DOI:
10.1039/C1CE05837A
(Paper)
CrystEngComm, 2012,
14, 116-123
Recurrent H-bond graph motifs between metal tris-ethylenediamine cations and uncoordinated oxalate anions: Fitting a three pin plug into a two pin socket†
Received
4th July 2011
, Accepted 3rd October 2011
First published on 13th October 2011
Abstract
The structures of four new compositionally related compounds are described; [Cu(en)2(H2O)2][ox] (1), [Cu(en)3][ox] (2), [Co(en)3]2[ox]3(H2O)3.61 (3), and [Co(en3)]2[ox]3(H2O)7 (4) [en = 1,2-ethylenediamine, ox = oxalate]. These materials all inhabit the broader structural landscape for compounds with a generic Mp(ox)q(en)r(H2O)s composition. Here the competing nature of the ligands; ethylenediamine, oxalate and water, results in complex solution chemistry. In addition the very different structure directing effects of each type of ligand yield a range of crystal architectures. In the present cases ethylenediamine displaces the oxalate dianion, which is non-coordinated in each of these compounds. Compound 3 has disordered water of crystallisation, and is a non-stiochoimetric hydrate, while compound 4 shows a correlated disorder in both ligand conformation and water site occupancies. The H-bond motifs linking pseudo 3-fold symmetric M(en)3n+ cations to pseudo 2-fold symmetric ox2− anions shows frequent occurrence of particular motifs; notably the R22(8) and R22(9) graphs and a tendency to form bifurcated hydrogen bonds. The oxalate geometric parameters of twist and C–C bond length in our compounds are correlated with data from related structures in the CSD.
Introduction
Certain organic functional groups have a strong predilection for particular packing arrangements in condensed phases. This is particularly evident in hydrogen bonded materials, where the particular geometric interactions can be characterised by graphset descriptors. For example the carboxylic acids tend to dimerise forming a cyclic structure which is labelled as a R22(6) motif.1 Such a predictable local interaction can be used to generate predictable extended features in a structure; thus 3,3′5,5′-biphenyl tetracarboxylic acid gives an entirely predictable 4-connected 2-D network structure.2 In addition knowledge about the relative strengths of competing motifs can be used to induce useful physical properties such as the induction of liquid crystalline behaviour.3–5Hydrogen bonding is not restricted to purely organic molecules, it can be important whenever it is found, and it certainly has important structure directing effects in many ionic molecular materials. Much like the degree of predictability seen in some hydrogen bonded systems, some areas of coordination chemistry also share an element of good structural predictability, notably coordination complexes and networks built from the dianionic oxalate ligand. We have been studying transition metal oxalate coordination chemistry for some time, and have found the addition of organic amines greatly complicates the solution chemistry. In such a system containing, a metal cation, the oxalate dianion, an amine and water, a large number of distinct molecular complexes can be formed. The solution speciation can be controlled by adjusting the proportions of the constituents directly, or through adjustment of the solution pH. The pKa of oxH2 and oxH−, and AH+ [ox = oxalate, A = amine] are very different, yet all accessible in aqueous solution. At low pH, a quaternary ammonium cation can be used to “template” a (Mp(ox)qm−)n anionic framework.6 With diamines increasing pH results first in monoprotonation and the unprotonated nitrogen is free to act as a monodentate ligand.7 Further increase in pH promotes the tendency of the free amine to coordinate as a ligand. However at even higher pH the coordination chemistry of the hydroxyl anion becomes much more important, and we find examples containing Mn+, ox2− and amine, such as Ni7(OH)8(ox)3(pip)38 and Co12(OH)18(ox)3(pip)9 (pip = piperazine).
Results
In water, transition metal oxalates typically have very low solubilities, a fact that is related to the bridging capacity of the oxalate ligand and its ability to form extended network structures. However transition metal oxalates are relatively soluble in many organic amines. These more strongly coordinating ligands can displace some or all of the oxalate from a metal complex. The change in solubility is a reflection of the changes in solution speciation, and can result in the formation of new phases containing the amine moiety.10,11 Dissolving copper(II) oxalate followed by slow evaporation allowed us to obtain crystals of Cu(ox)(en)2(H2O)2 (1) and Cu(ox)(en)3 (2); while a similar dissolution of cobalt(II) oxalate and subsequent crystallisation, yielded Co2(ox)3(en)6(H2O)3.61 (3) and Co2(ox)3(en)6(H2O)7 (4) (where en = 1,2-ethylenediamine). Compounds 1 and 2 are known12–14 but have not previously been structurally characterised.
Crystal structures
Compound 1 crystallises in the triclinic space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the molecular structure consists of a Cu(II) ion, situated on an inversion centre, chelated by two molecules of ethylenediamine and two water molecules in a trans-configuration (Fig. 1). Within the [Cu(en)2(H2O)2]2+ molecule, inversion symmetry at the metal dictates that one ethylenediamine has a δ conformation,15 and the other is λ. The coordination sphere of the Cu atom is approximately octahedral with a tetragonally elongated Jahn–Teller distortion. Thus the two chelating ethylenediamine ligands provide four short Cu–N contacts (1.997(3) and 2.032(5) Å), while the two water molecules occupy the very much longer axial site; Cu–O 2.599(4) Å. The oxalate anion also lies on a crystallographic inversion centre. There is some asymmetry in the carboxylate group, with C–O distances of 1.257(1) and 1.244(3) Å (Δ = 0.013(3) Å). All four N–H and two O–H potential H-bond donors in the crystallographic asymmetric unit are involved in hydrogen bonding. There is a pronounced 2-D character to this material, if we consider the shortest interactions we see that each oxalate is hydrogen bonded to four [Cu(en)2(H2O)2]2+ cations and vice-versa, giving layers in the bc-plane (Fig. 2). Within such layers we see a number of complex H-bonded ring and chain motifs. If we examine only O–H⋯O interactions we see a chain motif constructed from an alternation of a number of binary cyclic graphs; R21(5), R24(8), R21(5) running along the (010) direction. If we additionally consider N–H⋯O interactions we see two types of chain that comprise alternating cations and anions, linked by the R22(9) motif in the (011) direction and the R22(8) motif in the (01) direction. The weakest (and longest) interaction; N1–H1b⋯O1w is an interlayer interaction, and forms a stack of neighbouring cations along the a-axis. We note that H1w makes a bifurcated hydrogen bond to the oxalate.
and the molecular structure consists of a Cu(II) ion, situated on an inversion centre, chelated by two molecules of ethylenediamine and two water molecules in a trans-configuration (Fig. 1). Within the [Cu(en)2(H2O)2]2+ molecule, inversion symmetry at the metal dictates that one ethylenediamine has a δ conformation,15 and the other is λ. The coordination sphere of the Cu atom is approximately octahedral with a tetragonally elongated Jahn–Teller distortion. Thus the two chelating ethylenediamine ligands provide four short Cu–N contacts (1.997(3) and 2.032(5) Å), while the two water molecules occupy the very much longer axial site; Cu–O 2.599(4) Å. The oxalate anion also lies on a crystallographic inversion centre. There is some asymmetry in the carboxylate group, with C–O distances of 1.257(1) and 1.244(3) Å (Δ = 0.013(3) Å). All four N–H and two O–H potential H-bond donors in the crystallographic asymmetric unit are involved in hydrogen bonding. There is a pronounced 2-D character to this material, if we consider the shortest interactions we see that each oxalate is hydrogen bonded to four [Cu(en)2(H2O)2]2+ cations and vice-versa, giving layers in the bc-plane (Fig. 2). Within such layers we see a number of complex H-bonded ring and chain motifs. If we examine only O–H⋯O interactions we see a chain motif constructed from an alternation of a number of binary cyclic graphs; R21(5), R24(8), R21(5) running along the (010) direction. If we additionally consider N–H⋯O interactions we see two types of chain that comprise alternating cations and anions, linked by the R22(9) motif in the (011) direction and the R22(8) motif in the (01) direction. The weakest (and longest) interaction; N1–H1b⋯O1w is an interlayer interaction, and forms a stack of neighbouring cations along the a-axis. We note that H1w makes a bifurcated hydrogen bond to the oxalate.
 |
| | Fig. 1 Asymmetric unit, atom labelling scheme and selected symmetry equivalents of 1. Thermal ellipsoids at 70% probability. Symmetry operators: i) 1 − x, −y, −z; ii) 1 − x, 1 − y, 1 − z. | |
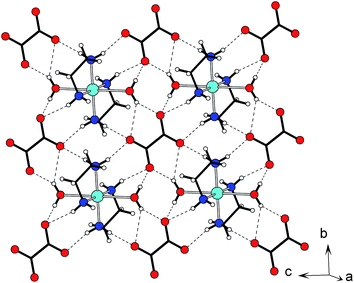 |
| | Fig. 2 The layered character of 1. Showing the hydrogen-bonded layer in the bc-plane. | |
The asymmetric unit of 2 consists of one Cu(II) ion chelated by three ethylenediamine molecules and one non-coordinated oxalate dianion (Fig. 3). In general tris-chelation of an octahedrally coordinated ion tends to result in a distortion to D3 symmetry on steric grounds. However the d9 configuration of Cu(II) provides the ion with a very strong tendency to show an electronically stabilized Jahn–Teller distortion, which is almost always observed as a tetragonal elongation of an octahedral coordination. While the [Cu(en)3]2+ cation in 2 lacks any symmetry, we note characteristics of both these distortions in the structure of this complex. Consideration of bond lengths shows the copper(II) ion to have an approximate tetragonal elongation, with four short Cu–N contacts between 2.032(4)–2.048(4) Å, and two long Cu–N contacts at 2.442(4) and 2.544(4) Å in the metal coordination sphere. The physical constraint imposed by the chelating ethylenediamine group results in a significant angular distortion with opposite groups giving trans N–Cu–N angles of 171.9, 170.2 and 159.1°. The three ethylenediamine ligands that chelate the Cu(II) ion give a pseudo 3-fold axis, which coincides with the crystallographic a-axis. The cation in the asymmetric unit is characterised by a chiral Δ(λλλ) ligand conformation.15 (The non-chiral space group, P21/c ensures that the crystal is comprised of equal numbers of Δ(λλλ) and Λ(δδδ) enantiomers). This particular conformation is the most commonly observed structure in crystalline materials of this type. The non-coordinated oxalate anion is twisted from a planar geometry with a torsion angle of 30.5(6)°. One carboxylate (C7) group is delocalised with equal distances between the carbon and oxygen atoms of 2.151(6) and 2.152(6) Å. The other carboxylate (C8) shows some localisation of the double bond with C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths of 1.251(6) and 1.232(6) Å respectively (Δ = 0.019(6) Å). In this ionic material hydrogen bonding is clearly very significant and we see N–H⋯O interactions where the N⋯O separation ranges from 2.895(6) to 3.436(5) Å. In fact all twelve potential N–H hydrogen bond donor groups are involved in hydrogen bonding. We note a clear bifurcated interaction for N6–H6a. However for N2–H2a although the geometry looks similar, any bifurcated interaction is much more asymmetric with the N2–H2a⋯O4 interaction being much shorter and more linear than the N2–H2a⋯O2 interaction. Looking at the molecular cation we note an approximate top/bottom symmetry along pseudo-3-fold axis, with three (axial) N–H bonds directed along this axis at each end of the molecule, and six N–H bonds with an equatorial disposition. Despite the lack of crystallographic symmetry, we see the same H-bond motif from the axial N–H groups at either end of the cation (Fig. 4), being comprised of R21(5), R12(6), R12(6), R22(9) binary graphs. The equatorial N–H bonds interact with three oxalate anions, and two of these show the R22(8) motif. Overall each oxalate anion is hydrogen bonded to five neighbouring [Cu(en)3]2+ cations, and vice-versa to form a 3-D network. We note that when viewed along the a-axis there are regions where alkyl groups are clustered to give “hydrophobic channels” (Fig. 5).
O bond lengths of 1.251(6) and 1.232(6) Å respectively (Δ = 0.019(6) Å). In this ionic material hydrogen bonding is clearly very significant and we see N–H⋯O interactions where the N⋯O separation ranges from 2.895(6) to 3.436(5) Å. In fact all twelve potential N–H hydrogen bond donor groups are involved in hydrogen bonding. We note a clear bifurcated interaction for N6–H6a. However for N2–H2a although the geometry looks similar, any bifurcated interaction is much more asymmetric with the N2–H2a⋯O4 interaction being much shorter and more linear than the N2–H2a⋯O2 interaction. Looking at the molecular cation we note an approximate top/bottom symmetry along pseudo-3-fold axis, with three (axial) N–H bonds directed along this axis at each end of the molecule, and six N–H bonds with an equatorial disposition. Despite the lack of crystallographic symmetry, we see the same H-bond motif from the axial N–H groups at either end of the cation (Fig. 4), being comprised of R21(5), R12(6), R12(6), R22(9) binary graphs. The equatorial N–H bonds interact with three oxalate anions, and two of these show the R22(8) motif. Overall each oxalate anion is hydrogen bonded to five neighbouring [Cu(en)3]2+ cations, and vice-versa to form a 3-D network. We note that when viewed along the a-axis there are regions where alkyl groups are clustered to give “hydrophobic channels” (Fig. 5).
 |
| | Fig. 3 Asymmetric unit of 2 and atom labelling scheme. Thermal ellipsoids are drawn at the 50% probability level. H atoms omitted for clarity. | |
 |
| | Fig. 4 Comparison of the molecular cation environments in 2, 3 and 4. Showing key hydrogen-bond interactions. | |
 |
| | Fig. 5
Hydrogen-bonded layer in 2. Showing the bc-plane and hydrophobic pockets. | |
Compound 3 crystallises in the triclinic space groupP and the asymmetric unit contains one [Co(en)3]3+ cation and 1.5 oxalate anions. There are also four sites which are assigned to water of crystallisation (Fig. 6). In this case, all three ethylenediamine molecules show the all-gauche conformation, with the crystal being composed of cations with the Δ(λλλ) and Λ(δδδ) conformations. The average Co–N distance is 1.967 Å. The two oxalate anions show different torsion angles, oxalate A (C7/C8) at 16.4° and oxalate B (C9/C9i) being 0°. The water of crystallisation is distributed over four general sites. A crystallographic inversion centre means that there is a cluster of eight nearby sites. Consideration of short inter-atomic distances means that we can discern a number of cases where the water site occupancy must be mutually exclusive; if O1w then not O3w, if O2w then not O4w, if O4w then not O4wii (ii = 1 − x, 1 − y, 1 − z). This still leaves seven possible patterns of occupancy, of which all (or any combination of) should, if maximally occupied, give a tetrahydrate stoichiometry; [Co(en)3]2[ox]3(H2O)4. A careful examination of the Fourier difference maps leads us to conclude that even with these mutually exclusive relationships, the site occupancies are significantly less than the maximum value. We refine occupancies; O1w, O2w, O3w and O4w of 52.6, 59.6, 26.6 and 41.6% respectively. This means that 3 is a non-stoichiometric hydrate with the formula [Co(en)3]2[ox]3(H2O)3.61. The disorder in the water sites is likely to be responsible for the large anisotropic displacement parameters associated with oxalate B. The hydrogen bonding between anions and cations give a complex 3-D network. Oxalate A links three cations into a ribbon structure, running along the b-axis (Fig. 7a). Oxalate B hydrogen bonds to four cations, forming a ribbon that runs along the a-axis (Fig. 7b). The same graph motifs are seen as found in the previous structures; most notably with oxalate A; R21(5), R22(9), R12(6), R12(6), R22(8), and R22(8); and oxalate B; R21(5), R12(6), R12(6), R22(9), R22(8) and R24(8). The disorder in the water molecule positions makes it very difficult to make any meaningful comment on its contribution to the hydrogen bonding of this network.
 |
| | Fig. 6 The asymmetric unit, selected symmetry equivalents and atom labelling scheme of 3, with thermal ellipsoids at 50% probability. H atoms omitted for clarity. Symmetry codes: i) 1 − x, −y, 1 − z. | |
 |
| | Fig. 7 Packing interactions in 3. a) The hydrogen-bonded network formed between cations and oxalate A. b) The hydrogen-bond network formed between the cations and oxalate B. | |
Compound 4 crystallises in the monoclinic space groupC2/c. The asymmetric unit is similar to 3 consisting of a tris-(ethylenediamino)-cobalt(III) complex cation, 1.5 oxalate dianions and 3.5 water molecules (Fig. 8). The coordination sphere about the Co atom is close to being a regular octahedron with Co–N bonds in the range 1.943(3) to 1.977(4) Å. The average bite angle for the three molecules is 85.60° with the opposite angle being on average 93.15°. The coordination of each ethylenediamine ligand results in a gauche conformation of the ligand. Careful refinement of the ligand containing N1 and N2 shows some disorder with two ligand conformations. Thus in the asymmetric unit the cation occurs with a Δ(λλδ) conformation 78(1)% of the time, and as a Δ(λλλ) conformation the remaining 22(1)%. It is argued16 that entropic contributions make the Δ(λλδ) conformation more stable than the Δ(λλλ) conformation, although the Δ(λλλ) conformation is consistently more frequently observed in crystal structures. The centrosymmetric space group means that there are equal populations of all enantiomers. Oxalate A (C7/C8) appears to be effectively delocalised with four very similar C–O bond lengths (1.241(5)–1.252(6) Å), while oxalate B (C9/C9i) shows longer C–O distances of 1.267(5) and 1.260(6) Å. The torsion angles are 6.8° and 36.7° for oxalate A and B respectively. There are five sites for the water of crystallisation, three of the water molecules are well defined and fully occupied, of which two are on general positions and one (O2w) is on a special 4e Wykoff site. The other two sites labelled O4Wa and O4Wb, are so close together as to require a mutually exclusive occupancy. Close examination of interatomic distances reveals a prohibitively close contact between O4Wb and proton H4d, which part of the partially occupied ethylenediamine molecule in cation. Thus the disorder in the position of this water molecule is correlated with the conformational disorder of the ligand molecule, and in our refinement the occupancies were tied to those of the disordered ligand. When the Δ(λλδ) cation conformer is present, O4wa is occupied, while for the Δ(λλλ) cation conformation, O4wb is occupied. The overall structure of 4 is complex, there are 23 hydrogen bond donor groups (N–H and O–H) in the asymmetric unit, and all are involved in crystallographically distinct hydrogen bond interactions. Thus a very complex three-dimensional H-bonded network is formed. Each cation is hydrogen bonded to five oxalate anions (Fig. 4), three type A oxalates and two type B oxalates. Oxalate A bridges between four cations (Fig. 9a), and forms a thick 2-D network in the bc-plane. Oxalate B also links four cations, forming ribbons that run parallel to the c-axis (Fig. 9b).
 |
| | Fig. 8 Asymmetric unit, atom labelling scheme and selected symmetry equivalents of 4. Thermal ellipsoids drawn at the 50% probability level, hydrogen atoms omitted for clarity. The minor component of the disordered conformation is superimposed with white atoms and dashed bonds. Symmetry codes: i) −x, y, 1½ − z. | |
 |
| | Fig. 9 Anion environments in 4. a)Hydrogen bonding to oxalate A, b)Hydrogen bonding to oxalate B. | |
Discussion
Compounds 1–4 are best considered in the wider family of compositionally related phases with the generic formula; Mp(ox)q(en)r(H2O)s, where M is a transition or transition-like metal, ox = oxalate and en = ethylenediamine. Table 1 lists the known phases of this compositional family (excluding simple oxalates, and their hydrates; i.e. where r = 0). Although not all of these compounds have known structures, there are some clear structural trends; the different binding strengths of these competing ligands mean that en tends to displace both ox and H2O, and ox tends to displace H2O. The extent of the observed displacement depends greatly on stiochoimetry. In addition these ligands show very differing characteristic coordination behaviour. H2O is small, monodentate, and rigid molecule that seldom bridges.17Oxalate has only a very limited conformational flexibility, which is effectively constrained in the normal chelating coordination mode. The ox2− anion is commonly found in a symmetric bridging mode, although it can be a terminal non-bridging ligand when there is a lack of any suitable coordination sites18–20 and it can form unsymmetric bridges.21,22 The ethylene diamine ligand has a much greater degree of conformational flexibility, nevertheless its coordination chemistry is dominated by a chelating mode. Examples of monocoordinate or bridging coordination are rare modes.23 If we look at the compounds where M = M(II) this sets the M:ox ratio as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. As ethylenediamine is progressively added to simple metal oxalate hydrates we see a displacement of O-donor ligands. For M(ox)(en) M = Co, Ni we preserved the linear M(ox) chain structure, seen in the metal oxalate dihydrates, the water is replaced by bridging en molecules giving a trans-MO4N2 octahedral coordination sphere.23 The related hydrate Zn(ox)(en)(H2O)2 also has a 1-D Zn(ox) chain motif, but chelation by en gives a cis-ZnO4N2 octaheadral coordination geometry.24 By contrast the compound Pt(ox)(en),25 (Pt(II) favours a 4-coordinate square planar geometry) contains discrete Pt(ox)(en) species with a cis-PtO2N2 coordination. The coordination preferences of Cu(II) are intermediate between the octahedral geometry seen in Co, Ni or Zn, and the square planar geometry associated with Pt(II). Copper(II) often shows 4 + 1 (square pyramidal) or 4 + 2 (tetragonally elongated octahedral) coordination geometry. The compound Cu(ox)(en)26 is better formulated as [Cu(ox)2][Cu(en)2]. Where complex cations and anions stack to give one square planar CuO4 environment, and one tetragonally elongated trans-CuO2N4 coordination environment with the O coming from an oxalate in a neighbouring [Cu(ox)2]2− unit. Increase the proportion of en and we observe [Cu(en)2(H2O)2][ox] (1) in which the oxalate ion is completely displaced. Thermogravimetric studies on 1 have been reported previously.12–14 Although the [Cu(en)2(H2O)2]2+ cation is well known. It is perhaps surprising to see water coordinate in the axial positions in preference to the dianionic oxalate ion. However the strong preference of Cu(II) for a square planner environment means that these axial interactions are very weak. Although the oxalate ions is not coordinated to the metal it is extensively hydrogen bonded to neighbouring complex cations. The fact that 1 and [Cu(en)3][ox] (2) are both crystallised from the same solution is evidence that both [Cu(en)2(H2O)2]2+ and [Cu(en)3]2+ are present as solution species. This tris-ethylenediamne copper(II) complex in 2 is a bit of a novelty. Data mining the CSD shows the bis-ethylenediamaine copper(II) unit to be much more common with more than 300 entries, by contrast only 8 compounds are reported containing the tris-ethylenediamne copper(II) complex. We note that the compositionally related compound [Ni(en)3][ox] has also been reported,27 as have more than 100 occurrences of the [Ni(en)3]2+ cation. The prevalence of these complex cations clearly relates to the particular coordination preference of CuII for a four coordinate square planar geometry. There are significant structural differences between [Ni(en)3][ox] and [Cu(en)3][ox] (2). Not only crystallising in different space groups; P
:1. As ethylenediamine is progressively added to simple metal oxalate hydrates we see a displacement of O-donor ligands. For M(ox)(en) M = Co, Ni we preserved the linear M(ox) chain structure, seen in the metal oxalate dihydrates, the water is replaced by bridging en molecules giving a trans-MO4N2 octahedral coordination sphere.23 The related hydrate Zn(ox)(en)(H2O)2 also has a 1-D Zn(ox) chain motif, but chelation by en gives a cis-ZnO4N2 octaheadral coordination geometry.24 By contrast the compound Pt(ox)(en),25 (Pt(II) favours a 4-coordinate square planar geometry) contains discrete Pt(ox)(en) species with a cis-PtO2N2 coordination. The coordination preferences of Cu(II) are intermediate between the octahedral geometry seen in Co, Ni or Zn, and the square planar geometry associated with Pt(II). Copper(II) often shows 4 + 1 (square pyramidal) or 4 + 2 (tetragonally elongated octahedral) coordination geometry. The compound Cu(ox)(en)26 is better formulated as [Cu(ox)2][Cu(en)2]. Where complex cations and anions stack to give one square planar CuO4 environment, and one tetragonally elongated trans-CuO2N4 coordination environment with the O coming from an oxalate in a neighbouring [Cu(ox)2]2− unit. Increase the proportion of en and we observe [Cu(en)2(H2O)2][ox] (1) in which the oxalate ion is completely displaced. Thermogravimetric studies on 1 have been reported previously.12–14 Although the [Cu(en)2(H2O)2]2+ cation is well known. It is perhaps surprising to see water coordinate in the axial positions in preference to the dianionic oxalate ion. However the strong preference of Cu(II) for a square planner environment means that these axial interactions are very weak. Although the oxalate ions is not coordinated to the metal it is extensively hydrogen bonded to neighbouring complex cations. The fact that 1 and [Cu(en)3][ox] (2) are both crystallised from the same solution is evidence that both [Cu(en)2(H2O)2]2+ and [Cu(en)3]2+ are present as solution species. This tris-ethylenediamne copper(II) complex in 2 is a bit of a novelty. Data mining the CSD shows the bis-ethylenediamaine copper(II) unit to be much more common with more than 300 entries, by contrast only 8 compounds are reported containing the tris-ethylenediamne copper(II) complex. We note that the compositionally related compound [Ni(en)3][ox] has also been reported,27 as have more than 100 occurrences of the [Ni(en)3]2+ cation. The prevalence of these complex cations clearly relates to the particular coordination preference of CuII for a four coordinate square planar geometry. There are significant structural differences between [Ni(en)3][ox] and [Cu(en)3][ox] (2). Not only crystallising in different space groups; P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1c and P21/c, but also showing significantly different packing interactions. The Ni containing compound shows a small distortion from octahedral coordination geometry, whilst the large Jahn–Teller distortion for CuII physically distorts the cation shape. Both Ni and Cu structures can be viewed as being built from stacks of alternating cations and anions. However, the relative orientation of the oxalate anion is rotated by ∼90°. Thus in [Ni(en)3][ox] the oxalate C–C bond is coincidental with the [Ni(en)3]2+ 3-fold axis (the stacking direction) and we observe the R22(8) H-bond motif. (Note that crystallographic symmetry imposes a three fold disorder on the anion). In [Cu(en)3][ox] (2), the oxalate C–C bond is roughly perpendicular to the [Cu(en)3]2+ pseudo-3-fold axis (the stacking direction) and we see the R22(9) H-bond motif.
1c and P21/c, but also showing significantly different packing interactions. The Ni containing compound shows a small distortion from octahedral coordination geometry, whilst the large Jahn–Teller distortion for CuII physically distorts the cation shape. Both Ni and Cu structures can be viewed as being built from stacks of alternating cations and anions. However, the relative orientation of the oxalate anion is rotated by ∼90°. Thus in [Ni(en)3][ox] the oxalate C–C bond is coincidental with the [Ni(en)3]2+ 3-fold axis (the stacking direction) and we observe the R22(8) H-bond motif. (Note that crystallographic symmetry imposes a three fold disorder on the anion). In [Cu(en)3][ox] (2), the oxalate C–C bond is roughly perpendicular to the [Cu(en)3]2+ pseudo-3-fold axis (the stacking direction) and we see the R22(9) H-bond motif.
Table 1 Known compounds with the composition Mp(ox)q(en)r(H2O)s where M is a transition metal, ox = oxalate, en = ethylenediamine
|
Reference: 12.
Reference: 23.
Reference: 13,14.
Reference: 26.
Reference: 25.
Reference: 24.
Reference: this work.
Reference: 27.
Reference: 28.
Reference: 29.
|
| M = M(II) |
p |
q |
r |
s |
|
Co,a,bNi,a,bCu,c,dZn,a Cd,aPte |
1 |
1 |
1 |
0 |
|
Zn
f |
1 |
1 |
1 |
2 |
|
Cu
c |
1 |
1 |
2 |
0 |
|
Cu
a,c,g (1) |
1 |
1 |
2 |
2 |
|
Co,aNi,a,hCua,g (2), Zn,a Cda |
1 |
1 |
3 |
0 |
|
Ni
a |
1 |
1 |
3 |
2 |
| M = M(III) |
|
Cr
i |
2 |
3 |
3 |
2 |
|
Co
j |
2 |
3 |
4 |
8 |
|
Co
g (3) |
2 |
3 |
6 |
3.61 |
|
Co
g (4) |
2 |
3 |
6 |
7 |
We see the same trends in behaviour for M = M(III), although there are fewer compounds with which to draw comparisons. With an M:ox ratio fixed at 2:3 we note two other relevant compounds. Cr2(ox)3(en)3(H2O) which is better formulated as [Cr(en)3][Cr(ox)3](H2O)2 being composed of molecular cations and anions.28 The higher proportion of en in Co2(ox)3(en)4(H2O)8 results in a displacement of more of the oxalate ions, and a dinuclear oxalate bridged cation.29 This compound is more usefully formulated as [(en)2Co(ox)Co(en)2][ox]2(H2O)8 being composed of an oxalate bridged dimer and uncoordinated oxalate anions. The structures of 3 and 4 show a continuation of this pattern. The higher en:M ratio results in all oxalate being displaced, and we see only the [Co(en)3]3+ cations, and uncoordinated oxalate anions. Compounds 3 and 4 differ in their degree of hydration. The distribution of water molecules in 3 is disordered and complex. The maximum permissible occupancy of all water molecule sites would make 3 a tetrahydrate. However, we see only partial occupancies and refine the compound as a non-stiochoimetric hydrate with a hydration level of 3.61. In 4 a degree of ligand conformational disorder also caused some disorder in the distribution of the water of crystallisation. However this does not affect the stiochoimetry, and 4 is a heptahydrate. Whilst there are many examples of compounds containing the [Co(en)3]3+ cation, very few additionally contain the oxalate anion. There is a family of structures with the composition [Co(en)3](ox)(X)(H2O)n where X = Cl, I, ClO4.30–32 The cation anion hydrogen-bond interaction in this family also contains the same R22(8) and R22(9) motif as seen in 3 and 4.
Despite the symmetry mismatch, that is a characteristic 2-fold symmetry to the oxalate dianion and a characteristic 3-fold symmetry to the tris-ethylenediamine metal molecular cation, it is remarkable that we see one of two patterns of hydrogen bonded interaction. These are shown schematically in Fig. 10. Either the oxalate tends to interact side-on and is characterised by a series of binary graphs; R22(9), R22(9), R22(9), R12(6), R12(6), R21(5) (Fig. 10a), this favours the formation of bifurcated H-bond interactions, or we see an end-on interaction (Fig. 10b) which is characterised by the series of graphs; R22(8), R22(8), R12(6) The frequent occurrence of these H-bond patterns must be a reflection of minimal energy structures.
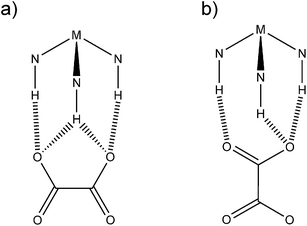 |
| | Fig. 10 Schematic representation of the observed H-bond motifs between the oxalate anion and the tris-ethylenediamine metal cation. a) side-on interaction, b) end-on interaction. | |
Both simple electrostatic considerations and more sophisticated high level calculations give the ground state conformation of the oxalate dianion as being staggered, with D2d point symmetry. Despite this gas phase conformational ground state, in solid state structures the observation of such a 90° torsion angle is exceedingly rare.33 The very common chelating interaction between an oxalate dianion and a metal cation massively stabilises a planar configuration and is the main reason why observed O–C–C–O torsion angles from solid state structures tend to a value of 0°. In compounds 1, 2, 3 and 4, the un-coordinated oxalate torsion angles ranges from 0 to 36.7°. Of all the geometric parameters that define the shape of an oxalate anion, we would expect the C–C bond length to be most likely correlated with the O–C–C–O torsion angles. We searched the Cambridge Structural Database (CSD, Conquest version 1.12)34 for non-coordinated oxalate structures in order to elicit any correlations between C–C bond length and O–C–C–O torsion angle. Of the structures that we found, four data sets were omitted; two with unfeasibly short C–C bond lengths and two with poor quality data/models, which left 91 crystal structures, some with more than one oxalate anion.
The mean C–C bond length is 1.548 Å, and a histogram of C–C bond length is shown in Fig. 11a. A plot of the C–C distance verses the O–C–C–O torsion angle (Fig. 11b) shows two things; a) a clustering of data with torsion angle at 0°, and b) an apparent correlation between d(C–C) and torsion O–C–C–O, with shorter C–C bonds associated with larger torsion angles. The geometric parameters of the oxalate ions from compounds 1–4 has been superposed on this data, and are consistent with the observed trend. Calculations indicate that there should be small changes in the C–O bond lengths with torsion angle,35 but this is likely to be more affected by hydrogen bonding as the strength of the hydrogen bond affects the electron density in the C–O bond.36
 |
| | Fig. 11
a) Histogram of the frequency of C–C bond lengths in non-coordinated; b) plot of C–C bond length against O–C–C–O torsion angle for compounds from the CSD (+) and for compounds 1–4 (see legend), showing a relationship between these two parameters. | |
Conclusions
Analysis of the structures of four new compounds presented here and of known structures of related compounds reveals preferred interaction geometries between tris-ethylenediamine metal cations and the oxalate dianion. These components have a mismatch in shape: the H-bond donor having three D–H groups and a three-fold character, while the acceptor can only present two accepting groups and has a two-fold character. This clear conflict of symmetry between the H-bond donor and the H-bond acceptor moieties results in a compromised interaction in which we frequently observe a side-on interaction with the oxalate and the formation of uncommon bifurcated H-bonds. This interaction ranges from being quite symmetric with two similar H⋯O interactions, to an asymmetric situation with one short and one long H⋯O interaction. In the symmetric case the motif is characterised by the series of binary graphs: R22(9), R22(9), R22(9), R12(6), R12(6), R21(5). If the interaction is sufficiently asymmetric as to become no longer bifurcated, three of the above graphs are lost, and the motif is characterised as R22(9), R22(9), R12(6). The presence of significantly twisted oxalate anions may be attributed the more isolated nature of this anion and the unusual absence of a metal-oxalate interactions. The observed long C–C bond lengths are consistent with the oxalate geometry, as analysis of oxalate in structures where the oxalate is “non-coordinated” show, on average, longer C–C bond lengths and a higher chance of large torsion angles between the carboxylate groups.
Experimental
Experimental techniques
Single crystal data were measured on an Enraf Nonius Kappa CCD area detector at −100 °C using Mo-Kα radiation. Data collection and unit cell refinement were managed by DENZO37 and absorption corrections were applied using SORTAV.38 Structure solutions were carried out by SIR9239 and the refinement by SHELXL-9740 in the WINGX41 environment. For crystallographic data see Table 2 and ESI.†
Table 2 Crystallographic parameters for 1–4
| Compound |
1
|
2
|
3
|
4
|
| Formula |
C6H20CuN4O6 |
C8H24CuN6O4 |
C18H55.2Co2N12O15.6 |
C18H62Co2N12O19 |
| Crystal system |
Triclinic |
Monoclinic |
Triclinic |
Monoclinic |
| Space group |
P
|
P21/c |
P
|
C2/c |
|
a/Å |
6.4347(6) |
9.8795(9) |
7.4525(19) |
28.964(2) |
|
b/Å |
6.9170(7) |
9.6336(9) |
8.971(2) |
10.3615(5) |
|
c/Å |
7.7596(6) |
14.4612(15) |
13.349(4) |
12.7579(8) |
|
α (°) |
90.268(4) |
90 |
71.49(2) |
90 |
|
β (°) |
107.581(4) |
91.718(5) |
89.61(2) |
114.446(2) |
|
γ (°) |
108.201(3) |
90 |
82.827(18) |
90 |
|
V/Å3 |
310.899(5) |
1375.7(2) |
839.2(4) |
3485.6(4) |
| Density/g cm−3 |
1.644 |
1.602 |
1.598 |
1.655 |
|
T/K |
120(2) |
120(2) |
120(2) |
120(2) |
|
μ/mm−1 |
1.780 |
1.609 |
1.073 |
1.046 |
| Reflections collected |
3144 |
12548 |
10682 |
12762 |
| Unique reflections (Rint) |
1407 (0.0811) |
3153 (0.1087) |
3799 (0.1896) |
3970 (0.1440) |
| Reflections F2 > 2σ(F2) |
1318 |
2020 |
3207 |
2516 |
| Data/restraints/parameters |
1407/3/85 |
3153/0/172 |
3799/0/216 |
3970/3/241 |
| Goodness of fit (S) |
1.080 |
1.038 |
1.071 |
1.051 |
|
R
1, wR2 [F2 > 2σ(F2)] |
0.0414/0.0951 |
0.0628/0.1265 |
0.0789/0.2028 |
0.0641/0.1532 |
|
R
1, wR2 [all data] |
0.0460/0.0982 |
0.1153/0.1430 |
0.0896/0.2147 |
0.1132/0.1759 |
| Residual peak/hole/e Å−3 |
0.531/−0.562 |
0.621/−0.431 |
1.725/−1.215 |
0.758/−0.742 |
Refinement details
[Cu(en)2(H2O)2][ox] (1).
All non H-atoms were refined anisotropically. All H-atoms on the ligand were placed in idealised geometries and refined in riding mode. H-atoms on the water molecule were found in the Fourier difference map, their geometry was restrained. All H-atom thermal parameters were constrained to 1.2 × Ueq of the parent atom.
[Cu(en)3][ox] (2).
All non H-atoms were refined anisotropically. All H-atom were placed in idealised geometries and refined in riding mode. H-atom thermal parameters were constrained to 1.2 × Ueq of the parent atom.
[Co(en)3]2[ox]3(H2O)3.61 (3).
All atoms, except H-atoms and the water of crystallisation, were refined anisotropically. All H-atom on the ligand were placed in idealised geometries and refined in riding mode. All H-atom thermal parameters were constrained to 1.2 × Ueq of the parent atom. No water protons were modelled. The thermal parameters of all water sites were left isotropic and fixed to a single parameter. The site occupancies were refined, giving a good flat part the Fourier difference map in this region. The site occupancies of O1w, O2w, O3w and O4w were refined as 49(1), 56(2), 25(1) and 40(2)% giving a composition [Co(en)3]2[ox]3(H2O)3.61.
[Co(en)3]2[ox]3(H2O)7 (4).
All non H-atoms were refined anisotropically. All H-atoms on the ligands were placed in idealised geometries and refined in riding mode with isotropic thermal parameters constrained to 1.2 × Ueq of the parent atom. H-atoms on the water molecule were not modelled. The conformational disorder in one of the ligand molecules, was refined with probabilities of 78(1) and 22(1)% for major and minor components. A number of constraints were applied to this disordered part of the structure: All comparable bond lengths in the two components were restrained to be the same; equivalent atoms were refined with the same anisotropic displacement parameters. Water molecule O4w is split over two nearby sites. Their occupancies were tied to that of the disordered ligand, with 78(1) and 22(1)% for O4wa and O4wb respectively.
Synthesis
[Cu(en)2(H2O)2][ox] (1) and [Cu(en)3][ox] (2).
Copper(II) oxalate was dissolved into 10 mL ethylenediamine to give a saturated solution. The resultant dark purple-blue solution left to evaporate over three days. A mixture of blue crystals of 1 and purple crystals of 2 were recovered, along with a small amount of copper oxalate.
[Co(en)3]2[ox]3(H2O)3.61 (3) and [Co(en)3]2[ox]3(H2O)7 (4).
Cobalt(II) oxalate dihydrate was dissolved in 10 mL ethylenediamine to give an orange solution. Upon evaporation, orange block-like crystals of 3 and 4 were recovered. The crystals of these two hydrates are very difficult to distinguish.
Acknowledgements
We are grateful to the UK EPSRC for financial support.
References
- M. C. Etter and J. C. MacDonald, Acta Cryst., 1990, B46, 256 CAS.
- S. J. Coles, R. Holmes, M. B. Hursthouse and D. J. Price, Acta Cryst., 2002, E58, o626 CAS.
- D. W. Bruce and D. J. Price, Adv. Mater. Opt. Electron., 1994, 4, 273 CrossRef CAS.
- D. J. Price, T. Richardson and D. W. Bruce, J. Chem. Soc., Chem. Commun., 1995, 1911 RSC.
- K. Willis, J. E. Luckhurst, D. J. Price, J. M. J. Fréchet, H. Kihara, T. Kato, G. Ungar and D. W. Bruce, Liq. Cryst., 1996, 21, 585 CrossRef CAS.
- Z. A. D. Lethbridge, A. F. Congreve, E. Esslemont, A. M. Z. Slawin and P. Lightfoot, J. Solid State Chem., 2003, 172, 212 CrossRef CAS.
- T. D. Keene, M. B. Hursthouse and D. J. Price, Acta Cryst., 2003, E59, m1129 Search PubMed.
- T. D. Keene, M. B. Hursthouse and D. J. Price, New J. Chem., 2004, 28, 558 RSC.
- T. D. Keene, M. E. Light, M. B. Hursrthouse and D. J. Price, Dalton Trans., 2011, 40, 2983 RSC.
- T. D. Keene, I. Zimmermann, A. Neels, O. Sereda, J. Hauser, M. Bonin, M. B. Hursrthouse, D. J. Price and S. Decurtins, Dalton Trans., 2010, 39, 4937 RSC.
- T. D. Keene, M. B. Hursthouse and D. J. Price, Acta Cryst., 2006, E62, m1373 CAS.
- J. M. Haschke and W. W. Wendlandt, Anal. Chim. Acta, 1965, 32, 386 CrossRef CAS.
- C. G. R. Nair, S. Mathew and K. N. Ninan, Thermochim. Acta, 1989, 150, 63 CrossRef CAS.
- S. Mathew, C. G. R. Nair and K. N. Ninan, Thermochim. Acta, 1991, 184, 269 CrossRef CAS.
- F. A. Jurnak and K. N. Raymond, Inorg. Chem., 1972, 11, 3149 CrossRef CAS.
-
F. A. Cotton, G. Wilkinson, Advanced Inorganic Chemistry, 5th ed, John Wiley and Sons Inc, 1988 Search PubMed.
- J. C. Goodwin, D. J. Price and S. L. Heath, Dalton Trans., 2004, 2833 RSC.
- T. D. Keene, M. B. Hursthouse and D. J. Price, Acta Cryst., 2004, E60, m378 CAS.
- T. D. Keene, M. B. Hursthouse and D. J. Price, Z. Anorg. Allg. Chem., 2004, 630, 350 CrossRef CAS.
- M. B. Hursthouse, M. E. Light and D. J. Price, Angew. Chem., Int. Ed., 2004, 43, 472 CrossRef CAS.
- D. J. Price, A. K. Powell and P. T. Wood, Dalton Trans., 2003, 2478 RSC.
- D. J. Price, A. K. Powell and P. T. Wood, J. Chem. Soc., Dalton Trans., 2000, 3566 RSC.
- T. D. Keene, M. B. Hursthouse and D. J. Price, Cryst. Growth Des., 2009, 9, 2604 CAS.
- G. A. van Albada, A. Mohamadou, I. Mutikainen, U. Turpeinen and J. Reedijk, Acta Cryst., 2004, E60, m1160 CAS.
- A. R. Battle, G. B. Deacon, R. C. Dolman and T. W. Hambley, Aust. J. Chem., 2002, 55, 699 CrossRef CAS.
- H. Oshio and U. Nagashima, Inorg. Chem., 1992, 31, 3295 CrossRef CAS.
- M. Padmanabhan, J. C. Joseph, X. Huang and J. Li, J. Mol. Struct., 2008, 885, 36 CrossRef CAS.
- X. Hua, K. Larsson, T. J. Neal, G. R. A. Wyllie, M. Shang and A. G. Lappin, Inorg. Chem. Commun., 2001, 4, 635 CrossRef CAS.
- J. Cai, J. Myrczek, H. Chun and I. Bernal, J. Chem. Soc., Dalton Trans., 1998, 4155 RSC.
- R. Bala, R. P. Sharma, U. Sharma, A. D. Burrows and K. Cassar, J. Mol. Struct., 2007, 832, 156 CrossRef CAS.
- A. Fuertes, C. Miravitlles, R. Ibanez, E. Martinez-Tamayo and A. Beltran-Porter, Acta Cryst., 1988, C44, 417 CAS.
- J. Cai, Y. Zhang, X. Hu and X. Feng, Acta Cryst., 2000, C56, 661 CAS.
- R. E. Dinnebier, S. Vensky, M. Panthofer and M. Jansen, Inorg. Chem., 2003, 42, 1499 CrossRef CAS.
- F. H. Allen, Acta Cryst., 2002, B58, 380 CAS.
- M. J. S. Dewar and Y.-J. Zheng, J. Mol. Struct. (Theochem), 1990, 209, 157 CrossRef.
- S. J. Grabowski, Annu. Rep. Prog. Chem., Sect. C, 2006, 102, 131 RSC and the references therein.
-
Z. Otwinowski, W. Minor in Methods in Enzymology, vol. 276: Macromolecular Crystallography (ed. C. W. Carter Jr, R. M. Sweet), Academic Press, 1997, Part A, pp 307 Search PubMed.
- R. H. Blessing, J. Appl. Crystallogr., 1997, 30, 421 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Cryst., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Cryst., 2008, A64, 112 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 829507–829510. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce05837a |
| ‡ Current address: School of Chemistry, University of Sydney, NSW 2006, Australia. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.