pH variation induced construction of a series of entangled frameworks based on bi- and tri-metallic cores as nodes†
Lian-Jie
Li
,
Chao
Qin
,
Xin-Long
Wang
,
Shuang
Wang
,
Liang
Zhao
*,
Guang-Sheng
Yang
,
Hai-Ning
Wang
,
Gang
Yuan
,
Kui-Zhan
Shao
and
Zhong-Min
Su
*
Institute of Functional Material Chemistry, Key Lab of Polyoxometalate Science of Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun, 130024, Jilin, People's Republic of China. E-mail: zmsu@nenu.edu.cn
First published on 14th October 2011
Abstract
Reactions of 1,4-bis(pyridin-4-ylmethoxy)benzene (L) and zinc acetate salts with different multi-caboxylate acids: 1,4-benzenedicarboxylic (p-H2BDC), isophthalic acid (m-H2BDC) and benzene-1,3,5-tricarboxylic acid (H3BTC) yield four entangled structures and one 3D supramolecular framework depending on pH variation. The structures of compounds 1–5 were elucidated by single crystal X-ray diffraction. Topology analysis revealed that the entangled structures of compounds 1–4 covered a range of poly-threading, self-penetrating and interpenetrating coordination. Interestingly, the poly-threaded arrays with cyclohexane-like windows in chair conformation in compound 2 was firstly observed. What's more, compound 4 exhibited a rare eight-connected self-penetrating network based on trinuclear zinc clusters as nodes with 42464 topology. Photoluminescent properties and thermogravimetric analyses for 1–5, were also investigated in detail.
Introduction
In recent years, coordination polymers have enjoyed extensive exploration not only stemming from their potential applications ranging from non-linear optics to magnetism, but also from their long-standing fascination of topologies.1 Entanglements, serving as a crucial subject in the area of supramolecular chemistry, are common in biology as seen in catenanes, rotaxanes, and molecular knots.2 A variety of attractive architectures ascribed to this category have been constructed and fully discussed in some excellent papers.3 Interpenetration4 recognized as one major theme of an entangled system, has been comprehensively investigated as shown by Batten and Robson.5 Differing from interpenetration, self-penetrating6 nets are characterized by the presence of the smallest topological rings or knots, which can be catenated by other shortest rings belonging to the same net. However, poly-threading7 coordination networks possess periodic analogues of the molecular rotaxanes or pseudo-rotaxanes, meanwhile, showing potential applications in the area of drug delivery vehicles and sensor devices.8 Unfortunately, only a limited number of cases with respect to this topological nature have been reported to date.9 Therefore, the exploration of new synthetic routes to this class of entanglement architectures is one of the most challenging issues in current synthetic chemistry. Indeed, a systematic investigation to establish useful relationships between structures and properties is unavoidable.Taking inspiration from previous work, ongoing research in our laboratory has been directed toward the design and synthesis of novel entangled networks.10 Conformationally, non-rigid ligands are usually the typical building elements for the assembly of interesting entangled structures, thanks to their varied geometries.11 In contrast, the rigid ligands usually result in larger voids that may lead to an intriguing variety of structures and potential applications.12 Based on an overall consideration, we chose the semi-rigid 1,4-bis(pyridin-4-ylmethoxy)benzene (L) ligand (Scheme 1) for the following reasons: (i) a rigid spacer of a phenyl ring could provide comparative stability as well as enough void within the whole framework; (ii) the freely rotating pyridyl arms may afford variable entanglement skeletons; and (iii) few cases with respect to entangled frameworks based on the L ligand have been reported.13 Fortunately, we obtained four entangled frameworks ranging from self-threading to poly-threading arrays by using three types of multi-caboxylate acids as auxiliary ligands. Compound 5 is synthesized under pH variation, which is a 3D supramolecular structure extended by weak interactions between adjacent layers.
 | ||
| Scheme 1 Molecular structure of 1,4-bis(pyridin-4-ylmethoxy)benzene ligand. | ||
Herein, we report the synthesis and characterization of four entangled structures and one 3D supramolecular framework, {[Zn2(L)2(HBTC)2]·H2O}n (1), {[Zn2(L)(HBTC)2]·H2O}n (2), {[Zn2(L)(m-BDC)2]H2O}n (3), [Zn3(L)(p-BDC)3]n (4), {[Zn2(L)(HBTC)2]·H2O}n (5), (L = 1,4-bis(pyridin-4-ylmethoxy)benzene; p-H2BDC = 1,4-benzenedicarboxylic; m-H2BDC = isophthalic acid; H3BTC = benzene-1,3,5-tricarboxylic acid). It should be mentioned that both compounds 1 and 2 exhibit the poly-threaded frameworks involving 2D coordination layers with dangling ligands, which are still rare in the system of entangled frameworks. Moreover, the poly-threaded arrays with cyclohexane-like windows in chair conformation in compound 2 are unprecedented. The structure of compound 3 possesses a three-fold interpenetrated 3D network structure with α-Po topology that is built from dinuclear zinc units with a paddle-wheel structure. While, compound 4 is an eight-connected self-penetrating framework based on trinuclear zinc clusters as node with a 42464 topology. Compound 5 shows a 3D supramolecular structure extended by hydrogen bonds interaction in the adjacent layers.
Experimental section
Materials and physical measurements
All reagents and solvents employed were commercially available and used as received without further purification. The ligand L was synthesized readily by the procedure reported in the literature.13 Elemental analyses (C, H, and N) were performed on a Perkin-Elmer 240C elemental analyzer. The Fourier transform infrared (FT-IR) spectra were recorded from KBr pellets in the range 4000–400 cm−1 on a Mattson Alpha-Centauri spectrometer. XRPD patterns were collected on a Rigaku Dmax 2000 X-ray diffractometer with graphite monochromatized Cu Kα radiation (λ = 0.154 nm) and 2θ ranging from 5 to 50°. Solid-state luminescent spectra were measured on a Cary Eclipse spectrofluorometer (Varian) equipped with a xenon lamp and quartz carrier at room temperature. Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer TG-7 analyzer heated from 40 to 800 °C under nitrogen.Synthesis of {[Zn2(L)2(HBTC)2]·H2O}n (1)
A mixture of Zn(Ac)2·2H2O (0.25 mmol, 0.055 g), L ligand (0.10 mmol, 0.029 g) and H3BTC (0.20 mmol, 0.042 g) in H2O (14 mL) was adjusted to approximately pH ≈ 3–4 with H2SO4 (6 M) stirred for 1 h and then transferred and sealed in a 25 mL Teflon-lined stainless steel container, which was heated at 150 °C for 72 h and then cooled down to room temperature at a rate of 5 °C h−1. Colorless crystals of 1 were collected and washed with distilled water and dried in air to give the product; yield, 42.5% (based on ZnII salts). Elemental analyses calcd (%) for C54H42N4O17Zn2 (1149.70): C, 56.36; H, 3.65; N, 4.87. Found C, 56.01; H, 3.10; N, 4.18.Synthesis of {[Zn2(L)(HBTC)2]·H2O}n (2)
Compound 2 was prepared in a manner similar to that used to prepare compound 1 at pH ≈ 5.5 instead of pH ≈ 3–4 with H2SO4 (6 M). Colorless crystals of 2 were collected and washed with distilled water and dried in air to give the product; yield, 32.5% (based on ZnII salts). Elemental analyses calcd (%) for C36H26N2O15Zn2 (857.37): C, 50.39; H, 3.03; N, 3.27. Found C, 48.56; H, 2.01; N, 2.92.Synthesis of {[Zn2(L)(m-BDC)2]·H2O}n (3)
A mixture of Zn(Ac)2·2H2O (0.20 mmol, 0.044 g), L ligand (0.10 mmol, 0.029 g) and m-H2BDC (0.15 mmol, 0.025g) in H2O (14 mL) was adjusted to approximately pH ≈ 6 with H2SO4 (6 M) and NaOH (1 M). Yield, 59.7% (based on ZnII salts). Elemental analyses calcd (%) for C34H26N2O11Zn2 (769.33): C, 53.03; H, 3.38; N, 3.64. Found C, 52.16; H, 3.07; N, 3.23.Synthesis of [Zn3(L)(p-BDC)3]n (4)
Compound 4 was synthesized in an analogous manner to compound 3 except that m-H2BDC (0.15 mmol, 0.025g) was replaced by p-H2BDC (0.30 mmol, 0.050 g). Yield, 50.4% (based on ZnII salts). Elemental analyses calcd (%) for C42H28N2O14Zn3 (980.83): C, 51.39; H, 2.85; N, 2.85. Found C, 52.15; H, 2.24; N, 2.43.Synthesis of {[Zn2(L)(HBTC)2]·H2O}n (5)
Compound 5 was prepared in a manner similar to that used to prepare compound 1 at pH ≈ 7 instead of pH ≈ 3–4 with H2SO4 (6 M) and NaOH (1 M). Colorless crystals of 5 were collected and washed with distilled water and dried in air to give the product; yield, 49.5% (based on ZnII salts). Elemental analyses calcd (%) for C36H26N2O15Zn2 (857.37): C, 50.39; H, 3.03; N, 3.27. Found C, 49.01; H, 2.01; N, 3.09.X-Ray crystallography study
Data collection of complexes 1–5 was performed on a Bruker Smart Apex II CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at room temperature. All absorption corrections were performed by using the SADABS program. The crystal structure was solved by direct methods and refined with full-matrix least-squares (SHELXTL-97)14 with atomic coordinates and anisotropic thermal parameters for all non-hydrogen atoms. The hydrogen atoms of aromatic rings were included in the structure factor calculation at idealized positions by using a riding model. Protonated hydrogen atoms attached to nitrogen atoms were bound at idealized positions. In compounds 1, 2 and 3, H atoms of water molecules could not be introduced in the refinement but were included in the structure factor calculation. The detailed crystallographic data and structure refinement parameters are summarized in Table S1 in the ESI.† The IR data of five compounds were provided in the ESI.† Selected bond lengths and angles for complexes 1–5 are given in Table S2 (in the ESI†).Results and discussion
Structure of {[Zn2(L)2(HBTC)2]·H2O}n (1)
Single-crystal X-ray diffraction analysis exhibits that compound 1 crystallizes in the monoclinic space groupP21/c and the asymmetric unit of 1 is constructed from two ZnII ions, two L ligands, two HBTC ligands and one free water molecule. Each ZnII ion in the dinuclear motif is coordinated by four carboxylic oxygen atoms of HBTC ligands (2.036–2.107 Å) and one nitrogen atom of an L ligand (2.025–2.043 Å)15 to furnish a square-pyramidal geometry as shown in Fig. 1a. Two crystallographically equivalent ZnII ions are bridged by four carboxylates bonded in the bridging bis(bidentate) fashion to give a paddle-wheel shaped [Zn2(CO2)4] fragment in which the Zn⋯Zn distance is 2.988 Å. The axis sites of each Zn2 paddle wheel are occupied by two additional L ligands. | ||
| Fig. 1 (a) Coordination environment of ZnII ions in 1, hydrogen atoms were omitted for clarity (symmetry transformations used to generate equivalent atoms: A, x, 0.5 − y, 0.5 + z). (b) The structure of a single 2D motif showing the dangling arms. (c) Schematic illustration of the mutual poly-threading of the 2D sheets in 1. (d) A schematic illustration of the mutual poly-threading of four layers. | ||
Compound 1 is an extended poly-threaded framework involving 2D coordination layers of square (4,4) topology with dangling ligands. The nodes within the layers are represented by dinuclear units that are bridged by the HBTC ligand and a 2D grid with large rhombic windows (dimensions 9.90 Å × 9.90 Å); dangling L ligands are coordinated in the axial sites of dinuclear nodes and disposed in a mutual anti orientation with respect to the layer plane (Fig. 1b, 1c). The layers are stacked in a parallel manner along the [0 0 1] direction in an ABAB sequence at a distance of approximately 15.18 Å. Each dangling arm has an effective length of about 18.55 Å. As a result, the dangling L ligands of each layer are threaded into the rhombic voids of the two adjacent layers above and below, thus every rhombic window of each layer is threaded by only two dangling ligands that come from opposite directions (this is expected considering that the dangling arms reside on the dinuclear nodes). Finally, this unique simultaneous threading fashion of the adjacent polymeric motifs gives the novel (2D → 3D) poly-threaded array observed for compound 1 (Fig. 1d).
Structure of {[Zn2(L)(HBTC)2]·H2O}n (2).
X-Ray crystallography shows that compound 2 crystallizes in the monoclinic space groupP21/c and the asymmetric unit of 2 contains two ZnII ions, one L ligand, two HBTC ligands and one isolated water molecules. The ZnII ions display two coordination modes. One ZnII (Zn2) ion is coordinated by four oxygen atoms from four HBTC ligands to furnish the distorted tetrahedral coordination geometry. While, the other ZnII (Zn1) ion is ligated with three oxygen atoms from three HBTC ligands and one nitrogen atoms from one L ligand to furnish the distorted tetrahedral coordination geometry, as shown in Fig. 2a. The Zn–O (1.945 Å–2.076 Å) and Zn–N (2.030 Å) bond distances are all in the normal ranges.15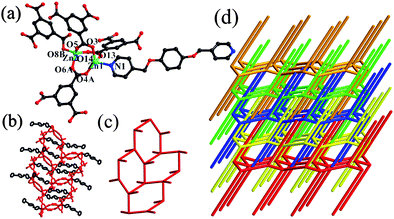 | ||
| Fig. 2 (a) Coordination environment of ZnII ions in 2, hydrogen atoms were omitted for clarity (symmetry transformations used to generate equivalent atoms: A, −x, 0.5 + y, 1.5 − z; B −x, −y, 1− z). (b, c) The structure of a single 2D motif showing the dangling arms in 2. (d) A schematic illustration of the mutual poly-threading of five layers. | ||
Compound 2 is an extended threaded framework involving 2D coordination layers of hexagonal (63) topology with dangling ligand. In these layers, two ZnII ions, related by a two-fold axis, are bridged by two pairs of HBTC carboxylate ends into a dinuclear unit with a Zn(1)⋯Zn(2) distance of 3.435 Å. Three factors can be envisaged to play a role in the generation of mutual poly-threading in 2: (i) the presence within the 2D motif of large hexagonal windows; (ii) the dangling Lgroups, occupying the axial position of each metal atom, are just like open arms nearly perpendicularly protruding from both sides of the sheets; and (iii) all the layers are stacked parallel and arranged in a staggered fashion, and the distance between the layer is shorter than the effective length of lateral arm, thus providing the possibility for the ultimate realization of poly-threading net (as shown in Fig. 2b, 2c). Under this premise, the dangling arms of each layer are threaded into the hexagonal voids of the adjacent layers, in a mutual relationship. Each hexagonal window is therefore penetrated by two L molecules that belong to two different sheets, one entering from one side and the other from the opposite one. This results in a novel 3D poly-threaded array (2D → 3D), originating from the entanglement of three adjacent polymeric units at a time (Fig. 2d).
On the basis of the aforementioned, the most fascinating feature of 2 is that also shows cyclohexane-like windows in chair conformation. Meanwhile, the dangling L ligand can be thought of as a hydrogen atom in the cyclohexane structure (Fig. S1 in the ESI†). Thus, the dangling L ligand exhibits the equatorial bonds with the chair of cyclohexane. Meanwhile, the chair of conformation stabilizes the solid-state structures.
Poly-threaded structures containing finite components are, at present, rare.9 The few species known include poly-threaded 0D rings with side arms that give 1D7b or 2D arrays,7c as well as molecular ladders with dangling arms that result in (1D → 2D)17d or (1D → 3D)7e poly-threaded arrays. Until now, the only known example of (2D → 3D) structures have been reported for poly-threading systems in metal–organic frameworks by us,9a,b in which the threading involves the two nearest layers only because of the shorter length of the dangling arms. Meanwhile, it should be pointed out that the structures of 1 and 2 are an extended threaded framework involving 2D coordination layers with dangling ligands. However, compound 2 is an unprecedented poly-threaded array with cyclohexane-like windows in chair conformation.
Structure of {[Zn2(L)(m-BDC)2]·H2O}n (3)
The structure of 3 contains two ZnII ions, one L ligand, two m-BDC ligands and one isolated water molecule in the asymmetric unit. Compound 3 is a three-fold interpenetrated 3D network structure of α-Po topology that is built from dinuclear Zn2 units with a paddle-wheel configuration. Fig. 3a illustrates the coordination environment of the ZnII ions and the six connectivity of the bimetallic unit. Each ZnII ion in the dinuclear motif is coordinated by three carboxylic oxygen atoms of m-BDC ligands (Zn–O 1.952–1.977 Å) and one nitrogen atom of an L ligand (Zn–N1 2.025 Å)15 to furnish a distorted tetrahedral geometry. Two crystallographically equivalent ZnII ions are bridged by two carboxylates with bis(bidentate) fashion to furnish the [Zn2(CO2)4N2] fragment in which the Zn⋯Zn distance is 3.634 Å. The axis sites of each Zn2 paddle wheel are occupied by two additional L ligands via a nitrogen atom. Each binuclear unit is connected to six others through four bridging m-BDC and two L ligands to generate an extended neutral 3D network (Fig. 3b), which can also be considered to be constructed from distorted 2D square-grid (4,4) layers of composition [Zn(m-BDC)] pillared by the long L ligand (Fig. S2 in the ESI†). The overall topology of the 3D frame is best described as a compressed α-Po net based on three intersecting (4,4) nets that possesses large distorted cube-like cavities of approximately 10.34 × 10.34 × 21.48 Å3 (Fig. 3c). The large voids formed by a single 3D network allow incorporation of two other identical networks, thus giving a three-fold interpenetrated α-Po-related network as shown in Fig. 3d.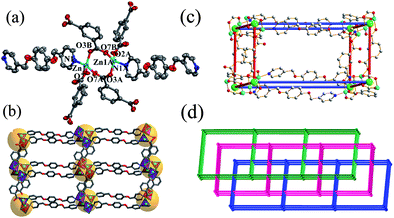 | ||
| Fig. 3 (a) Coordination environment of ZnII ions in 3 with the ellipsoids drawn at the 30% probability level, hydrogen atoms were omitted for clarity (symmetry transformations used to generate equivalent atoms: A, −1 − x, 1 − y, −z; B, x, −y, −0.5 + z). (b) View of one independent primitive cubic net. (c) A single distorted cube-like unit of the α-Po net with the relative dimensions (10.34 × 10.34 × 21.48 Å) of α-Po topology. (d) A schematic illustration of the 3-fold interpenetrated structure. | ||
Structure of [Zn3(L)(p-BDC)3]n (4)
Single-crystal X-ray structure analysis reveals that compound 4 crystallizes in the triclinic space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and the fundamental building unit of 4 consists of one-and-a-half ZnII ions, one L ligand with half occupation and one-and-a-half p-H2BDC ligands. Meanwhile, the structure of 4 consists of trinuclear zinc clusters, in which 1.5 crystallographically independent ZnII ions exhibit different coordination spheres. One ZnII center (Zn1) lies on a center of symmetry and is coordinated by six carboxylic oxygen atoms from six different p-H2BDC ligands (Zn–O 2.074–2.150 Å) (Fig. 4a). The other ZnII center (Zn2) is ligated with three carboxylic oxygen atoms (Zn–O 1.930–2.150 Å) from three p-H2BDC ligands, one nitrogen atom from an L ligand (Zn–N 2.047 Å).15 The Zn1 is connected to two adjacent Zn2 centers in a corner-sharing mode to form a [Zn3O12N2C6] core via carboxylate oxygen atoms with a non-bonding Zn⋯Zn distance of 3.633 Å (Fig. S3 in the ESI†). As illustrated in Fig. 4b, 4c, there are eight organic ligands (six p-H2BDC and two L) surrounding each Zn3 unit. This, therefore, defines an eight-connected node which is further linked to eight nearest neighbors with distances of 10.660–25.660 Å through eight bridging ligands. This process, when repeated, results in a unique 3D uninodal eight-connected self-penetrating network as shown in Fig. 4d.16 If the trinuclear zinc clusters take the place of balls, compound 4 shows that it is three types of links of different lengths of type I (20.331 Å), II (11.823 Å) and III (11.476 Å). If we can imagine removing type I (20.331 Å) or II (11.823 Å), then compound 4 shows that it is a three-dimensional six-connected framework with the distorted α-Po topology. Further insight into the nature of this intricate architecture can be obtained by considering the net constructed from two interpenetrating primitive cubic nets (α-Po), which are cross-linked by two extra connections from each node along the cube diagonals (Fig. 4e), and the extension of this structure results in the eight-connected self-penetrating coordination net, in which trinuclear zinc clusters act as eight-connected nodes.
and the fundamental building unit of 4 consists of one-and-a-half ZnII ions, one L ligand with half occupation and one-and-a-half p-H2BDC ligands. Meanwhile, the structure of 4 consists of trinuclear zinc clusters, in which 1.5 crystallographically independent ZnII ions exhibit different coordination spheres. One ZnII center (Zn1) lies on a center of symmetry and is coordinated by six carboxylic oxygen atoms from six different p-H2BDC ligands (Zn–O 2.074–2.150 Å) (Fig. 4a). The other ZnII center (Zn2) is ligated with three carboxylic oxygen atoms (Zn–O 1.930–2.150 Å) from three p-H2BDC ligands, one nitrogen atom from an L ligand (Zn–N 2.047 Å).15 The Zn1 is connected to two adjacent Zn2 centers in a corner-sharing mode to form a [Zn3O12N2C6] core via carboxylate oxygen atoms with a non-bonding Zn⋯Zn distance of 3.633 Å (Fig. S3 in the ESI†). As illustrated in Fig. 4b, 4c, there are eight organic ligands (six p-H2BDC and two L) surrounding each Zn3 unit. This, therefore, defines an eight-connected node which is further linked to eight nearest neighbors with distances of 10.660–25.660 Å through eight bridging ligands. This process, when repeated, results in a unique 3D uninodal eight-connected self-penetrating network as shown in Fig. 4d.16 If the trinuclear zinc clusters take the place of balls, compound 4 shows that it is three types of links of different lengths of type I (20.331 Å), II (11.823 Å) and III (11.476 Å). If we can imagine removing type I (20.331 Å) or II (11.823 Å), then compound 4 shows that it is a three-dimensional six-connected framework with the distorted α-Po topology. Further insight into the nature of this intricate architecture can be obtained by considering the net constructed from two interpenetrating primitive cubic nets (α-Po), which are cross-linked by two extra connections from each node along the cube diagonals (Fig. 4e), and the extension of this structure results in the eight-connected self-penetrating coordination net, in which trinuclear zinc clusters act as eight-connected nodes.
 | ||
| Fig. 4 (a) Coordination environment of ZnII ions in 4 with the ellipsoids drawn at the 30% probability level, hydrogen atoms were omitted for clarity (symmetry transformations used to generate equivalent atoms: A, −x, 1 − y, −z). (b) Perspective views of the eight-connecting trinuclear zinc clusters as nodes. (c) The symbols of three types of linkers are I (25.66 Å), II (10.66 Å), and III (11.47 Å). (d) Topological representation of 4 showing the 42464 topology. (e) Two cross-linked interpenetrating α-Po nets. | ||
Structure of {[Zn2(L)(HBTC)2]·H2O}n (5)
Single-crystal X-ray diffraction analysis reveals that compound 5 crystallizes in the triclinic space groupP. In the asymmetric unit of 5, there are two kinds of crystallographically unique ZnII ions, one L ligand, two HBTC ligands and one water molecule. The ZnII ion shows two coordination modes. One ZnII (Zn1) ion is coordinated by three carboxylic oxygen atoms from three HBTC ligands and one nitrogen atom from an L ligand. Meanwhile, the other ZnII ion (Zn2) is coordinated by four oxygen atoms from three HBTC ligands and one water molecule to furnish a distorted tetrahedral coordination geometry. The Zn–O (1.913 Å–1.977 Å) and Zn–N (2.046 Å) bond distances are all in the normal ranges,15 as shown in Fig. 5a. In addition, each HBTC ligand bridges two ZnII ions to form a cationic [{Zn4(HBTC)}4] square with dimensions of 9.66 × 9.40 Å (Fig. S4 in the ESI†). Meanwhile, the coordinated L ligands thread into the void which is constructed by the four HBTC ligands and four ZnII ions. The extension of the void results in infinite 1D tube structures (Fig. S5 in the ESI†) which are linked in a parallel fashion with the neighboring ones to give rise to a 2D layer (Fig. 5b). In summary, compound 5 can be described as a 3D supramolecular architecture further formed via the hydrogen bonds in the adjacent layers. Moreover, there are the hydrogen-bonding interactions (Fig. 5c) between the coordinated water molecules from one entangled network and carboxylic oxygen atoms from the other in the adjacent 2D layers (dOw⋯O8A, 2.705 Å, A, −x, 2 − y, 1 − z; dOw⋯O18B, 2.675 Å, A, −x, 2 − y, 1 − z; B, −1 + x, y, z), which extend the 2D layers into a 3D supramolecular structure (Fig. 5d). Both the hydrogen bonds and the special pattern make the whole structure of the title compound much tighter. The hydrogen bonding data are summarized in Table S3 (in the ESI†).
 | ||
| Fig. 5 (a) Coordination environment of ZnII ion in 5 with the ellipsoids drawn at the 30% probability level, hydrogen atoms were omitted for clarity (symmetry transformations used to generate equivalent atoms: A, 2 − x, 1 − y, −z). (b) Polyhedral representation of the 2D metal–organic framework of compound 5. (c) The O–H⋯O hydrogen bonds in the compound 5. (d) Schematic representation of crystal structures in 5 along a axis direction. | ||
Effect of coordination modes of carboxylate ligands
Multidentate carboxylate ligands have been extensively applied in the construction of a rich variety of MOFs because of their diverse coordination modes and structural stability.17 It is instructive to compare the coordination modes of carboxylate ligands. In 1, the rigid H3BTC exhibits a bis(bidentate) coordination mode (Fig. S6a in the ESI†). Whereas in 2, the H3BTC exists bidentate and monodentate modes (Fig. S6b in the ESI†). Notably, both of them have a free carboxylate which does not bridge the metal ions, and this coordination mode of the H3BTC ligand is very important for the poly-threading structure. In 5, apart from in the bis(monodentate) only binding mode, H3BTC ligand coordination tends to favor the formation of a dimeric unit, due to coordinating in both bis(monodentate) and bidentate modes, with no free carboxylate (Fig. S6d,e in the ESI†). However, the bidentate and monodentate modes of m-H2BDC in 3 and the bis(bidentate) modes of p-H2BDC in 4 are very important in chelating metal ions and can lock their positions into M–O–C clusters and is a key factor for the formation of the high dimension structures18 (Fig. S6f,g in the ESI†). Thus the coordination modes of carboxylate ligands influence the metal nuclearity and hence the connectivity of the resulting net.The simulated and experimental PXRD patterns of 1–5 are shown in Fig. S7–S11 (in the ESI†). The simulated and experimental PXRD patterns are in good agreement with each other, indicating the phase purity of the products. The differences in intensity may be due to the preferred orientation of the powder samples.
Luminescent properties
Taking into account the excellent luminescent properties of ZnII, the solid-state luminescent spectra of 1–5 have been studied at room temperature. Complexes 1, 2 and 3 show the emission maxima at 423 nm (λex = 365 nm), 423 nm (λex = 362 nm), 424 nm (λex = 369 nm). Meanwhile, complex 4 shows the emission maxima at 422 nm (λex = 365 nm), and complex 5 shows the emission maxima at 422 nm (λex = 365 nm). In order to understand the nature of the emission, we analyzed the photoluminescence property of the L ligand and found a strongest emission peak at 394 nm (λex = 325 nm) for L. The emission of the ligands may be assigned to π*→n and π*→π transitions of the intraligands. In comparison with that of the free ligands as shown in Fig. S12 (in the ESI†), the maximum emission wavelengths of complexes 1–5 occur slightly red shifted, which is probably due to intraligand charge transitions.19 In summary, compounds 1–5 show the potential fluorescence properties in the solid state at room temperature.Thermal properties
To study the thermal stability of the complexes, thermogravimetric analyses (TGA) were performed on polycrystalline samples under a nitrogen atmosphere with a heating rate of 10 °C min−1. The TG diagram of 1 displays three distinct weight losses, the first one from 70 to 150 °C and corresponds to the loss of the lattice water molecules. The observed weight loss of 1.36% is in agreement with the calculated value of 1.56%. The dehydrated complex showed a weight loss of 7.38%, which is in agreement with the calculated value of 7.65%, corresponding to the loss of two CO2 molecules in the L ligand. Then, the dehydrated complex is stable up to 329 °C. The frameworks collapsed in the temperature range of 329–626 °C. The remaining weight may correspond to the final products of ZnO in 1 (obsd 14.51%, calcd 13.92%). Compound 2 displays three distinct weight losses. The first one from 40 to 102 °C and corresponds to the loss of the lattice water molecules. The observed weight loss of 1.31% is in agreement with the calculated value of 2.09%. The dehydrated complex weight loss of 3.89% observed is in agreement with the calculated value of 5.13%, corresponding to the loss of one CO2 molecule the in the L ligand. The frameworks collapsed in the temperature range of 313–519 °C. The remaining weight may correspond to the final products of ZnO in 2 (obsd 18.38%, calcd 18.66%). The TG curve of 4 shows two main weight losses, respectively. The first, of 50.01% from 303 to 331 °C, corresponds to the loss of three p-H2BDC molecules (obsd 50.77%, calcd 50.16%). The frameworks collapsed in the temperature range of 439–475 °C. The remaining weight may correspond to the final products of ZnO in 4 (obsd 24.51%, calcd 24.46%). The TG curves of 3 and 5 both show one main weight loss. Respectively, the frameworks collapsed in the temperature ranges of 372–623 °C and 374–618 °C. The remaining weight may correspond to the final products of ZnO in 3 (obsd 21.12%, calcd 20.79%) and ZnO in 5 (obsd 19.01%, calcd 18.66%) as shown in Fig. S13 (in the ESI†).Conclusions
In summary, we have synthesized and characterized five novel compounds utilizing the 1,4-bis(pyridin-4-ylmethoxy)benzene ligand, multi-caboxylate acids and zinc acetate salts. Compounds 1–4 display entangled structures ranging from interpenetration to self-penetrating all the way to poly-threading modes. By adjusting the pH value, the poly-threading architecture in compound 1 was transferred to an unprecedented poly-threading structure of 2, and further pH variation resulted in a 3D supramolecular framework with no entangled motifs as seen in compound 5. The successful isolation of compounds 1–5 not only provides fascinating instances of chemical topology but also enriches the new born poly-threading family.Acknowledgements
The authors gratefully acknowledge the financial support from the NNSF of China (No. 21001022), The National Grand Fundamental Research 973 Program of China (2010CB635114), Program for New Century Excellent Talents in Chinese University (NCET-10-0282), PhD Station Foundation of Ministry of Education (20100043110003), The Foundation for Author of National Excellent Doctoral Dissertation of P.R. China (FANEDD) (No. 201022), The Science and Technology Development Planning of Jilin Province (201001169, 20100182), The Fundamental Research Funds for the Central Universities (09QNJJ018, 09ZDQD003, 10CXTD001), Open Project Program of Key Laboratory of Functional Inorganic Material Chemistry (Heilongjiang University), Ministry of Education.Notes and references
- (a) T. F. Liu, D. Fu, S. Gao, Y. Z. Zhang, H. L. Sun, G. Su and Y. J. Liu, J. Am. Chem. Soc., 2003, 125, 13976 CrossRef CAS; (b) R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428 CrossRef CAS; (c) M. Eddaoudi, H. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391 CrossRef CAS; (d) A. J. Fletcher, E. J. Cussen, T. J. Prior, M. J. Rosseinsky, C. J. Kepert and K. M. Thoms, J. Am. Chem. Soc., 2001, 123, 10001 CrossRef CAS; (e) S. I. Noro, S. Kitagawa, M. Kondo and K. Seki, Angew. Chem., Int. Ed., 2000, 39, 2082 CrossRef CAS; (f) M. J. Zaworotko, Angew. Chem., Int. Ed., 2000, 39, 3052 CrossRef CAS; (g) X. Y. Wang, L. Wang, Z. M. Wang, G. Su and S. Gao, J. Am. Chem. Soc., 2006, 128, 674 CrossRef CAS.
- Molecular Catenanes, Rotaxanes and Knots, A Journey Through the World of Molecular Topology, ed. J. P. Sauvage and C. Dietrich-Buchecker, Wiley-VCH, Weinheim, 1999 Search PubMed.
- (a) K. Liang, H. G. Zheng, Y. G. Song, M. F. Lappert, Y. Z. Li, X. Q. Xin, Z. X. Huang, J. T. Chen and S. F. Lu, Angew. Chem., Int. Ed., 2004, 43, 5776 CrossRef CAS; (b) X. H. Bu, M. L. Tong, H. C. Chang, S. Kitagawa and S. R. Batten, Angew. Chem., Int. Ed., 2004, 43, 192 CrossRef CAS; (c) S. A. Bourne, J. J. Lu, B. Moulton and M. J. Zaworotko, Chem. Commun., 2001, 861 Search PubMed.
- (a) Q. M. Wang, G. C. Guo and T. C. W. Mak, Chem. Commun., 1999, 1849 RSC; (b) P. Jensen, D. J. Price, S. R. Batten, B. Moubaraki and K. S. Murray, Chem.–Eur. J., 2000, 6, 3186 CrossRef CAS; (c) S. H. Chiu, S. J. Rowan, S. J. Cantrill, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem. Commun., 2002, 2948 RSC; (d) M. Eddaoudi, J. Kim, N. L. Rosi, D. T. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS; (e) B. Moulton, H. Abourahma, M. W. Bradner, J. J. Lu, G. J. McManus and M. J. Zaworotko, Chem. Commun., 2003, 1342 RSC; (f) A. Galet, V. Niel, M. C. Munoz and J. A. Real, J. Am. Chem. Soc., 2003, 125, 14224 CrossRef CAS; (g) R. Vaidhyanathan, S. Natarajan and C. N. R. Rao, Cryst. Growth Des., 2003, 3, 47 CrossRef CAS; (h) T. J. Prior and M. J. Rosseinsky, Inorg. Chem., 2003, 42, 1564 CrossRef CAS.
- (a) S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef; (b) S. R. Batten, CrystEngComm, 2001, 3, 67 RSC.
- (a) B. F. Abrahams, M. J. Hardie, B. F. Hoskins, R. Robson and E. E. Sutherland, J. Chem. Soc., Chem. Commun., 1994, 1049 RSC; (b) L. Carlucci, G. Ciani, D. M. Proserpio and F. Porta, Angew. Chem., Int. Ed., 2003, 42, 317 CrossRef CAS; (c) B. F. Abrahams, S. R. Batten, M. J. Grannas, H. Hamit, B. F. Hoskins and R. Robson, Angew. Chem., Int. Ed., 1999, 38, 1475 CrossRef CAS; (d) M. A. Withersby, A. J. Blake, N. R. Champness, P. A. Cooke, P. Hubberstey and M. Schröer, J. Am. Chem. Soc., 2000, 122, 4044 CrossRef CAS; (e) M. L. Tong, X. M. Chen and S. R. Batten, J. Am. Chem. Soc., 2003, 125, 16170 CrossRef CAS; (f) L. Carlucci, G. Ciani, D. M. Proserpio and S. Rizzato, J. Chem. Soc., Dalton Trans., 2000, 3821 RSC; (g) D. L. Long, R. J. Hill, A. J. Blake, N. R. Champness, P. Hubberstey, C. Wilson and M. Schröer, Chem.–Eur. J., 2005, 11, 1384 CrossRef CAS; (h) X. J. Wang, C. H. Zhan, Y. L. Feng, Y. Z. Lan, J. L. Yin and J. W. Cheng, CrystEngComm, 2011, 13, 684 RSC.
- (a) L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247 CrossRef CAS; (b) S. Banfi, L. Carlucci, E. Caruso, G. Ciani and D. M. Proserpio, J. Chem. Soc., Dalton Trans., 2002, 2714 RSC; (c) G. F. Liu, B. H. Ye, Y. H. Ling and X. M. Chen, Chem. Commun., 2002, 1442 RSC; (d) L. Carlucci, G. Ciani and D. M. Proserpio, Chem. Commun., 1999, 449 RSC; (e) M. L. Tong, H. J. Chen and X. M. Chen, Inorg. Chem., 2000, 39, 2235 CrossRef CAS.
- (a) J. P. Sauvage, Acc. Chem. Res., 1998, 31, 611 CrossRef CAS; (b) S. I. Jun, J. W. Lee, S. Sakamoto, K. Yamaguchi and K. Kim, Tetrahedron Lett., 2000, 41, 471 CrossRef CAS.
- (a) X. L. Wang, C. Qin, E. B. Wang, Y. G. Li, Z. M. Su, L. Xu and L. Carlucci, Angew. Chem., Int. Ed., 2005, 44, 5824 CrossRef CAS; (b) C. Qin, X. L. Wang, L. Carlucci, M. L. Tong, E. B. Wang, C. W. Hu and L. Xu, Chem. Commun., 2004, 1876 RSC; (c) L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2003, 5, 269 RSC.
- (a) X. L. Wang, C. Qin, E. B. Wang, Z. M. Su, L. Xu and C. W. Hu, Angew. Chem., Int. Ed., 2004, 43, 5036 CrossRef CAS; (b) X. L. Wang, C. Qin, E. B. Wang, Z. M. Su, Y. G. Li and L. Xu, Angew. Chem., Int. Ed., 2006, 45, 7411 CrossRef CAS; (c) X. L. Wang, C. Qin, Y. Q. Lan, K. Z. Shao, Z. M. Su and E. B. Wang, Chem. Commun., 2009, 410 RSC.
- (a) J. Yang, J. F. Ma, Y. Y. Liu and S. R. Batten, CrystEngComm, 2009, 11, 151 RSC; (b) G. J. Xu, Y. H. Zhao, K. Z. Shao, Y. Q. Lan, X. L. Wang, Z. M. Su and L. K. Yan, CrystEngComm, 2009, 11, 1842 RSC; (c) L. J. Li, G. Yuan, L. Chen, D. Y. Du, X. L. Wang, G. J. Xu, H. N. Wang, K. Z. Shao and Z. M. Su, J. Coord. Chem., 2011, 64, 1578 CrossRef CAS.
- (a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS; (b) G. Ferey, C. M. Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217 CrossRef CAS; (c) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS.
- (a) L. P. Zhang, C. K. Lam, H. B. Song and T. C. W. Mak, Polyhedron, 2004, 23, 2413 CrossRef CAS; (b) H. W. Hou, T. Y. Fan, L. P. Zhang, C. X. Du and Y. Zhu, Inorg. Chem. Commun., 2001, 4, 168 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97, Program for X-ray Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- (a) E. C. Yang, H. K. Zhao, B. Ding, X. G. Wang and X. J. Zhao, Cryst. Growth Des., 2007, 7, 2009 CrossRef CAS; (b) Y. Y. Liu, J. F. Ma, J. Yang, J. C. Ma and G. J. Ping, CrystEngComm, 2008, 10, 565 RSC; (c) K. Z. Shao, Y. H. Zhao, Y. Xing, Y. Q. Lan, X. L. Wang, Z. M. Su and R. S. Wang, Cryst. Growth Des., 2008, 8, 2986 CrossRef CAS; (d) X. M. Zhang, M. L. Tong, M. L. Gong and X. M. Chen, Eur. J. Inorg. Chem., 2003, 138 CrossRef; (e) A. D. Burrows, R. W. Harrington, M. F. Mahon and S. J. Teat, Eur. J. Inorg. Chem., 2003, 766 CrossRef CAS.
- (a) X. J. Wang, C. H. Zhan, Y. L. Feng, Y. Z. Lan, J. L. Yin and J. W. Cheng, CrystEngComm, 2011, 13, 684 RSC; (b) G. S. Yang, Y. Q. Lan, H. Y. Zang, K. Z. Shao, X. L. Wang, Z. M. Su and C. J. Jiang, CrystEngComm, 2009, 11, 274 RSC; (c) X. L. Wang, C. Qin, E. B. Wang, Z. M. Su, L. Xu and S. R. Batten, Chem. Commun., 2005, 4789 RSC.
- (a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS; (b) G. Ferey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217 CrossRef CAS.
- C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS.
- (a) L. Y. Zhang, J. P. Zhang, Y. Y. Lin and X. M. Chen, Cryst. Growth Des., 2006, 6, 1685 Search PubMed; (b) Q. Chu, G. X. Liu, Y. Q. Huang, X. F. Wang and W. Y. Sun, Dalton Trans., 2007, 4302 RSC; (c) G. H. Wang, Z. G. Li, H. Q. Jia, N. H. Hu and J. W. Xu, Cryst. Growth Des., 2008, 8, 1932 CrossRef CAS; (d) Y. Y. Qin, J. Zhang, Z. J. Li, L. Zhang, X. Y. Cao and Y. G. Yao, Chem. Commun., 2008, 2532 RSC; (e) Y. H. He, Y. L. Feng, Y. Z. Lan and Y. H. Wen, Cryst. Growth Des., 2008, 8, 3586 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structural information, thermogravimetric analysis, spectroscopy details (Fig. S1–S7, Tables S1–S3). CCDC reference numbers 804835–804839. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ce05810j |
| This journal is © The Royal Society of Chemistry 2012 |