DOI:
10.1039/C1CE05728F
(Paper)
CrystEngComm, 2012,
14, 138-146
Different crystal forms of a rich hydrogen bond acceptor compound resulting from alternative C–H⋯O and orthogonal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) O⋯CO molecular interaction patterns†‡
O⋯CO molecular interaction patterns†‡
Received
15th June 2011
, Accepted 9th September 2011
First published on 14th October 2011
Abstract
The racemic compound 2,4,6,8-tetracarbomethoxybicyclo[3.3.0]octa-2,6-diene-3,7-diol 3 is known to form a solvent-free crystal structure (Polymorph I) through repetition of a centrosymmetric dimeric unit that is constructed by concave face to concave face association of two shallow dish-shaped tetraester 3 molecules. Compounds employing such awkwardly shaped repeat units are known to be prone to yielding more than one crystal form. Systematic screening has now revealed a second solvent-free crystal form (Polymorph II), and also a family of essentially isostructural lattice inclusion compounds (Inclusion form I), of 3. The X-ray crystal structures of Polymorph II, and the p-xylene and cyclohexanol compounds of the inclusion crystal form, are described. The predominant weak interactions in all three crystal forms comprise C–H⋯O and orthogonal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CO attractive forces and their different arrangements are compared and contrasted.
O⋯CO attractive forces and their different arrangements are compared and contrasted.
Introduction
The ability of a given compound to produce more than one crystal form is a topic of much contemporary interest, and there is a growing realisation that this solid state diversity is far more frequent than formerly believed.2–4 Hence a substance might be capable of yielding multiple clathrates,5,6hydrates,7–9hydrogen bonded cocrystalline complexes,10–12 or polymorphs,13–15 under different crystallisation conditions. Kitaigorodsky argued that organic molecules should normally adopt close-packing in their crystals,16 but early work on clathrate compounds suggested that their formation was related to molecules that would pack awkwardly in their pure state.17–19
What exactly is meant, however, by ‘awkward’ in this context? Proteins are examples of molecules whose overall shape frequently does not permit them to close-pack efficiently. In consequence, protein crystal structures often contain large quantities of disordered solvent, though this outcome is generally ignored in the much more important context of the protein structure. On the other hand, a molecule like hydroquinone 1, despite having a very simple molecular structure and being highly symmetrical, yields several different crystal forms.20 The solvent-free α-form has an exceptionally complex crystal structure and contains an astonishing 54 hydroquinone molecules in its unit cell.21
The enigmatic property of awkwardness therefore can arise from either the shape of the molecule itself, or from directional orientations imposed by the intermolecular forces (such as hydrogen bonding). The outcomes from these characteristics have been termed packing frustration22 and supramolecular synthon frustration,23 respectively. In both cases, the incorporation of solvent molecules with appropriate complementarity could result in denser crystal packing of lower energy.16 It has been argued recently that this awkward concept extends more widely to encompass formation of the other crystal form types mentioned above, and also Z′ > 1 crystal structures.23,24 Although prediction is difficult at the present time, the present paper describes an example of awkward shape where this can be done.
Dishes are stacked simply in the kitchen in a concave face to convex face manner. Therefore dish-shaped molecules that do not pack in this way are exhibiting awkwardness, and are candidates for producing clathrate structures and/or yielding more than one crystal form. A familiar example is the steroidal bile acidcholic acid 2, which has a fairly rigid shallow dish-like topology with a hydrophobic convex face and a concave face carrying hydrophilic substituents capable of hydrogen bonding. This molecule crystallises by means of concave face to concave face hydrogen bonded associations which thereby create an awkwardly shaped repeat unit. Cholic acid is a very effective inclusion host and also yields more than one crystal form.25,26 This report explores the crystallisation behaviour of the simple tetraester 3 that also has a dish-shaped topology.
Results
Preparation of the tetraester 3
The racemic C2-symmetric tetraester 3 is obtained from reduction of Schroeter and Vossen's Red Salt27,28 using sodium amalgam29 or, more simply, by condensation of one equivalent of glyoxal with two equivalents of dimethyl 3-ketoglutarate.30–32Crystallisation of 3 from methanol33 or acetone34 gave identical crystals that showed indications of inefficient packing in the solid state. We have a long-standing interest in the crystal structures of alicyclic diols and especially those that exhibit inclusion properties.35–41 Consequently we felt that the crystallisation properties of compound 3 warranted more detailed examination.
The P21/n crystal structure of 3 (Polymorph I)
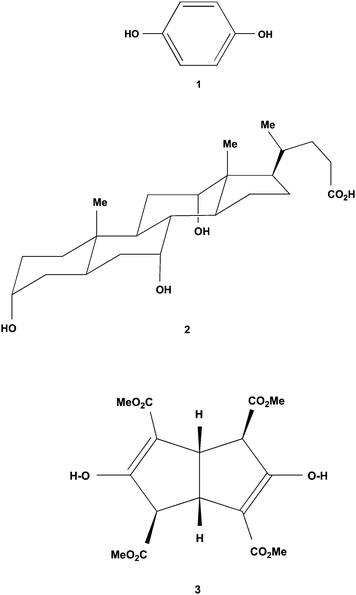
Crystallisation of racemic 3 from either methanol33 or acetone34 at room temperature gave highly crystalline material in space groupP21/n that is free of included solvent (Polymorph I). The molecule 3 adopts a shape like a shallow dish with a handle. Although 3 has C2-symmetry in solution, this is not carried through to the solid state where one ester group C12 (the handle) is orientated quite differently to the other three, as seen in Fig. 1. There is cyclic intramolecular hydrogen bonding O1–H⋯O3C9 and O2–H⋯O7C13, between each enol hydroxy group and the carbonyl oxygen of its adjacent conjugated ester group. These motifs are formed even though polar solvents containing hydrogen bond acceptor atoms had been used in both recrystallisations. Numerical details of the molecular attractions present in this crystal form are listed in Table 1. These data are those for the published structure.34
 |
| | Fig. 1 The molecular conformation adopted by the tetraester 3 in Polymorph I crystals. This perspective highlights its shallow dish-like shape and the one protruding handle-like C12 ester group. The crystallographic numbering shown applies to all the crystal structures of 3 discussed here. The intramolecular hydrogen bonding (dashed lines) is present in both Polymorph I and Inclusion form I, and a highly modified variant of it in the Polymorph II structure. | |
Table 1 Numerical details of the molecular interactions present in the Polymorph I structure of tetraester 334,a
| Type |
Donor (D)–H⋯Acceptor (A) |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
Angle (°) |
|
Equivalent position indicators: a 1/2 − x, 1/2 + y, 1/2 − z b 1 − x, 1 − y, 1 − z c 1/2 + x, 3/2 − y, 1/2 + z. Intra indicates an interaction within the dimeric unit, and inter between dimers.
|
| Intra |
O1–H⋯O3[C9] |
0.89(3) |
1.89(3) |
2.672(2) |
146(2) |
| Intra |
O2–H⋯O7[C13] |
0.85(2) |
1.91(3) |
2.660(2) |
146(2) |
| Intra |
C5–H⋯O6[C11] |
1.00 |
2.62 |
2.981(2) |
101 |
| Inter |
C1–H⋯O7a[C13] |
1.00 |
2.30 |
3.147(2) |
142 |
| Inter |
C4–H⋯O2b–[C7] |
1.00 |
2.53 |
2.237(2) |
128 |
| Type |
CO⋯CO |
O⋯C (Å) |
CO⋯C (°) |
O⋯CO (°) |
| Inter |
C9O3⋯C13 cO7 |
2.962(2) |
165.1(2) |
108.5(2) |
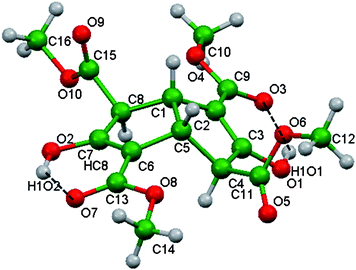
Hunter has drawn attention to molecules that are deficient in hydrogen bonding acceptor groups and the consequences this may have on intermolecular association.42 The tetraester 3 represents the reverse situation. There is a large difference between the number of hydrogen bond donor (two) and acceptor (ten) atoms present in its molecular structure, and this imbalance is exacerbated by the intramolecular hydrogen bonding already described. No strong hydrogen bonding occurs between different molecules of 3 in Polymorph I, and only weaker supramolecular synthons43 operate. The C1–H and C5–H hydrogen atoms, plus the exo-C4 and exo-C8 carbomethoxy groups, of 3 also preclude the stacking of the dish-like molecules in a concave face to convex face manner.24 Instead, opposite pairs of enantiomers of 3 associate and produce a cyclic centrosymmetric double-dish unit with the two concave surfaces facing each other (Fig. 2, upper). Formation of this unit utilises two identical C–H⋯O weak hydrogen bonds44,45 between C4–C4H of one molecule and O2 of its enantiomer. These double-dish units constitute an awkwardly shaped molecular combination, but nonetheless their repetition generates the relatively simple overall crystal structure. Pure enantiomers of 3, also linked by C–H⋯O interactions, surround twofold screw axes, and the resulting helices are linked by means of two orthogonal CO⋯CO contacts (Fig. 2 lower).
 |
| | Fig. 2 Upper: The centrosymmetric double-dish formed by opposite enantiomers of 3 in Polymorph I crystals. Lower: repetition of the double-dish unit generates the overall crystal structure of solvent-free 3. Five of these units are shown, together with the inter-unit associations. The weak intermolecular attractions are indicated by dashed lines: C–H⋯O (thin black dashes) and orthogonal CO⋯CO (thick blue dashes). Atom code: O red, H light blue, C green (opposite enantiomers light or dark). | |
Allen and coworkers have reported that both parallel and orthogonal carbonyl - carbonyl interactions can be competitive with hydrogen bonds in crystal assembly.46 The antiparallel carbonyl - carbonyl motif occurs in a series of polymorphs of indanetrione 1,2-dioxime solvates recently described by Kobayashi,47 and sheared parallel arrangements have been discussed by Mak.48 The non-planar geometry of tetraester 3, however, precludes the involvement of these interaction types. The orthogonal CO⋯CO supramolecular synthon has been surveyed and analysed by Diederich and his colleagues who obtained 16546 Cambridge Structural Database hits for O⋯C distances in the range 2.25–3.75 Å.49,50 This dipolar orthogonal CO⋯CO interaction is therefore widespread amongst carbonyl-containing compounds where it often plays an important role in crystal packing.46,51 This is certainly the case for the carbonyl-rich tetraester 3.
A new solvent-free crystal form of 3 (Polymorph II)
We screened the crystallisation properties of racemic 3 by growing X-ray quality crystals from a range of test solvents, and then examining these by using X-ray diffractometry. Several further examples of the original solvent-free structure in space groupP21/n were observed at room temperature, such as the crystals obtained from cyclopentane, dichloromethane or t-butylbenzene. However, crystallisation of racemic 3 from methanol at 0 °C yielded a second solvent-free structure in space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Polymorph II). Numerical details of the solution and refinement of this X-ray structure, and also those of two inclusion compounds (Inclusion form I, described later) are presented in Table 2.
(Polymorph II). Numerical details of the solution and refinement of this X-ray structure, and also those of two inclusion compounds (Inclusion form I, described later) are presented in Table 2.
Table 2 Numerical details of the solution and refinement of the crystal structures
| Compound |
3
|
(3)2·(p-xylene) |
(3)2·(cyclohexanol) |
| Crystal form |
Polymorph II |
Inclusion form I |
Inclusion form I |
| Formula |
C16H18O10 |
(C16H18O10)·(C8H10)0.5 |
(C16H18O10)·C6H11O)0.5 |
| Formula mass |
370.30 |
423.38 |
419.88 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P
|
P21/c |
P21/c |
|
a/Å |
7.2550(2) |
11.0883(5) |
11.3769(5) |
|
b/Å |
9.2807(3) |
8.5689(3) |
8.3198(4) |
|
c/Å |
13.0344(4) |
21.5332(9) |
21.4511(10) |
|
α (°) |
77.359(1) |
90 |
90 |
|
β (°) |
78.816(1) |
93.104(1) |
98.021(3) |
|
γ (°) |
88.811(1) |
90 |
90 |
|
V/Å3 |
839.87(4) |
2042.97(14) |
2010.56(16) |
|
Z
|
2 |
4 |
4 |
|
μ/mm−1 |
0.12 |
0.11 |
0.11 |
| Crystal size/mm |
0.23 × 0.20 × 0.19 |
0.46 × 0.44 × 0.37 |
0.45 × 0.12 × 0.10 |
|
T
min, Tmax |
0.973, 0.977 |
0.951, 0.960 |
0.951, 0.989 |
| No. measured, |
11935 |
14126 |
14448 |
| independent, and |
2945 |
3587 |
3522 |
| obsd. [I > 2σ(I)] reflections |
2743 |
3242 |
2586 |
|
R
int
|
0.041 |
0.045 |
0.054 |
|
R [F2 > 2σ(F2)] |
0.033 |
0.036 |
0.059 |
|
wR
(F2) |
0.089 |
0.100 |
0.181 |
|
S
|
1.05 |
1.05 |
1.03 |
| No. of reflections |
2945 |
3587 |
3522 |
| No. of parameters |
247 |
276 |
289 |
| No. of restraints |
0 |
0 |
52 |
| Δ > max, Δ > min/e Å−3 |
0.23, −0.22 |
0.30, −0.22 |
0.55, −0.95 |
| CCDC number |
829076
|
829075
|
829074
|
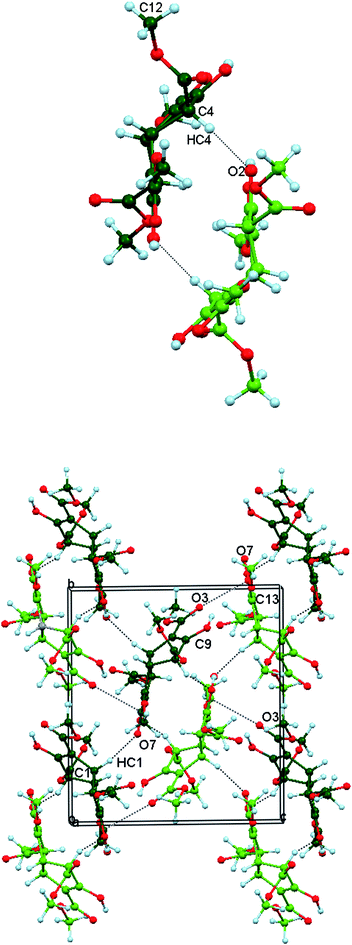
Organisation of the molecules in Polymorph II involves the combination of intra- and inter-molecular O–H⋯O hydrogen bonding shown in Fig. 3 (upper). (Only the intramolecular type is present in the other two crystal forms). Molecules along the b axis are linked into chains by four-membered cycles, in which the diagonally opposite hydroxy oxygens O1–H1O1 and O2–H1O2 make bifurcated contacts with the carbonyl oxygens O3 and O7. Polymorph II contains homochiral layers of 3 in the ab plane. The hydrogen bonded chains are connected along the a direction by a bifurcated interaction involving O9. This comprises a CO⋯CO dipolar interaction (C15O9⋯C13O7) and an associated C–H⋯O contact (C4–H4⋯O9), as seen in Fig. 3 (lower).
 |
| | Fig. 3 Upper: Part of a hydrogen bonded chain running along b in crystalline Polymorph II and showing the combination of intra- and inter-molecular hydroxy group hydrogen bonding between the tetraester 3 molecules. Lower: Part of a homochiral layer of tetraester 3 molecules in Form II projected onto the ab plane. The hydrogen bonded chains along b are rotated relative to the representation shown above. They are joined to their neighbours by C4–H4⋯O9 weak hydrogen bonds (black dots) and orthogonal C15O9⋯C13O7 interactions (blue dashes). | |
Layers of opposite handedness alternate along c and are weakly associated by means of C10–H10C⋯O3 and CH3⋯π interactions (involving the C12 methyl and C2/C9) (Fig. 4). Numerical values are listed in Table 3 for these attractive interactions present in this crystal form.
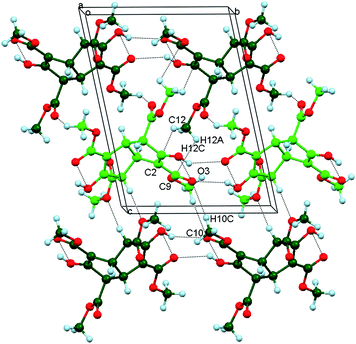 |
| | Fig. 4 The arrangement of molecules in Polymorph II projected onto the bc plane and showing the edge-on layers of alternating handedness (light or dark green) along the c direction. Adjacent layers are linked by C10–C10H⋯O3 and CH3⋯π weak hydrogen bonds. | |
Table 3 Numerical details of the molecular interactions present in Polymorph II of tetraester 3a
| Type |
Donor (D)–H⋯Acceptor (A) |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
Angle (°) |
|
Equivalent position indicators: a x, 1 + y, z b x, −1 + y, z c 1 − x, 1 − y, 2 − z d 1 + x, y, z.
|
| Intra |
O1–H⋯O3[C9] |
0.88(2) |
2.04(2) |
2.758(2) |
138(2) |
| Intra |
O2–H⋯O7[C13] |
0.91(2) |
2.00(2) |
2.762(4) |
140(2) |
| Intra |
C1–H3⋯O9[C15] |
1.00 |
2.52 |
2.920(2) |
104 |
| Intra |
C5–H⋯O5[C11] |
1.00 |
2.46 |
2.861(2) |
103 |
| Inter |
O1–H⋯O7a[C13] |
0.88 (2) |
2.24(2) |
2.933(1) |
135(2) |
| Inter |
O2–H⋯O3b[C9] |
0.91(2) |
2.21(2) |
2.891(1) |
130(2) |
| Inter |
C10–H⋯O3c[C9] |
0.98 |
2.42 |
3.367(2) |
161 |
| Type |
CO⋯CO |
O⋯C (Å) |
CO⋯C (°) |
O⋯CO (°) |
| Inter |
C15O9⋯C13dO7 |
3.015(2) |
146.0(1) |
90.7(1) |
| C–H⋯pi |
| C12–H12A⋯C9 |
d = 2.80 |
C12–H12C⋯C2 |
d = 2.76 |
|
|
Clathrate crystal form of 3 (Inclusion form I)
Crystallisation screening of tetraester 3 at room temperature also revealed a series of essentially isostructural clathrate compounds in space groupP21/c (Inclusion form I). These materials contained, for example, carbon disulfide, diethyl ether, hexane, dioxane or chlorobenzene in the host–guest ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Numerical details of the solution and refinement of the compounds (3)2.(p-xylene) and (3)2.(cyclohexanol) are presented in Table 2. A thorough analysis of the molecular inclusion properties of 3 has been carried out and will be reported shortly.
:1. Numerical details of the solution and refinement of the compounds (3)2.(p-xylene) and (3)2.(cyclohexanol) are presented in Table 2. A thorough analysis of the molecular inclusion properties of 3 has been carried out and will be reported shortly.
Host molecules of the same chirality assemble around a crystallographic 21 screw axis along b as shown in Fig. 5. This molecular association involves assembly of the shallow dishes of 3 in a convex face to concave face manner. In doing so, however, the molecules are tilted away from the screw axis to avoid unfavourable steric contacts from the protruding C1–H and C5–H atoms. This orientation also optimises attractive inter-host contacts. Adjacent pairs of molecules of 3 are bridged via two different C–H⋯O and two different CO⋯CO interactions, the numerical details of which are listed in Table 4 for each of (3)2.(p-xylene) and (3)2.(cyclohexanol). These values lie within the expected ranges. Both carbonyl interactions are near orthogonal but exhibit dissimilar CO⋯C angles. The Polymorph I and Inclusion form I utilise only intramolecular enol - ester hydrogen bonding, unlike the different arrangement present in the Polymorph II structure.
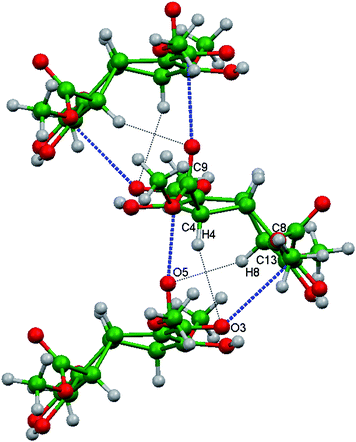 |
| | Fig. 5 Part of the crystal structure of (3)2.(p-xylene) showing the helical assembly of homochiral host molecules around the crystallographic 21 screw axis running along the b direction in the Inclusion form I structure. Adjacent pairs of molecules are bridged via two different C–H⋯O (black ⋯) and two different orthogonal CO⋯CO (blue ---) interactions. | |
Table 4 Numerical details of the molecular interactions present in Inclusion form I
|
Host
-
host
interactions for (33)2·(p-xylene) |
| Type |
Donor (D)–H⋯Acceptor (A) |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
Angle (°) |
| Intra |
O1–H⋯O3[C9] |
0.84 |
2.00 |
2.700(1) |
140 |
| Intra |
O2–H⋯O7[C13] |
0.84 |
1.95 |
2.655(1) |
141 |
| Inter |
C16–H16A⋯O9a[C15] |
0.98 |
2.52 |
3.406(2) |
150 |
| Type |
CO⋯CO |
O⋯C (Å) |
CO⋯C (°) |
O⋯CO (°) |
|
| Inter |
C9O3⋯C13O7b |
3.096(2) |
138.6(1) |
88.1(1) |
|
| Inter |
C11O5⋯C9O3b |
2.956(2) |
167.3(1) |
87.9(1) |
|
| Equivalent position indicator: a 1 − x, 1 − y, −z b 1 − x, 1/2 + y, 1/2 − z |
|
Host
-
host
interactions for (33)2·(cyclohexanol) |
| Intra |
O1–H⋯O3[C9] |
0.84 |
2.06 |
2.752(3) |
139 |
| Intra |
O2–H⋯O7[C13] |
0.84 |
2.05 |
2.744(3) |
139 |
| Inter |
O1–H⋯O1a |
0.84 |
2.21 |
2.847(8) |
133 |
| Inter |
C12–H12A⋯O5b[C11] |
0.98 |
2.54 |
3.517(4) |
177 |
| Inter |
C12–H12B⋯O10c–[C15] |
0.98 |
2.53 |
3.474(4) |
161 |
| Inter |
C16–H16A⋯O3d[C9] |
0.98 |
2.59 |
3.510(4) |
156 |
| Type |
CO⋯CO |
O⋯C (Å) |
CO⋯C (°) |
O⋯CO (°) |
| Inter |
C15O9⋯C13O7e |
2.850(4) |
165.9(2) |
85.7(2) |
| Inter |
C13O7⋯C9O3e |
3.107(4) |
135.3(2) |
88.6(2) |
| Equivalent position indicators: a 1 − x, 1 − y, 1 − z b −x, −y, 1 − z c x, 1/2 − y, 1/2 + zd 1 − x, −1/2 + y, 1/2 − z e −x, −1/2 + y, 1/2 − z |
|
Host
-guest interactions for (33)2·(p-xylene) |
|
Host O2⋯H3X2–C3X guest |
d = 2.709, D = 3.081 Å |
103.01° |
|
Host
-guest interactions for (33)2·(cyclohexanol) |
|
Host O1–O1H⋯O1X guest |
D = 2.847 Å |
133.08° |
|
Host = C2⋯H–C3X guest |
d = 2.81 Å |
|
The 21 helices pack parallel to each other as homochiral host layers in the ab plane. Layers of alternating chirality pack along the c direction and parallel channels with a roughly rectangular cross-section (approximately 5.0 × 6.1 Å) are generated between the neighbouring helices (Fig. 6, upper). These channels run along b and are occupied by the guest molecules. Each ordered guest in (3)2.(p-xylene) is located on a centre of inversion and is anchored at each end by means of identical host (enol) O2⋯H3X2–C3X (methyl) guest interactions (Fig. 6, centre).
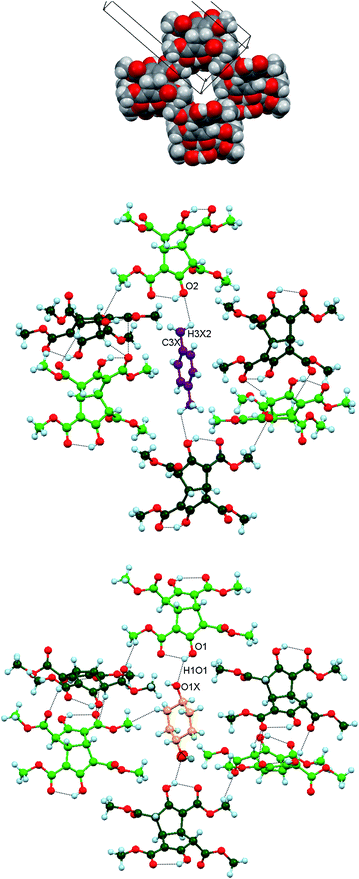 |
| | Fig. 6 Upper: Space filling representation of Inclusion form I projected near the ac plane and showing the cross-section of one empty guest channel along the b direction. Atom code: O red, C grey, H off-white. Centre: Part of the crystal structure of (3)2.(p-xylene) showing one ordered p-xylene molecule (C magenta) within the guest channel. The two identical host O2⋯H3X2–C3X guest interactions are indicated. Lower: The comparable view of (3)2.(cyclohexanol) with the two equivalent guest positions superimposed. The cyclohexanol oxygen atom is shown as two ellipsoids, both coloured red to represent each of these alternative orientations. The guest C atoms are shown in yellow. | |
In (3)2.(cyclohexanol), however, the guest molecule is disordered over two equivalent positions that are shown superimposed on a host inversion centre. One host–guest attraction is shown in Fig. 6 (lower), namely a host (enol) O1–O1H1⋯O1X guest interaction. There is also a C2C3(OH) host enol π⋯H–C3X guest interaction, involving a different molecule of 3 located below those shown. The latter hydrogen atom is α- to the CH(OH) group and consequently is slightly more acidic than the other guest methylene hydrogens. In the second guest orientation, the equivalent H atom interacts with a tetraester molecule above those shown. Numerical values for these host–guest interactions appear in Table 4.
Discussion
The calculated densities of the four structures are 1.401 (Polymorph I), 1.464 (Polymorph II), 1.377 (p-xylene compound), and 1.387 g cm−3 (cyclohexanol compound). It is notable that both apohost polymorphs were obtained from methanol solution, but at slightly different temperatures, and that the density of Polymorph II (obtained at 0 °C) is significantly higher than that of Polymorph I (consistently obtained at room temperature, 20 °C). This observation indicates that factors additional to close-packing play an important role in the behaviour of tetraester 3. The packing coefficients of Polymorph I, Polymorph II, and the p-xylene compound are 67.7, 70.4, and 68.7%, respectively. Once again, this indicates that Polymorph II is a better-packed structure than Polymorph I.
The three crystal forms of 3 involve different ester group conformations. Such conformational change is a common means of a molecule achieving alternative crystal forms.14 A recent related example involves an annulene structure containing two dimethyl phthalate groups, the ester groups of which can adopt different orientations to yield three conformational polymorphs.52 The conformations observed for tetraester 3 in the three crystal forms are generally similar apart from the orientation of one ester group O5C11–O6–C12H3, which causes significant changes in the molecular shape (Fig. 7). This substituent protrudes outwards like a handle in Polymorph I, rotated by around 180° relative to the more compact Inclusion form I. The Polymorph II arrangement is closer in overall shape to Polymorph I than the inclusion structure. The torsion angle C3–C4–C11–O6 is 57.13°, −90.16°, 115.13° (p-xylene) and 118.00° (cyclohexanol) in Polymorph I, Polymorph II and the two inclusion compounds, respectively.
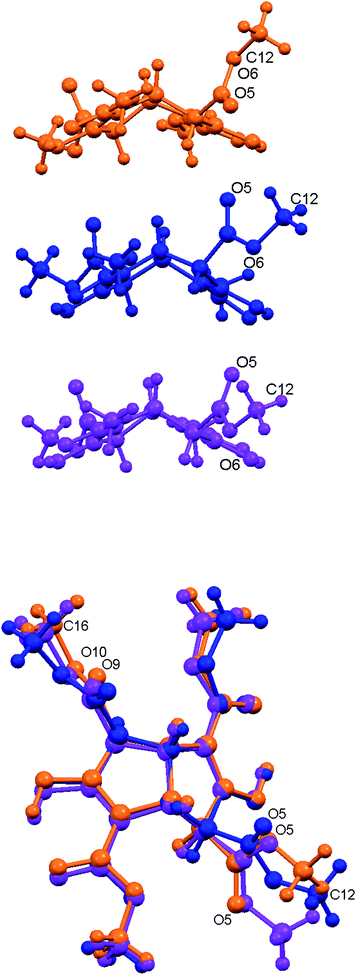 |
| | Fig. 7 Upper: Side view of tetraester 3 in the three crystal forms; Polymorph I (orange), Polymorph II (blue) and Inclusion form I (p-xylene; magenta). Lower: Overlap of these three conformations. | |
Approximately half of the test recrystallisation solvents yielded the apohost Polymorph I, and half the Inclusion form I. These experiments were conducted at the same temperature and pressure, and hence their outcomes depend solely on the solvent. There appears to be low correlation of outcome with solvent polarity or chemical functionality, but guest size is an important parameter. These, and other, behavioural aspects of 3 will be the subject of a later publication.
The Polymorph I and Inclusion form I show some structural similarity with both containing molecules of 3 arranged around crystallographic twofold screw axes. These homochiral 21 helices in Polymorph I pack parallel to each other and run along the b direction. Helices of opposite handedness alternate along c and do not leave any space for guest inclusion. These two crystal forms are also similar in using only intramolecular hydroxy group hydrogen bonding, whereas Polymorph II utilises a combination of intra- and inter-molecular associations. This change results in layers, instead of helices, of 3 being produced. Despite their different packing arrangements all three structures undergo a considerable degree of enantiomer separation53 during their crystal formation, as illustrated in Fig. 8. In the inclusion structure, however, the domains of opposite handedness become separated due to the insertion of guest molecules.
 |
| | Fig. 8 Relative arrangements of the enantiomers of 3 in the three crystal forms. Upper: Polymorph I projected onto the bc plane. Centre: Polymorph II also projected on the bc plane. Lower: The Inclusion form I p-xylene compound. Colour code: molecules of 3 green (opposite enantiomers dark or light), and the p-xylene guests blue. | |
Conclusions
The published crystal structure of tetraester 3 (Polymorph I)34 involves an awkwardly shaped concave face to concave face dimer. Repetition of this unit results in a simple crystal packing arrangement in which only two types of weak intermolecular attractions, C–H⋯O and orthogonal CO⋯CO, are involved. These characteristics led us to suspect that alternative crystal structures might be possible. Our views were vindicated by discovery of a second solvent-free structure of 3 (Polymorph II) and a family of essentially isostructural lattice inclusion compounds (Inclusion form I). It is noteworthy that the awkward repeat unit of Polymorph I, which alerted us to the packing difficulties of 3,1,24 is abandoned in these new structures. The prevalence of hydrogen bond acceptor groups in the molecular structure of 3 makes it ideal for generation of different arrangements and combinations of C–H⋯O weak hydrogen bonds and orthogonal CO⋯CO dipolar attractions in its alternative crystal forms.
These results support our contention that awkwardly shaped molecules can sometimes be identified and that these have a high probability of yielding multiple crystal forms under ambient laboratory conditions. On the other hand, prediction of exactly how this will occur remains problematic at our current level of understanding.
Experimental
NMR data were recorded using a Bruker DPX300 instrument (1H 300 MHz, 13C 75.4 MHz) at 25 °C and are reported as chemical shifts (δ) relative to SiMe4. Carbon substitution information was determined using the DEPT procedure, and the IR spectrum recorded using a Perkin-Elmer PE298 spectrophotometer.
The tetraester was prepared by reduction of the Schroeter and Vossen Red Salt27,28 using 2% sodium amalgam following the procedure of Yates et al.29 Identical material was also synthesised using the method of Bertz et al.32 Mp 103–105 °C (from methanol), lit.29 104–107.5 °C. IR (paraffin mull): 1730s, 1650s, 1620m, 1320m, 1250s, 1190m, 1150m, 1040w, 1010w, 980w, 780m cm−1. 1H NMR (CDCl3): δ 3.56 (s, 2H), 3.61 (t, J = 2.6 Hz, 2H), 3.75 (s, 6H), 3.77 (s, 6H), 3.84 (t, J = 2.6 Hz, 2H). 13C NMR (CDCl3): δ 43.8 (CH), 51.6 (CH3), 52.3 (CH3), 55.2 (CH), 103.8 (C), 169.0 (C), 170.6 (C), 170.8 (C). Crystals were grown by dissolving pure 3 in each test solvent and allowing slow evaporation to occur. These were then examined by diffractometry, and solution continued for those crystals which proved to be different to the apohost structure in space groupP21/n (Polymorph I).33,34
Structure determinations
A suitable single crystal was selected under a polarizing microscope (Leica M165Z) and was mounted on a cryoloop with paraffin oil, which fixed onto it upon freezing at the temperature of data collection. The intensities were measured on a Bruker kappa APEX-II CCD diffractometer equipped with graphite monochromated Mo Ka radiation (λ = 0.71073 Å) and operating at a low temperature of 150(2) K maintained using an Oxford Cryostream 700 system. Upon obtaining an initial refinement of unit cell parameters, the data collection strategy achieved a redundancy of at least 4 throughout the resolution range (inf-0.80 A°) at 10 s exposure time per frame making use of the kappa offsets on the four-circle goniometer geometry. The data integration and reduction with the multiscan absorption correction method54 was carried out using the APEX2 suite of software.55 The structure was solved by Direct methods using SHELXS-9756 and was refined by the full-matrix least-squares refinement program SHELXL to the final R value. All non-hydrogen atoms were refined anisotropically. All the H-atoms could be located in the difference Fourier map, but in the least-squares all H-atoms were fixed stereochemically using the riding model option in SHELXL, excepting those belonging to the hydroxy groups. The guest molecule of cyclohexanol is disordered around centre of inversion, creating a statistical centre of symmetry that the original molecule lacks. The two-fold (orientationally) disordered model was used in the least-squares refinement, with molecular geometry and thermal parameters restrained using the restraints DFIX, DELU and SIMU available in SHELXL. Molecular graphics were generated using ORTEP-3v2. Key crystallographic data and refinement details are shown in Table 1.
Acknowledgements
We gratefully acknowledge financial support from the Australian Research Council.
References
- Part 7. I. Y. H. Chan, M. M. Bhadbhade and R. Bishop, Design of a new inclusion host: 3,7-diphenylbicyclo[3.3.0]octane- endo-3, endo-7-diol, CrystEngComm, 2011, 13, 3162 RSC.
- S. R. Vippagunta, H. G. Brittain and D. J. W. Grant, Adv. Drug Delivery Rev., 2001, 48, 3 CrossRef CAS.
-
S. Aldridge, The Shape Shifters, Chemistry World, April 2007, pp. 64–70 Search PubMed.
-
D. Braga, M. Curzi, S. L. Giaffreda, F. Grepioni, L. Maini, A. Pettersen and M. Polito, Nanoporous crystals, co-crystals, isomers and polymorphs from crystals, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH Verlag, Weinheim, 2008, pp. 155–177 Search PubMed.
-
Comprehensive Supramolecular Chemistry, Vol. 6 Solid-state Supramolecular Chemistry: Crystal Engineering, ed. D. D. MacNicol, F. Toda and R. Bishop, Pergamon Press, Oxford, 1996 Search PubMed.
-
F. H. Herbstein, Crystalline Molecular Complexes and Compounds: Structures and Principles, Oxford University Press, Oxford, 2005 Search PubMed.
- R. Custelcean, C. Afloroaei, M. Vlassa and M. Polverejan, Angew. Chem., Int. Ed., 2000, 39, 3094 CrossRef CAS.
- B. C. R. Sansam, K. M. Anderson and J. W. Steed, Cryst. Growth Des., 2007, 7, 2649 CAS.
- L. Infantes, L. Fabian and W. D. S. Motherwell, CrystEngComm, 2007, 9, 65 RSC.
- F. Lara-Ochoa and G. Espinosa-Pérez, Supramol. Chem., 2007, 19, 553 CrossRef CAS.
- M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 4 CAS.
-
P. Vishweshwar, J. A. McMahon and M. J. Zaworotko, Crystal Engineering of Pharmaceutical Co-Crystals, in Frontiers in Crystal Engineering, ed. E. R. T. Tiekink and J. J. Vittal, Wiley, Chichester, 2006, Ch. 2, pp 25–49 Search PubMed.
- J. Bernstein, R. J. Davey and J.-O. Hencke, Angew. Chem., Int. Ed., 1999, 38, 3440 CrossRef.
-
J. Bernstein, Polymorphism in Molecular Crystals, Oxford Science Publications, Oxford, 2002 Search PubMed.
- D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1 RSC.
-
A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- H. M. Powell, J. Chem. Soc., 1948, 61 RSC.
- L. Mandelcorn, Chem. Rev., 1959, 59, 827 CrossRef CAS.
- D. D. MacNicol, J. J. McKendrick and D. R. Wilson, Chem. Soc. Rev., 1978, 7, 65 RSC.
-
T. C. W. Mak and B. R. F. Bracke, Hydroquinone Clathrates and Diamondoid Host Lattices, in Comprehensive Supramolecular Chemistry, Vol. 6 Solid-state Supramolecular Chemistry: Crystal Engineering, ed. D. D. MacNicol, F. Toda and R. Bishop, Pergamon Press, Oxford, 1996, Ch. 2, pp. 23–60 Search PubMed.
- S. C. Wallwork and H. M. Powell, J. Chem. Soc., Perkin Trans. 2, 1980, 641 RSC.
- P. M. Duesing, R. H. Templer and J. M. Seddon, Langmuir, 1997, 13, 251 Search PubMed.
- K. M. Anderson, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2008, 8, 2517 CAS.
- I. Y. H. Chan, V. T. Nguyen, R. Bishop, D. C. Craig and M. L. Scudder, Cryst. Growth Des., 2010, 10, 4582 CAS.
- K. Nakano, E. Mochizuki, N. Yasui, K. Morioka, Y. Yamauchi, N. Kanehisa, Y. Kai, N. Yoswathananont, N. Tohnai, K. Sada and M. Miyata, Eur. J. Org. Chem., 2003, 2428 CrossRef CAS.
- K. Nakano, K. Aburaya, I. Hisaki, N. Tohnai and M. Miyata, Chem. Rec., 2009, 9, 124 CrossRef CAS.
- P. Yates and G. Bhat, Chem. Ind. (London), 1954,(40), 1237 CAS.
- D. Djaidi, R. Bishop, D. C. Craig and M. L. Scudder, New J. Chem., 2002, 26, 614 RSC.
- P. Yates, E. S. Hand and G. B. French, J. Am. Chem. Soc., 1960, 82, 6347 CrossRef CAS.
- U. Weiss and J. M. Edwards, Tetrahedron Lett., 1968, 9, 4885 CrossRef.
- S. H. Bertz, G. Rihs and R. B. Woodward, Tetrahedron, 1982, 38, 63 CrossRef CAS.
- S. H. Bertz, J. M. Cook, A. Gawish and U. Weiss, Org. Synth., 1990, Coll. Vol. VII, 50 Search PubMed.
-
D. Djaidi, Ph.D. Dissertation, The University of New South Wales, Sydney, 2006.
- A. Vega, O. Donoso-Tauda, A. Ibañez and C. A. Escobar, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2008, 64, o199 Search PubMed . CCDC refcode WIZHUJ.
- R. Bishop, S. Choudhury and I. Dance, J. Chem. Soc., Perkin Trans. 2, 1982, 1159 RSC.
- R. Bishop, I. G. Dance and S. C. Hawkins, J. Chem. Soc., Chem. Commun., 1983, 889 RSC.
- S. C. Hawkins, M. L. Scudder, D. C. Craig, A. D. Rae, R. B. Abdul Raof, R. Bishop and I. G. Dance, J. Chem. Soc., Perkin Trans. 2, 1990, 855 RSC.
- R. Bishop, D. C. Craig, I. G. Dance, S. Kim, M. A. I. Mallick, K. C. Pich and M. L. Scudder, Supramol. Chem., 1993, 1, 171 CrossRef CAS.
- R. Bishop, D. C. Craig, M. L. Scudder, A. P. Marchand and Y. Wang, J. Chem. Soc., Perkin Trans. 2, 1993, 937 RSC.
- S. C. Hawkins, R. Bishop, I. G. Dance, T. Lipari, D. C. Craig and M. L. Scudder, J. Chem. Soc., Perkin Trans. 2, 1993, 1729 RSC.
- R. Bishop, Acc. Chem. Res., 2009, 42, 67 CrossRef CAS.
- L. R. Hanton, C. A. Hunter and D. H. Purvis, J. Chem. Soc., Chem. Commun., 1992, 1134 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 1991, 24, 290 CrossRef CAS.
-
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford Science Publications: Oxford, 1999 Search PubMed.
- F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320 CrossRef.
- M. Suzuki and K. Kobayashi, Cryst. Growth Des., 2011, 11, 1814 CAS.
- C.-Q. Wan and T. C. W. Mak, Cryst. Growth Des., 2011, 11, 832 CAS.
- R. Paulini, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788 CrossRef CAS.
- F. R. Fischer, P. A. Wood, F. H. Allen and F. Diederich, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17290 CrossRef CAS.
- C. Fäh, L. A. Hardegger, M.-O. Ebert, W. B. Schweizer and F. Diederich, Chem. Commun., 2010, 46, 67 RSC.
- I. Hisaki, Y. Sakamoto, H. Shigemitsu, N. Tohnai and M. Miyata, Cryst. Growth Des., 2009, 9, 414 CAS.
- V. T. Nguyen, I. S. H. Chan, R. Bishop, D. C. Craig and M. L. Scudder, New J. Chem., 2009, 33, 1736 RSC.
-
G. M. Sheldrick, Sadabs, Empirical Absorption and Correction Software, University of Gottingen, Germany, 1999–2003 Search PubMed.
-
APEX2 and SAINT, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.