Facile synthesis and observation of discontinuous red-shift photoluminescence of CdTe/CdS core/shell nanocrystals†
Weiwei
Xu
ac,
Huaibin
Shen
a,
Jin Zhong
Niu
a,
Changhua
Zhou
a,
Cailan
Yu
b,
Xiaomin
Li
a,
Yuan
Hang
c,
Hongzhe
Wang
a,
Lan
Ma
*c and
Lin Song
Li
*a
aKey Laboratory for Special Functional Materials of Ministry of Education, Henan University, Kaifeng, 475004, P. R. China. E-mail: lsli@henu.edu.cn; Fax: +86-378-3881358; Tel: +86-378-3881358
bKey Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100080, P. R. China
cLife Science Division, Graduate School at Shenzhen, Tsinghua University, Shenzhen, 518055, P. R. China. E-mail: malan@sz.tsinghua.edu.cn
First published on 25th October 2011
Abstract
A simple method was introduced to synthesize CdTe/CdS core/shell nanocrystals (NCs) by using a new kind of “green” tellurium precursor. Both absorption and photoluminescence (PL) spectra of CdTe/CdS NCs exhibited a discontinuous shift as follows: slight (<10 nm), large (∼30 nm), and slight (<10 nm) red-shift during the growth of CdS shell. Different sized CdTe cores were introduced to synthesize CdTe/CdS core/shell NCs and similar discontinuous red-shift phenomena in the PL spectra were observed. These characteristics indicated that the structure of CdTe/CdS NCs gradually evolved from type-I to type-II, in agreement with transmission electron microscopy (TEM), the tuning of PL quantum yields (QYs), and PL lifetime results.
Introduction
Recently, semiconductor nanocrystals (NCs) with core/shell structures have attracted tremendous attention in fundamental studies as well as for potential applications, such as light emitting diodes,1–4 lasers,5–7 and solar cells.8–10 Such core/shell structures are usually divided into two categories, type-I and type-II, depending on the relative alignment of conduction and valence band edges of the materials that are combined at the heterointerface. For type-I core/shell NCs, both electrons and holes are confined in the same core or shell, and these have been preferentially investigated to achieve high photoluminescence (PL) quantum yields (QYs).11 In the case of type-II core/shell NCs, the energy gradient exists at the interfaces and tends to spatially separate electrons and holes on different sides of the heterojunction. Due to the giant bending effect12,13 and long radiative lifetime,14–16 type-II NCs should have a great deal of potential applications. Until now, various type-II semiconductor materials, such as CdS/ZnSe,17ZnSe/CdS,18CdSe/ZnSe,19 and CdTe/CdSe20–22 have been discussed in detail. However, few studies have been focused on the optical properties and band offsets of CdTe/CdS core/shell NCs. Yang's group reported that CdTe/CdS NCs formed type-I structures under certain conditions by using Te with trioctylphosphine (TOP) as the Te precursor.23 Zhu's group reported evidence that CdTe/CdS behaved as a mixture of a type-I and type-II structure.24 Zhang and co-workers reported that type-II CdTe/CdS NCs had been synthesised via successive ion layer adsorption and reaction in the aqueous phase.25 Even with such progress, the relationship between optical properties and the band offsets of CdTe/CdS core/shell NCs, and how the optical properties are tuned by manipulating the band offset, is still not entirely clear.Herein, we focus on the study of optical properties and adjusting the band offsets by the synthesis of CdTe/CdS core/shell NCs. To study the optical properties upon consecutive growth of CdS shells in detail, 0.02 mmol S or Cd precursors have been added alternately. With the increase of CdS shell thickness step by step, both the absorption onsets in the absorption spectra and the band-edges of emission in the PL spectra exhibited discontinuous red-shift. Meanwhile, different sizes of CdTe cores were introduced to synthesize CdTe/CdS core/shell NCs and similar PL red-shift phenomena were observed. In particular, we have investigated the effect of CdS shell thickness on PL QY and PL decay. It was found that the structure of CdTe/CdS NCs gradually evolved from type-I to type-II with the increase of CdS shell thickness.
Experimental
Chemicals
All reagents were used as received without further experimental purification. Cadmium oxide (CdO, 99.998%), tri-n-octylphosphine oxide (TOPO, 90%), sulfur powder (S, 99.9%), tellurium powder (Te, 99.8%), oleic acid (OA, 90%), and 1-octadecene (ODE, 90%) were purchased from Aldrich. Hexanes (analytical grade) and methanol (analytical grade) were obtained from Beijing Chemical Reagent Ltd, China.Stock solutions for Cd, Te and S precursors
Synthesis of CdTe/CdS NCs
A mixture of Cd precursor I (1 mL, 0.2 mmol mL−1) and paraffin oil (3 g) was heated to 280 °C under a nitrogen environment in a 25 mL three-necked flask. Next, Te precursor (1 mL, 0.1 mmol mL−1) was injected and the temperature was decreased to 250 °C for NC growth.26 The as-synthesized CdTe NCs were purified by precipitation with methanol and redispersed in hexanes. A typical CdS shell growth was performed as follows: 6 mL of ODE was loaded into a 50 mL reaction flask, the purified CdTe NCs in hexanes were added, and the system was kept at 100 °C under nitrogen flow for 30 min to remove hexanes. Subsequently, the solution was heated to 230 °C for shell growth. Then, the S precursor stock solution (0.2 mL) was added to the reaction flask. After 10 min, once the S precursor was fully overcoated on the CdTe surface, an equimolar amount of Cd precursor stock solution was added to the reaction system. After that, the S/Cd precursor solution was added at intervals of 10 min. With the addition of the S/Cd precursors, the original slightly red solution gradually turned deep-red and finally into a black solution. For purification, 10 mL of hexanes was added and the unreacted compounds and byproducts were removed by successive methanol extraction.Characterization
UV-vis absorption spectra and PL spectra were recorded using an Ocean Optics spectrophotometer (mode PC2000-ISA). PL spectra were taken using an excitation wavelength of 365 nm. PL QYs of NCs were measured relative to organic dyes with known emission efficiencies. All the QYs data of NCs and dyes were collected through SPEX F212. PL QYs of NCs were calculated using the method reported previously.26–28 All optical measurement was performed at room temperature under ambient conditions. X-ray diffraction (XRD) studies of NCs were carried out with a Philips X'Pert Pro X-ray diffractometer using Cu-Kα radiation. Drop casted films of the NCs were deposited on glass substrates. To obtain better signal-to-noise ratios, the XRD data were collected at a scan rate of 16 s for each step and step intervals of 0.2°. Transmission electron microscopy (TEM) studies were performed using a 100 CX microscope.Results and discussion
For the synthesis of CdTe NCs, Te powder was directly dissolved in TOPO at 400 °C to form a stable precursor solution. Because of the high chemical stability of TOPO and Te in air, the Te precursor is stable for several weeks and can be used effectively for the NCs synthesis under nitrogen flow. This method does not involve the use of a glove box throughout the entire synthesis. Using this precursor, high quality CdTe NCs with an approximate PL range of 563–643 nm can be synthesized. In order to study the changes in optical properties and band offsets of CdTe/CdS NCs in detail, we employed a CdS shell growth method by alternatively injecting 0.2 mL (0.1 mmol mL−1) of S or Cd precursors into the reaction flask.The most direct and immediate evidence for the CdS shell growth comes from the absorption and PL spectra, which show a discontinuous red-shift phenomenon. Fig. 1 shows the temporal evolution of the absorption and PL spectra of aliquots which were taken from the reaction solution during the overgrowth of the CdS shell on CdTe cores at 230 °C. The PL peak of CdTe cores is 601 nm with full width at half maximum (FWHM) of 41 nm. With the increase of CdS shell thickness, both the absorption onsets in the absorption spectra and the PL emission peaks in the PL spectra shift systematically to the red (or lower bandgap energy). After injecting two times of S and Cd precursors, the PL peaks only had a slight red-shift (∼7 nm) and FWHM remained unchanged. This phenomenon was consistent with the main feature of type-I NCs, and indicated that the CdTe core was covered by a thin CdS shell or a possible CdTeS structure was formed in the shell. When 0.6 mL, 0.8 mL, and 1 mL S with Cd precursors were injected, the corresponding PL peaks of CdTe/CdS red-shifted to 634 nm, 671 nm, and 705 nm, respectively. Table 1 shows that the corresponding PL peaks red-shifted 26, 27, and 34 nm, and the FWHM of the corresponding PL peaks increased from 40 nm to 90 nm. This was caused by the valence band of CdTe and CdS being closer, and electrons were localized in the CdTe core and CdS shell. This is a process of the adjustment from a type-I to type-II structure. Finally, there is a 33 nm red-shift with the growth of the shell, which is consistent with the main feature of a type-II structure. Overall, the red-shift process of the PL peaks has three steps; slight, large, and slight again with the increase of CdS shell thickness. We call this a “PL jump” and attribute it to the change of core/shell band offsets with the increase of the CdS shell. Such a PL red shift for CdTe/CdS NCs was different from other core/shell structures. For example, when we use a similar method (injecting 0.2 mL of Se and Cd precursors alternatively) to synthesize type-II CdTe/CdSe NCs, the PL peak has a slight red-shift each time and did not show the red-shift jump (see Fig. S1, ESI).
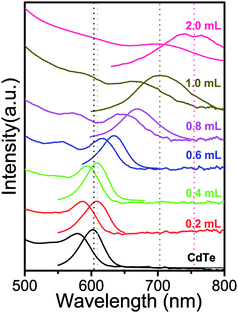 | ||
| Fig. 1 Absorption (left) and PL (right) spectra upon consecutive growth of CdTe/CdS NCs. | ||
| Sample | Precursor (mL) | λ em (nm) | Δλem (nm) | FWHM (nm) | Φ PL (%) |
|---|---|---|---|---|---|
| CdTe | 0 | 601 | 7 | 41 | 20 |
| CdTe/CdS | 0.4 | 608 | 26 | 40 | 23 |
| CdTe/CdS | 0.6 | 634 | 27 | 45 | 21 |
| CdTe/CdS | 0.8 | 671 | 34 | 62 | 18 |
| CdTe/CdS | 1 | 705 | 34 | 90 | 15 |
| CdTe/CdS | 2 | 738 | 112 | 13 |
Generally, the bandgap energy of the core/shell system depends on the radius of the core, the shell thickness, the conduction-band offset between two materials, and the effective masses of the charge carriers in the respective materials.24,29–32 For the energy bands of bulk CdTe and CdS in our system, CdS is in principle a good candidate as the shell material for CdTe cores not only because its band gap (2.5 eV) is wider than that of CdTe (1.5 eV) but also because its lattice parameter mismatch (3.6%) to CdTe is relatively small compared with ZnSe (12.5%) and ZnS (16.5%), and therefore it gives quite a small band offset for the bulk conduction bands and a large band offset for the bulk valence bands as shown in Fig. 2A.33,34 Concerning the band alignments at the bulk interface between CdTe/CdS, the electronic band potentials for both CdS and CdTe are pulled up by the quantum confinement for the NCs and the current literature provides different data sets, both from theory and experiments.24,34,35 Consequently, we simply assume that the band edges of nanosized CdS and CdTe make three situations with the increase of CdS shell thickness (Fig. 2B). The conduction band edge of the CdTe core was situated lower than that of the CdS thin shell, so electrons and holes were primarily localized in the core and a type-I structure was formed. Then the conduction band edge of the shell was pulled down to closer to the conduction band energy of the core with the increase of shell thickness, which means tuning from a type-I structure. Finally, the conduction band edge of the shell was lower than that of the core and a type-II structure was formed. This showed the possibility of the structure of CdTe/CdS NCs gradually evolving from type-I to type-II with the increase of CdS shell thickness from theory.
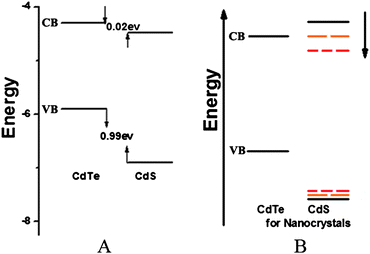 | ||
| Fig. 2 A) Band offsets of bulk CdTe and CdS. B) Schematic of tunable band offsets of CdTe/CdS NCs with different shell thicknesses. | ||
When a core/shell system evolves from a type-I to a type-II structure, the electron–hole recombination process, which is responsible for the PL, also changes from direct recombination to indirect recombination. Such a change leads to the decline of the QY. Upon coating the CdTe cores with thinner CdS shells, the QYs of the core/shell NCs has not changed obviously which was caused by the passivation of the core surface by the shell. However, the QYs of the CdTe/CdS NCs decreased steadily from about 23% to approximately 13% with the addition of further CdS shells. This QY decrease was taken as the feature of type-II NCs which usually led to a decline of PL QY. A similar conclusion was also drawn for this system in previous studies.24,25
Typical TEM images for the as-prepared CdTe and CdTe/CdS NCs are shown in Fig. 3, and the corresponding size distribution histograms obtained from the TEM images are shown in Fig. S2. It is quite evident that these NCs, which have a typical size distribution of approximately ±10%, are dot-shaped with excellent monodispersity. The diameters of the NCs were determined to be 2.84 nm for the CdTe cores and 3.19 nm, 3.24 nm and 4.65 nm after the CdTe cores were covered with the designed CdS shells using 0.4, 0.6 and 2 mL S and Cd precursors, respectively. The size difference of the CdTe/CdS NCs between Fig. 3b and c was only about 0.1 nm, and the difference in the PL peak was 27 nm. This proved that the PL jump was not caused by the increased size of CdTe/CdS NCs, but because the position of the conduction bands was pulled down with the growth of the CdS shell. This indicated that the CdTe/CdS NCs gradually evolve from type-I to type-II NCs. The HRTEM image of CdTe/CdS core/shell NCs is shown in Fig. 3e and the (400) planes are separated by d = 0.145 nm, which is consistent with that of zinc blende CdS. The HRTEM images of core/shell NCs show lattice planes extend straight across the particle, with no evidence of an interface between the core and shells. This implies the coherent epitaxial growth of CdS shells along the lattice planes of the CdTe core.
 | ||
| Fig. 3 TEM images of a) CdTe and b–d) CdTe/CdS NCs after injecting 0.4 mL, 0.6 mL, and 2 mL of S and Cd precursors. e) high-resolution TEM image of the corresponding samples. | ||
To further characterize the crystal structures of the CdTe cores and CdTe/CdS core/shell NCs, their crystallographic properties were determined using powder X-ray diffraction (XRD) (Fig. 4). For comparison, the standard powder diffraction patterns of zinc blende CdTe and CdS bulk crystals are provided. According to the X-ray powder diffraction pattern, the peak positions of the CdTe NCs are in agreement with bulk CdTe in the zinc blende structure (JCPDS, 75–2086). Three obvious diffraction peaks located at 24.0, 39.7, and 46.4 degrees were indexed as the (111), (220), and (311) planes of zinc blende phased CdTe, respectively. When overcoated with CdS, the XRD patterns shifted from a zinc blende CdTe-like one to a zinc blende CdS-like one upon the increase of the shell thickness. The peak positions for CdTe/CdS core/shell NCs were located between those of pure CdTe and pure CdS (Fig. 4).
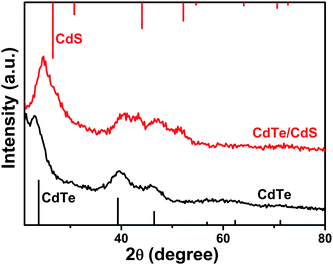 | ||
| Fig. 4 X-ray diffraction patterns of CdTe (601 nm) and CdTe/CdS NCs (after the injection of 2 mL S and Cd precursors). The lines show the peak positions for bulk zinc blende CdTe (bottom, JCPDS, 75–2086) and CdS (top, JCPDS, 80–0019). | ||
To verify radiation mechanisms, the radiative lifetime of CdTe/CdS PL emission was introduced.25,36–38 The emission decay curves were fitted by dual exponentials, following I(t) = A + B1*exp(−t/T1) + B2*exp(−t/T2), where I(t) is PL intensity, T1 (T2) represents the decay time of the PL emission; B1 (B2) represents the amplitude of the decay components at t = 0. The average lifetime T is calculated by an expression T = B1*T1 + B2*T2 and the radiative lifetime of the NCs is calculated by using Tr = ΦPL/T, where ΦPL is the PL QY. Table 2 displays the PL decay parameters of CdTe and CdTe/CdS NCs having λem of 601, 608, 634, and 705 nm, and their corresponding lifetimes are 27.34, 25.7, 32.12, and 37.91 ns, respectively. The lifetime shortens for thin shells and lengthens again for thicker shells, which accorded with the PL peaks jump red-shift (see Fig. S3, ESI). The type-I structure of NCs existed for a short period with the growth of the CdS thin shell and the type II structure of NCs existed for a long period with the growth of the CdS thick shell. This result would be related to the systematic gradual evolution from type-I to type-II NCs. The radiative lifetime for CdTe/CdS NCs was changed gradually from 136.7 ns for the bare core to 111.7, 153, and 252.7 ns for the shells, which is also consistent with systematic gradual evolution from type-I to type-II core/shell NCs.
| Sample | PL/nm | T 1/ns | A 1 (%) | T 2/ns | A 2 (%) | T/ns | T r/ns |
|---|---|---|---|---|---|---|---|
| CdTe | 601 | 8.284 | 9.46 | 29.34 | 90.54 | 27.34 | 136.7 |
| CdTe/CdS | 608 | 6.312 | 12.5 | 28.48 | 87.50 | 25.70 | 111.7 |
| CdTe/CdS | 634 | 52.09 | 27.75 | 24.45 | 72.25 | 32.12 | 153 |
| CdTe/CdS | 705 | 7.275 | 8.11 | 40.62 | 91.89 | 37.91 | 252.7 |
To investigate the effect of the CdTe core on the PL jump red-shift of core/shell NCs, different sized CdTe cores were selected. Fig. 5 shows the temporal evolution of CdTe/CdS NCs PL spectra, which were prepared by using different sized CdTe cores. The PL peaks of CdTe/CdS NCs with thinner shells have a slight red-shift, a large red-shift with the growth of thicker shells, and then a slight red-shift for the growth of even thicker shells using CdTe cores with PL between 563 and 643 nm (Fig. 6). Unfortunately, the exact value cannot be determined for this case because of insufficient data on the interface of CdTe/CdS. However, the band offset between type-I and type-II structures with different CdTe core or CdS shell thickness can still be judged according to the temporal evolution of the CdTe/CdS NCs optical properties.
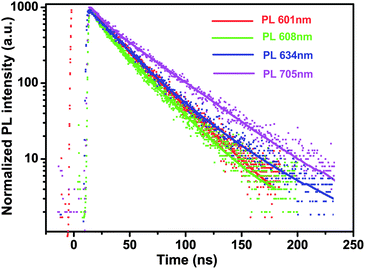 | ||
| Fig. 5 PL decays of CdTe and CdTe/CdS NCs detected at 601 nm, 608 nm, 634 nm, and 705 nm, respectively. | ||
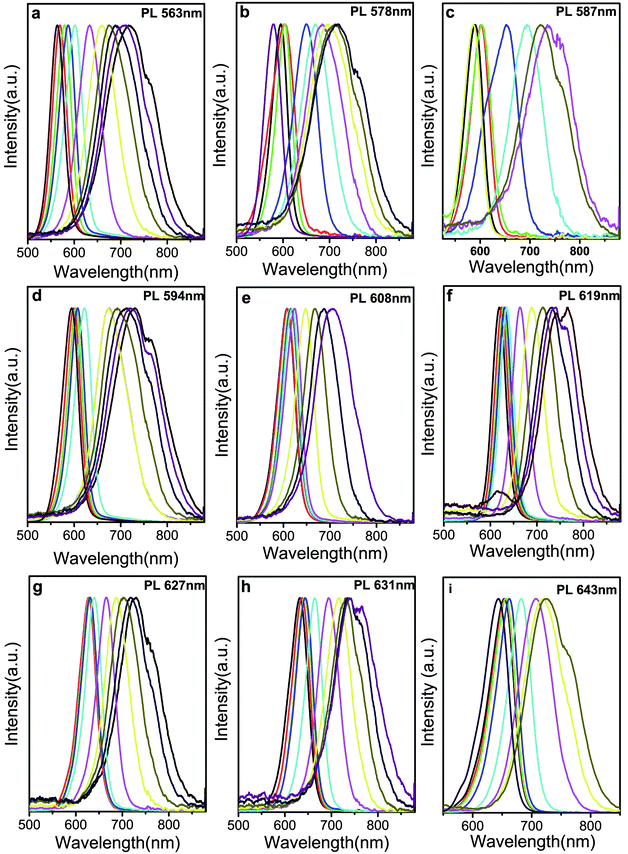 | ||
| Fig. 6 PL spectra of CdTe/CdS NCs with different sized CdTe cores, the emission for CdTe cores are: (a) 563 nm; (b) 578 nm; (c) 587 nm; (d) 594 nm, (e) 608 nm, (f) 619 nm, (g) 627 nm, (h) 631 nm, (i) 643 nm. | ||
Conclusions
High quality CdTe/CdS core/shell NCs have been successfully synthesized using a new kind of “green” tellurium precursor to avoid the use of a glove box. The thickness of the CdS shell was accurately controlled by injecting equivalent precursors. The absorption spectra and photoluminescence peaks of CdTe/CdS NCs exhibited a discontinuous red-shift as follows: slight, large, and slight red-shift during the growth of CdS shells. TEM, PL QY, and PL decay results supported that this phenomenon was due to the transition from type-I to type-II structures with the gradual growth of the CdS shell. Different sized CdTe cores were introduced to synthesize CdTe/CdS and also show similar discontinuous red-shift phenomena in the PL spectra.Acknowledgements
This work was financially supported by the research project of the National Natural Science Foundation of China (21071041) and Program for New Century Excellent Talents in University of Chinese Ministry of Education.References
- S. Coe, W.-K. Woo, M. Bawendi and V. Bulovic, Nature, 2002, 420, 800 CrossRef CAS.
- N. Tessler, V. Medvedev, M. Kazes, S. Kan and U. Banin, Science, 2002, 295, 1506 CrossRef.
- J. Lee, V. C. Sundar, J. R. Heine, M. G. Bawendi and K. F. Jensen, Adv. Mater., 2000, 12, 1102 CrossRef CAS.
- Q. Sun, Y. A. Wang, L. S. Li, D. Wang, T. Zhu, J. Xu, C. Yang and Y. Li, Nat. Photonics, 2007, 1, 717 CrossRef CAS.
- L. P. Balet, S. A. Ivanov, A. Piryatinski, M. Achermann and V. I. Klimov, Nano Lett., 2004, 4, 1485 CrossRef CAS.
- V. I. Klimov, S. A. Ivanov, J. Nanda, M. Achermann, I. Bezel, J. A. McGuire and A. Piryatinski, Nature, 2007, 447, 441 CrossRef CAS.
- J. Nanda, S. A. Ivanov, M. Achermann, I. Bezel, A. Piryatinski and V. I. Klimov, J. Phys. Chem. C, 2007, 111, 15382 CAS.
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354 CrossRef CAS.
- M. Kazes, D. Y. Lewis, Y. Ebenstein, T. Mokari and U. Banin, Adv. Mater., 2002, 14, 317 CrossRef CAS.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425 CrossRef CAS.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463 CrossRef CAS.
- M. Larsson, A. Elfving, P. O. Holtz, G. V. Hansson and W.-X. Ni, Appl. Phys. Lett., 2003, 82, 4785 CrossRef CAS.
- Y. S. Chiu, M. H. Ya, W. S. Su and Y. F. Chen, J. Appl. Phys., 2002, 92, 5810 CrossRef CAS.
- K. Das and S. K. De, J. Phys. Chem. C, 2009, 113, 3494 CAS.
- D. Dorfs, A. Salant, I. Popov and U. Banin, Small, 2008, 4, 1319 CrossRef CAS.
- D. Dorfs, T. Franzl, R. Osovsky, M. Brumer, E. Lifshitz, T. A. Klar and A. Eychmüller, Small, 2008, 4, 1148 CrossRef CAS.
- S. A. Ivanov, A. Piryatinski, J. Nanda, S. Tretiak, K. R. Zavadil, W. O. Wallace, D. Werder and V. I. Klimov, J. Am. Chem. Soc., 2007, 129, 11708 CrossRef CAS.
- A. Nemchinov, M. Kirsanova, N. N. Hewa-Kasakarage and M. Zamkov, J. Phys. Chem. C, 2008, 112, 9301 CAS.
- M. Danek, K. F. Jensen, C. B. Murray and M. G. Bawendi, Chem. Mater., 1996, 8, 173 CrossRef CAS.
- C. H. Wang, T. T. Chen, K. W. Tan, Y. F. Chen, C. T. Cheng and P. T. Chou, J. Appl. Phys., 2006, 99, 123521 CrossRef.
- P.-T. Chou, C.-Y. Chen, C.-T. Cheng, S.-C. Pu, K.-C. Wu, Y.-M. Cheng, C.-W. Lai, Y.-H. Chou and H.-T. Chiu, ChemPhysChem, 2006, 7, 222 CrossRef CAS.
- J. Bang, B. Chon, N. Won, J. Nam, T. Joo and S. Kim, J. Phys. Chem. C, 2009, 113, 6320 CAS.
- C. Jia-Yaw, S.-R. Wang and C.-H. Yang, Nanotechnology, 2007, 18, 345602 CrossRef.
- J. Wang, Y. Long, Y. Zhang, X. Zhong and L. Zhu, ChemPhysChem, 2009, 10, 680 CrossRef CAS.
- Q. Zeng, X. Kong, Y. Sun, Y. Zhang, L. Tu, J. Zhao and H. Zhang, J. Phys. Chem. C, 2008, 112, 8587 CAS.
- H. Shen, H. Wang, X. Chen, J. Z. Niu, W. Xu, X. M. Li, X.-D. Jiang, Z. Du and L. S. Li, Chem. Mater., 2010, 22, 4756 CrossRef CAS.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991 CrossRef CAS.
- H. Shen, H. Wang, Z. Tang, J. Z. Niu, S. Lou, Z. Du and L. S. Li, CrystEngComm, 2009, 11, 1733 RSC.
- A. R. Kortan, R. Hull, R. L. Opila, M. G. Bawendi, M. L. Steigerwald, P. J. Carroll and L. E. Brus, J. Am. Chem. Soc., 1990, 112, 1327 CrossRef CAS.
- J. W. Haus, H. S. Zhou, I. Honma and H. Komiyama, Phys. Rev. B: Condens. Matter, 1993, 47, 1359 CrossRef CAS.
- D. Schooss, A. Mews, A. Eychmüller and H. Weller, Phys. Rev. B: Condens. Matter, 1994, 49, 17072 CrossRef CAS.
- A. Sitt, F. D. Sala, G. Menagen and U. Banin, Nano Lett., 2009, 9, 3470 CrossRef CAS.
- M. A. Hines and P. Guyot-Sionnest, J. Phys. Chem., 1996, 100, 468 CrossRef CAS.
- S.-H. Wei, S. B. Zhang and A. Zunger, J. Appl. Phys., 2000, 87, 1304 CrossRef CAS.
- D. W. Niles and H. Höchst, Phys. Rev. B: Condens. Matter, 1990, 41, 12710 CrossRef CAS.
- X. Li, H. Shen, S. Li, J. Z. Niu, H. Wang and L. S. Li, J. Mater. Chem., 2010, 20, 923 RSC.
- Y. Nonoguchi, T. Nakashima and T. Kawai, Small, 2009, 5, 2403 CrossRef CAS.
- H. Shen, C. Zhou, S. Xu, C. Yu, H. Wang, X. Chen and L. S. Li, J. Mater. Chem., 2011, 21, 6046 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ce05757j |
| This journal is © The Royal Society of Chemistry 2012 |