.008, aD(1) = 7.3 MHz and aCl(2) = 11.2 MHz, Fig. 4. It should be noted that the aD was estimated by dividing the value of aH by 6.5, i.e., the ratio of the magnetogyric ratios for the H and D nuclei.32
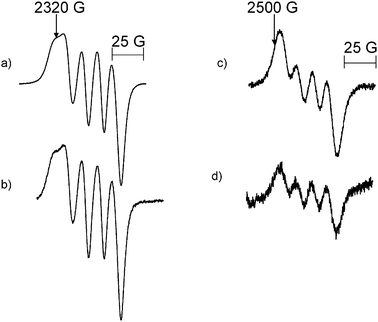 | ||
| Fig. 3 The MI = 1/2 transition of species 35A/37A formed in (a) the reaction of Al atoms with CH2Cl2 (T = 115 K, m.p. = 20 mW and ν = 9120 MHz) (b) the reaction of Al atoms with CD2Cl2 (T = 115 K, m.p. = 20 mW and ν = 9121 MHz). The MI = 1/2 transition of species 35B/37B formed in (c) the reaction of Al atoms with CH2Cl2 (T = 115 K, m.p. = 20 mW and ν = 9120 MHz) (d) the reaction of Al atoms with CD2Cl2 (T = 115 K, m.p. = 20 mW and ν = 9120 MHz). | ||
 | ||
| Fig. 4 (a) The central region of the EPR spectrum of the Al- CD2Cl2 reaction mixture recorded at 195 K (9119 MHz, m.p. = 20 mW). (b) Simulation of the central feature assuming g = 2.008, aD(1) = 7.3 MHz and aCl(2) = 11.2 MHz. | ||
Computational results
We have been able to determine two stationary points for ClCH2AlCl, namely, the Cl1–Cl2gauche and Cl1–Cl2 anti conformers. The geometry of these conformers was optimized using three different levels of theory, Table 1. The DFT calculations indicate that the Cl1–Cl2gauche conformer is slightly more stable than the anti conformer in the case of the B3LYP (0.7 kcal/mole) and the B3P86 (1.1 kcal/mole) calculations while the Cl1–Cl2 anti conformer is more stable in the QCISD calculations (0.8 kcal/mole). These small energy differences suggest that both of the two conformers are equally probable.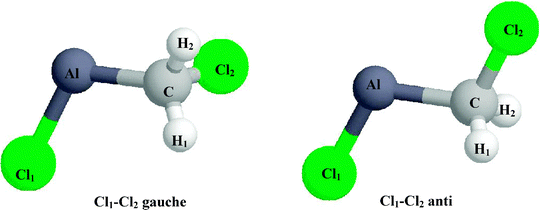
| QCISD/6-31G(df,p) | B3LYP/6-31G(df,p) | B3P86/6-31G(df,p) | QCISD/6-31G(df,p) | B3LYP/6-31G(df,p) | B3P86/6-31G(df,p) | |
|---|---|---|---|---|---|---|
| Cl1–Al (Å) | 2.099 | 2.134 | 2.124 | 2.098 | 2.139 | 2.126 |
| Al–C (Å) | 1.962 | 1.990 | 1.973 | 1.985 | 2.005 | 1.993 |
| C–Cl2 (Å) | 1.845 | 1.847 | 1.844 | 1.788 | 1.815 | 1.799 |
| C–H1 (Å) | 1.085 | 1.090 | 1.088 | 1.091 | 1.093 | 1.093 |
| C–H2 (Å) | 1.086 | 1.089 | 1.089 | 1.091 | 1.093 | 1.093 |
| ∠AlCCl2 (°) | 82.4 | 94.3 | 87.0 | 111.2 | 111.5 | 110.7 |
| ∠AlCH1 (°) | 121.2 | 117.2 | 120.1 | 111.1 | 111.0 | 111.2 |
| ∠AlCH2 (°) | 121.1 | 118.0 | 119.5 | 111.1 | 111.0 | 111.2 |
| ∠Cl1AlC (°) | 117.7 | 116.9 | 116.7 | 115.9 | 114.2 | 114.5 |
| D(Cl2CAlCl1) (°) | −93.0 | −91.1 | −91.9 | 180.0 | 180.0 | 180.0 |
| D(H2CAlCl1) (°) | 161.7 | 157.1 | 160.2 | 59.7 | 60.1 | 60.1 |
| D(H1CAlCl1) (°) | 12.5 | 20.8 | 16.2 | −59.7 | −60.1 | −60.1 |
| Energy (hartrees) | −1199.954657 | −1202.177273 | −1203.202415 | −1199.955855 | −1202.176135 | −1203.200598 |
Single point calculations, using the computational methods listed in column 1 of Table 2 were used to determine the nuclear hfis of the gauche and anti conformers of ClCH2AlCl. In the case of the Cl1–Cl2gauche conformer the variation in the Al and Cl hfi is ≈6–7% and ≈10–12%, respectively, for all four methods tested. Similarly, the variation in the Al and Cl hfi is ≈2–3% and ≈9–10%, respectively, for the Cl1–Cl2 anti conformer. This indicates that the effect on the value of the Al hfi of using different levels of theory to optimize the geometry of the conformers is relatively small. In comparing the results for the two conformers optimized at the same level of theory we note that the Al and Cl hfi are larger for the Cl1–Cl2gauche conformer in all cases. The increase in the value of the Al hfi in going from the anti to the gauche conformer is between 12–14%, 4–6% and 9–11% for the geometries optimized using the QCISD, B3LYP and B3P86 levels of theory, respectively, while the value of the Cl hfi increases by 8–13%, 7–12% and 8–14%, respectively. Although the variation in the H hfi is large for both conformers, it is interesting to note that (a) the hfi for H1 and H2 are approximately the same for the anti conformer while for the gauche conformer the H1 hfi is larger than that of H2; (b) the H1 hfi of the anti conformer (1.3–5.6 MHz) is significantly smaller than that of the gauche conformer (10.5–16.1 MHz).
| Geometry | ||||
|---|---|---|---|---|
| Method | Nuclei | QCISD/6-31G(df,p) | B3LYP/6-31G(df,p) | B3P86/6-31G(df,p) |
| (a) Cl1–Cl2gauche conformation | ||||
| B3LYP/6-311+G(2df,p) | Al | 1264.45 | 1182.53 | 1241.90 |
| Cl | 37.42 | 33.30 | 34.66 | |
| H1, H2 | 11.31, −5.80 | 14.43, −7.85 | 12.52, −6.76 | |
| BHandHLYP/6-311+G(2df,p) | Al | 1298.68 | 1223.70 | 1280.22 |
| Cl | 39.95 | 36.12 | 37.38 | |
| H1, H2 | 10.51, −6.68 | 12.92, −9.22 | 11.43, −7.87 | |
| BHandHLYP/6-311G(d,p) | Al | 1311.45 | 1234.80 | 1291.81 |
| Cl | 39.45 | 35.64 | 36.84 | |
| H1, H2 | 11.21, −6.61 | 13.55, −9.22 | 12.09, −7.84 | |
| mPWP86/IGLO-III | Al | 1231.43 | 1145.07 | 1206.16 |
| Cl | 34.15 | 30.22 | 31.53 | |
| H1, H2 | 12.24, −4.80 | 16.10, −6.49 | 13.7, −5.59 | |
| (b) Cl1–Cl2 anti conformation | ||||
| B3LYP/6-311+G(2df,p) | Al | 1099.25 | 1124.90 | 1117.31 |
| Cl | 33.57 | 30.24 | 30.91 | |
| H1, H2 | 3.11, 3.10 | 3.58, 3.58 | 3.31, 3.32 | |
| BHandHLYP/6-311+G(2df,p) | Al | 1146.41 | 1177.04 | 1168.04 |
| Cl | 36.94 | 33.70 | 34.38 | |
| H1, H2 | 1.31, 1.30 | 1.58, 1.58 | 1.41, 1.42 | |
| BHandHLYP/6-311G(d,p) | Al | 1155.39 | 1186.72 | 1177.11 |
| Cl | 36.47 | 33.18 | 33.84 | |
| H1, H2 | 1.32, 1.31 | 1.60, 1.60 | 1.43, 1.43 | |
| mPWP86/IGLO-III | Al | 1059.09 | 1079.14 | 1073.69 |
| Cl | 29.69 | 26.58 | 27.2 | |
| H1, H2 | 4.92, 4.92 | 5.55, 5.54 | 5.18, 5.19 | |
Discussion
Spectrum 35A/37A
Spectrum 35A/37A can be described as two overlapping sextets of quartets corresponding to two mononuclear Al compounds containing 35Cl and 37Cl, respectively. The isotropic Al hfi, of 35A/37A, calculated from the equation (Aiso = (a1+a2+a3)/3),32 is 1284 MHz. A comparison of this value to those reported in the literature for divalent Al radicals, Table 3, suggests that the C–Cl insertion products, ClCH2Al35Cl and ClCH2Al37Cl have generated Spectrum35A/37A. More specifically, the value of Aiso for ClCH2AlCl falls between that of HAlCl and ClAlCl. This is consistent with the observation33 that the magnitude of the Al hfi increases as the electronegativity (or the electron-withdrawing ability) of the ligands increases. This is obvious in comparing the Aiso for HAlH, HAlCl and ClAlCl. Replacing H by Cl in going from HAlH to HAlCl causes the Al hfi to increase by 25% and by 30% in going from HAlCl to ClAlCl. As the ligands become more electron-withdrawing the ionic character of the σ-bonds to Al increases. This is translated to spx hybrid orbitals with greater p-character and a semi-occupied molecular orbital (SOMO) with higher s character. Therefore we would expect Aiso for ClCH2AlCl to be greater than that of HAlCl but less than that of ClAlCl because the electron-withdrawing ability of ClCH2 is in between that of H and Cl.| Radical | a Al | a H | a X | ρ 3s | Ref. |
|---|---|---|---|---|---|
| CH3AlH | 772 | 152 | — | 0.20 | 9 |
| HAlH | 834 | 128 | — | 0.21 | 33 |
| HAlOH | 911 | 286 | — | 0.23 | 5, 6 |
| HOAlOH | 1220 | — | — | 0.31 | 33 |
| HAlSH | 984 | 210 | — | 0.25 | 6 |
| HAlSeH | 941 | 134 | 20 (H) | 0.24 | 6 |
| HAlCl | 1115 | 279 | 31 (Cl) | 0.29 | 12, 13 |
| ClAlCl | 1598 | — | 31 (Cl) | 0.41 | 12, 13 |
| ClCH2AlCl | 1284 | — | 34 (Cl) | 0.33 | This work |
| HAlNH2 | 923 | 229 | 27 (N) | 0.24 | 3 |
| 27 (H) | |||||
| CH3AlOCH3 | 1002 | — | — | 0.26 | 8 |
| CpAl79Br | 1041 | — | 176 (79Br) | 0.27 | 14 |
| CpAl81Br | 1042 | — | 190 (81Br) | 0.27 | 14 |
The Al 3s-spin contribution, ρ3s, to the SOMO, estimated by dividing the Aiso values for the divalent Al radicals by the one-electron atomic parameter for an Al 3s (3911 MHz)32 orbital, has been included in Table 3. While the values range from 0.20–0.41, the ρ3s for ClCH2AlCl is 0.33 indicating that the σ-bonds involving Al are slightly more ionic than those of HAlCl.
DFT methods have been very useful in predicting the Al hfi of a number of divalent organoaluminium radicals.14,34,35 Many different density functionals are available. It is not obvious which are the best suited for use in the DFT calculation of nuclear hfi. One of the main factors influencing the selection of a functional is its reported effectiveness in predicting the nuclear hfi of well-characterized systems.36 We have had success in predicting the Al hfi of several divalent Al compounds by optimizing their geometries with the split valence 6-31(d,p) basis set and determining the nuclear hfi with the B3LYP functional and the 6-311+G(2df,p) basis set.35 This method predicted values that deviated from the experimental values by no more that 8%. In a theoretical study carried out by Fängström et al.13 on the Al–atom insertion products of HCl and Cl2, similar agreement was obtained between experiment and theory at the B3LYP/6-311+G(2df,p)//6-31G(d,p) and mPWP86/IGLO-III//QCISD/6-31G(d,p) levels. The Becke half and half functional (BHandH)27,28 with the nonlocal correlation of Lee, Yang and Parr25 (LYP) has been found to be effective in calculating the geometries and nuclear hfi of open shell systems.37 Consequently, the nuclear hfi of the two conformers of ClCH2AlCl were calculated at the B3LYP/6-311+G(2df,p), BHandHLYP/6-311+G(2df,p), BHandHLYP/6-311G(d,p) and mPWP86/IGLO-III levels of theory, Table 2.
In general, the deviation between the experimental Al (1284 MHz) and 35Cl (34 MHz) hfi and the calculated values is smaller for the gauche conformer; the Al and 35Cl hfi deviate from the experimental values by 0.2–11% and 1–18%, respectively for the gauche conformer and by 8–17% and 1–22% for the anti conformer. The Al and 35Cl hfi values calculated at the B3LYP/6-311+G(2df,p)//B3P86/6-31G(df,p) level of theory for the gauche isomer agree the best with the experimental values; i.e., the deviation between the calculated and experimental values is 3% for the Al hfi and 2% for the Cl hfi. This is well within the expected error for these types of calculations.14,34,35 At the same level of theory, the Al and Cl hfi calculated for the anti conformer differ from experiment by 13% and 9%, respectively. Experimentally the H hfi is too small to detect. The variation in the H hfi of the gauche conformer predicted by the different theoretical models is large making it difficult to draw any definitive conclusions.
Spectrum 35B/37B
The spectral pattern labeled 35B/37B in Fig. 1 is identical to that of 35A/37A, i.e., a sextet of quartets. As in the case of 35A/37A, one can deduce that the unpaired electron is interacting with an Al (I = 5/2) and a 35/37Cl (I = 3/2) nucleus. The Al hfi (1086 MHz) extracted from the 35B/37B spectrum is 15% smaller than that of ClCH2AlCl while the 35Cl and 37Cl hfi are identical, i.e., a35Cl = 34 MHz and a37Cl = 28 MHz.As previously mentioned, the difference in the calculated energy for the Cl1–Cl2gauche versus the anti conformer is small suggesting that both conformers are equally possible. In addition, the theoretical methods predict a decrease of up to 14% in the Al hfi in going from the gauche to the anti conformer. This is close to the 15% difference in Al hfi found between the Al hfi of Species 35A/37A and 35B/37B. In a study involving the reaction of Al atoms with dimethyl ether, Kasai7 found that both the cis and trans conformers of CH3OAlCH3 were formed. In this case the Al hfi for the cis conformer was greater than that for the trans conformer by 14%. Although theory predicted only a marginal difference in stability, the trans conformer was more abundant indicating that it was in fact more stable. 35B/37B could therefore be the Cl1–Cl2 anti conformer of ClCH2AlCl. The fact that both the gauche and anti conformers are observed on the EPR time scale would suggest that the barrier for the interconversion of the two conformers is relatively high.
Alternately, a decrease in the Al hfi could be due to the coordination of the Al atom of ClCH2AlCl with a molecule containing a lone pair of electrons. For instance, in the reactions of Al atoms with NH3 in Ar at 4 K,4cyclopropylamine in adamantane at 77 K,35 and methoxymethane in Ar at 4 K7 radicals producing identical spectral patterns differing only in the magnitude of the values of the nuclear hfi were detected. This phenomenon was attributed to complexation of a vacant coordination site of the divalent Al insertion products by a molecule containing a lone pair of electrons, Table 4. Coordination of NH3 to HAlNH2 results in the 18% decrease in the Al hfi.4 The H hfi is estimated to decrease by 55% while the N hfi increases by about 4%. For [CpNH(AlH)]:CpNH2, the Al, H and N hfi are 17, 38 and 28% smaller than that found for CpNH(AlH).35 Kasai suggested that the radical with an Al hfi that was 4% smaller than that of CH3AlOCH3 was the complexed insertion product [CH3AlOCH3]:O(CH3)2. This suggests that 35B /37B could be a mixture of [ClCH2Al35Cl]:X and [ClCH2Al37Cl]:X where X is a molecule with a lone pair of electrons. X could be H2O from adventitious water present in the cryostat. This possibility was tested by carrying out an exploratory DFT calculation. The geometry of the [ClCH2AlCl]:OH2 complex was optimized using the B3LYP and B3P86 functionals and the 6-31G(df,p) basis set, Table 5. The Al and Cl hfi were obtained from a single point calculation using the B3LYP/6-311+G(2df,p), BHandHLYP/6-311+G(2df,p) and BHandHLYP/6-311G(d,p) levels of theory, Table 6. The Al hfi and Cl hfi for [ClCH2AlCl]:OH2 differed from those of the gauche conformation of ClCH2AlCl by 10–16% and 15–19%, respectively, at the same level of theory (Table 2 and 6). The calculation shows that complexation of a water molecule to the Al atom of ClCH2AlCl causes the Al hfi to decrease by approximately the same amount as that observed between 35A/37A and 35B/37B.
| Nucleus | HAlNH2a | [HAlNH2]:NH3a | %Δ in nuclear hfi |
|---|---|---|---|
| a Ref. 4. b Ref. 35. c Ref. 7. d This work. | |||
| Al | 950 | 779 | −18% |
| H1 | 213 | 95 | −55% |
| H2 | 27 | 28 | +4% |
| N | 27 | 28 | +4% |
| CpNH(AlH) b | [CpNH(AlH)]:CpNH2b | %Δ in nuclear hfi | |
| Al | 962 | 801 | −17% |
| H | 216 | 133 | −38% |
| N | 32 | 23 | −28% |
| CH3OAlCH3c | [CH3OAlCH3]:O(CH3)2c | %Δ in nuclear hfi | |
| Al | 873 | 838 | −4% |
| ClCH2AlCl d | [ClCH2AlCl]:Xd | %Δ in nuclear hfi | |
| Al | 1284 | 1086 | −15% |
| 35Cl | 34 | 34 | 0% |

| B3LYP/6-31G(df,p) | B3P86/6-31G(df,p) | |
|---|---|---|
| Cl1–Al (Å) | 2.185 | 2.174 |
| Al–C (Å) | 1.998 | 1.987 |
| C–Cl2 (Å) | 1.831 | 1.814 |
| C–H1 (Å) | 1.093 | 1.093 |
| C–H2 (Å) | 1.092 | 1.092 |
| O–Al (Å) | 2.065 | 2.050 |
| ∠AlCCl2 (°) | 107.1 | 105.5 |
| ∠AlCH1 (°) | 113.7 | 114.1 |
| ∠AlCH2 (°) | 114.2 | 114.5 |
| ∠Cl1AlC (°) | 114.2 | 113.9 |
| ∠OAlC (°) | 102.2 | 103.1 |
| D(Cl2CAlCl1) (°) | 84.0 | 83.9 |
| D(Cl2CAlO) (°) | −179.6 | −179.3 |
| D(H2CAlCl1) (°) | −158.6 | −159.0 |
| D(H1CAlCl1) (°) | −33.3 | −33.1 |
| Energy (hartrees) | −1278.630514 | −1279.835873 |
| Geometry | |||
|---|---|---|---|
| Method | Nuclei | B3LYP | B3P86 |
| B3LYP/6-311+G(2df,p) | Al | 1054.67 | 1044.43 |
| Cl | 28.47 | 28.79 | |
| H1 | 17.30 | 16.88 | |
| H2 | −5.90 | −5.71 | |
| BHandHLYP/6-311+G(2df,p) | Al | 1099.64 | 1087.93 |
| Cl | 29.98 | 30.21 | |
| H1 | 16.21 | 15.89 | |
| H2 | −6.70 | −6.45 | |
| BHandHLYP/6-311G(d,p) | Al | 1117.11 | 1104.95 |
| Cl | 29.89 | 30.14 | |
| H1 | 17.19 | 16.87 | |
| H2 | −6.81 | −6.57 | |
Spectrum C
The 8-lined spectrum, separated by 64 MHz, that dominates the central region at 77 K, disappears at temperatures >77 K leaving behind a doublet of septets with magnetic parameters, g = 2.011, aH = 47.6 MHz and a35Cl = 11.2 MHz, similar to those reported for CHCl2 produced by exposing CH2Cl2 to 60Co γ-rays, i.e., g = 2.007, aH = 56 MHz and a35Cl = 11 MHz.38 The difference in the aH may be due to the fact the spectra were recorded under different conditions. In fact, in another study39 involving the γ-radiolysis of CH2Cl2, slightly different magnetic parameters were obtained for CHCl2, i.e., g = 2.013, aH = 56 MHz and a35Cl = 18 MHz. The magnetic parameters are sensitive to the structure of the radical which can be distorted depending upon the nature of the matrix used in the experiment, i.e., adamantanevs.CH2Cl2.There is some suggestion that the formation of Al-atom insertion products is a multistep process. Interaction of the Al atoms with the substrate promotes bond cleavage and the resulting radicals recombine with the lone pair of electrons on the Al atom to form the insertion product. In the case of CH3AlOCH3, Kasai7 proposed that Al atoms interact with a lone pair of electrons on the oxygen atom of dimethyl ether. Next, the C–O bond cleaves producing methyl radicals. The unpaired electron of the methyl radical interacts with the lone pair electrons of the Al atom to form the insertion product, CH3AlOCH3.
As in the case of the Al atom activation of the C–Br bond in CpBr we decided to carry out a theoretical investigation of the mechanism of the Al–CH2Cl2 insertion reaction. The Al atom was made to approach the CH2Cl2 molecule. We chose to investigate the energy cross-section over the (C–Al, C–Cl1) plane using geometries in which the dihedral angle defined by Cl2, C, Al, and Cl1 was approximately 90°. The geometry of structures with fixed Al–C and C–Cl1 bond lengths were optimized at the B3LYP level with a 6-31G(d,p) basis set and a three dimensional plot of C–Al versus C–Cl1versus energy was constructed, Fig. 5. As in the case of the interaction of Al atoms and CpBr, the Al atom–CH2Cl2 reaction favours the formation of AlCl and CH2Cl. There is no low energy channel that leads directly to the gauche conformer. The energy profile suggests that its formation is barrierless. Similarly the decomposition of ClCH2AlCl follows a barrierless channel that leads to AlCl and CH2Cl. Presumably the recombination of AlCl and CH2Cl trapped in an adamantane matrix could lead to the formation of the insertion product, ClCH2AlCl.
 | ||
| Fig. 5 Born–Oppenheimer potential energy surface of the lowest energy doublet for the reaction of Al atoms with CH2Cl2. The contour lines represent the energy in kcal/mole and are spaced by +5 kcal/mole. The minimum energy point was set to 0 kcal/mole and is denoted by *. The other minima (for AlCl + ClCH2 and Al + CH2Cl2) are asymptotic and hence not indicated on the diagram. | ||
We did not detect the CH2Cl radical; however this may have something to do with its stability. In the study39 involving the γ-radiolysis of CH2Cl2 at 77 K, the resulting EPR spectrum was attributed to a mixture of CH2Cl and CHCl2 in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. From this we could speculate that the CH2Cl may have formed in our case but in concentrations too low to detect.
:3. From this we could speculate that the CH2Cl may have formed in our case but in concentrations too low to detect.
Conclusions
Two main radicals were detected in the Al–CH2Cl2 reaction, namely, ClCH2AlCl and CHCl2. The large isotropic Al hfi for ClCH2AlCl falls between those reported for HAlCl and ClAlCl supporting the hypothesis that the Al hfi increases as the electron-withdrawing ability of the ligands increases. At higher annealing temperatures, a weak spectrum, identical to that of ClCH2AlCl, with the exception of the Al hfi, was observed. The smaller Al hfi led us to attribute the spectrum to either the anti conformer of ClCH2AlCl or to ClCH2AlCl:X where X possesses a lone pair of electrons, e.g., as in the case of H2O. The nuclear hfi calculated using a DFT method support the assignments. Exploration of a cross section of the potential energy surface for the Al–CH2Cl2 reaction shows that AlCl and CH2Cl are favoured. The recombination of AlCl and CH2Cl, trapped in an adamantane matrix, could lead to the formation of the insertion product, ClCH2AlCl.Acknowledgements
The Natural Sciences and Engineering Research Council of Canada (NSERCC) and Laurentian University are gratefully acknowledged for financial support. We would like to thank Ms Julie Feola and Mr Jean Pierre Rank for their technical assistance. We express our gratitude to Drs Gustavo Arteca, Sabine Montaut and Tony Howard for helpful discussions. The authors also wish to thank the reviewers for comments as they helped improve the paper.References
- C. L. Geiger, K. Carvalho-Knighton, S. Novaes-Card, P. Maloney and R. DeVor, in Environmental Applications of Nanoscale and Microscale Reactive Metal Particles, ACS Symp. Ser., 2009, vol. 1027, p. 1 Search PubMed.
- C. Douvris, C. M. Nagaraja, C.-H. Chen, B. M. Foxman and O. V. Ozerov, J. Am. Chem. Soc., 2010, 132, 4946 CrossRef CAS.
- J. A. Howard, H. A. Joly, P. P. Edwards, R. J. Singer and D. E. Logan, J. Am. Chem. Soc., 1992, 114, 474 CrossRef CAS.
- P. H. Kasai and H.-J. Himmel, J. Phys. Chem. A, 2002, 106, 6765 CrossRef CAS.
- L. B. Knight, Jr., B. Gregory, J. Cleveland and C. A. Arrington, Chem. Phys. Lett., 1993, 204, 168 CrossRef.
- H. A. Joly, J. A. Howard, M. Tomietto and J. S. Tse, J. Chem. Soc., Faraday Trans., 1994, 90, 3145 RSC.
- P. H. Kasai, J. Phys. Chem. A, 2002, 106, 83 CrossRef CAS.
- J. H. B. Chenier, J. A. Howard, H. A. Joly, M. LeDuc and B. Mile, J. Chem. Soc., Faraday Trans., 1990, 86, 3321 RSC.
- J. M. Parnis and G. A. Ozin, J. Phys. Chem., 1989, 93, 1204 CrossRef CAS.
- H. A. Joly, J. A. Howard and G. A. Arteca, Phys. Chem. Chem. Phys., 2001, 3, 760 RSC.
- J. A. Howard, H. A. Joly and B. Mile, J. Chem. Soc., Faraday Trans., 1990, 86, 219 RSC.
- R. Köppe and P. H. Kasai, J. Am. Chem. Soc., 1996, 118, 135 CrossRef.
- T. Fängström, S. Lunell, P. H. Kasai and L. A. Eriksson, J. Phys. Chem. A, 1998, 102, 1005 CrossRef.
- H. A. Joly, M. Y. Levesque, F. Koudra and J. P. Rank, Chem. Phys. Lett., 2006, 420, 140 CrossRef CAS.
- P. S. Skell and J. E. Girard, J. Am. Chem. Soc., 1972, 94, 5518 CrossRef CAS.
- B. S. Ault, J. Am. Chem. Soc., 1980, 102, 3480 CrossRef CAS.
- H.-G. Cho, L. Andrews, B. Vlaisavljevich and L. Gagliardi, Organometallics, 2009, 28, 6871 CrossRef CAS.
- A. J. Buck, B. Mile and J. A. Howard, J. Am. Chem. Soc., 1983, 105, 3381 CrossRef CAS.
- K. F. Preston and J. R. Morton, ISOPLOT and ESRLSQ, National Research Council of Canada, Ottawa Search PubMed.
- J. A. Weils, EPR-NMR, University of Saskatchewan, Saskatoon Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.1), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. A. Pople, M. Head-Gordon and K. Raghavachari, J. Chem. Phys., 1987, 87, 5968 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- J. P. Perdew and W. Yue, Phys. Rev. B, 1986, 33, 8800 CrossRef.
- J. P. Perdew, Phys. Rev. B, 1986, 34, 7406 CrossRef.
- W. Kutzelnigg, U. Fleischer and M. Schindler, NMR: Basic principles and Progress, Springer-Verlag, 1990 Search PubMed.
- W. Weltner, Jr., Magnetic Atoms and Molecules, Dover, 1989 Search PubMed.
- L. B. Knight, Jr., J. R. Woodward, T. J. Kirk and C. A. Arrington, J. Phys. Chem., 1993, 97, 1304 CrossRef.
- H. A. Joly, J. A. Howard and G. A. Arteca, Phys. Chem. Chem. Phys., 2001, 3, 750 RSC.
- H. Joly, J. Ashley, M. Levesque and J. Rank, J. Phys. Chem. A, 2006, 110, 3911 CrossRef CAS.
- H. M. Tuononen, T. Chivers, A. Armstrong, C. Fedorchuk and R. T. Boeré, J. Organomet. Chem., 2007, 692, 2705 CrossRef CAS.
- J.-F. Xiao, Z.-S. Li, Y.-H. Ding, J.-Y. Liu, X.-R. Huang and C.-C. Sun, J. Phys. Chem. A, 2001, 106, 320 CrossRef.
- S. P. Mishra, G. W. Neilson and M. C. R. Symons, J. Chem. Soc., Faraday Trans. 2, 1973, 69, 1425 RSC.
- S. Truszkowski and W. Szymański, J. Radioanal. Nucl. Chem., 1994, 177, 415 CrossRef CAS.
Footnote |
| † Present address: Gowlings, Ottawa, ON, K1P 1C3, Canada. |
| This journal is © the Owner Societies 2012 |