Crystal structure of a hybrid salt–cocrystal and its resolution by preferential crystallization: ((±)trans-N,N′-dibenzyldiaminocyclohexane)(2,3-dichlorophenylacetic acid)4†
Julien
Mahieux
,
Silvia
Gonella
,
Morgane
Sanselme
and
Gérard
Coquerel
*
SMS laboratory, University of Rouen, rue Lucien Tesnière, Mont Saint Aignan, 76130, France. E-mail: gerard.coquerel@univ-rouen.fr; Fax: +33(0)2 35 52 29 59; Tel: +33(0)2 35 52 29 27
First published on 11th October 2011
Abstract
trans-N,N′-Dibenzyldiaminocyclohexane (B) crystallizes with 2,3-dichlorophenylacetic acid (AH); the crystal structure, resolved by using crystal X-ray diffraction, revealed an odd stoichiometry composed of H2B2+, two A− and two AH forming an unexpected hybrid salt–cocrystal. As this compound is a stable conglomerate (i.e. every single crystal contains the enantiomerically pure cation H2B2+, RR or SS), several preferential crystallization attempts (AS3PC) were performed in methanol and in THF and gave unexpected final enantiomeric excesses greater than 20% for the entrainment in methanol. These results suggest that the crystal growth mechanism preferentially involves building units composed of [H2B2+; 2A− and 2AH] or reconstruction of some crystal interfaces rather than a layer by layer construction.
Introduction
The demand for chiral compounds is steadily increasing both in academic research and for industrial applications. Although efficient routes of asymmetric syntheses are now available, chiral resolutions of racemic mixtures keep being the preferred options in many large-scale applications. These processes can be run by chromatography on chiral stationary phases,1 biocatalytic processes2 (involving enzymes or microorganisms) and crystallization.3 This latter mode offers several possibilities to resolve a racemic mixture: diastereomeric entities (by formation of salts in the Pasteur resolution4,5 or the enantioselective encapsulation into a chiral macrocycle in the host–guest association6), preferential nucleation,7 simultaneous crystallization,8 symmetry breaking9,10 and preferential crystallization3 (PC). PC itself can be subdivided into several variants according to the nature of the crystallization process11 (e.g. seeding procedures: SIPC (Seeded Isothermal Preferential Crystallization), S3PC (Seeded Polythermic Programmed Preferential Crystallization) and AS3PC12 (Auto-Seeded Polythermic Programmed Preferential Crystallization)) and the possibility or not of in situ racemisation, SOAT13 (Second Order Asymmetric Transformation). Whatever those variants, the pre-requisite condition for application of preferential crystallization is the existence of a stable conglomerate11 (see ref. 14–16 for counter-examples associated with metastable conglomerates). That is to say the racemic mixture of crystals is composed of enantiomerically pure solid particles even if partial solid solutions might exist without impairing preferential crystallization.17 Thus the screening of conglomerates corresponds to the identification of crystallized phases providing full chiral discrimination. When the molecule to be resolved contains an acidic or a basic function, this is usually performed by numerous attempts to crystallize salts. At the top of that, in order to spot conglomerates of solvated or heterosolvated salts,18 it is recommended to make a screening with different solvents or mixture of solvents for every counter-ion.The conglomerates can be spotted by using various methods. The detection of the Second Harmonic Generation (SHG) effect19 is a pre-screening technique, which has recently proved to be fast, cheap and efficient. Nevertheless, complementary analyses are necessary to confirm the existence of a conglomerate by, for instance, comparisons of spectroscopic data such as X-ray powder diffraction (XRPD) or IR or ss-NMR or Raman patterns of the pure enantiomer and of the racemic mixture.
This article aims at showing that odd stoichiometries can sometimes provide the full chiral discrimination in the solid state, necessary for the application of PC. Therefore no a priori restriction should be considered in the screening of associations between partners of crystallization.
The molecule to be resolved is the diaminetrans-N,N′-dibenzyldiaminocyclohexane (compound B) (see the Syntheses section and Fig. 15) synthesized in our laboratory. This diamine crystallizes as a conglomerate with 2,3-dichlorophenylacetic acid (compound AH) (see the Screening of conglomerate section and Fig. 16). It belongs to a series of resolution agents, easy to obtain, and cheap, that are being developed within the European project: IntEnant.‡ An alternative process consists of synthesizing the pure enantiomer of compound B from the pure enantiomer of trans-1,2-diaminocyclohexane. The pure enantiomer of trans-1,2-diaminocyclohexane ((±) DACH) can be resolved by two distinct methods: preferential crystallization20 or formation of diastereomeric salt.21 These resolutions circumvent the asymmetric synthesis of (+) or (−) DACH. Moreover, DACH being simultaneously CO2, O2, hν and moisture sensitive, it is preferably stored as a salt (e.g.citrate monohydrate20).
Results and discussion
Single crystal structural description
The complete structure of the phase between B and AH was determined by single crystal X-ray diffraction. Despite numerous attempts on different single crystals, the R-factors remained greater than 5% probably due to the poor crystal quality (Fig. 1). Nevertheless, the reliability of the results is high enough for an error free determination of the stoichiometry. Crystallographic data are summarized in Table 1. | ||
| Fig. 1 SEM picture of a single crystal of a hybrid salt–cocrystal obtained by smooth evaporation in methanol. Defects at the extremities can be observed on many single crystals. | ||
| Chemical formula | [C6H10–(NH2–CH2–C6H5)2]2+, 2[C6H3Cl2–CH2–COO]−, 2[C6H3Cl2–CH2–COOH] |
| CSD number | CCDC 818797 |
| Molecular weight/g mol−1 | 1114.54 |
| Crystal system | Monoclinic |
| a/Å | 25.444(2) |
| b/Å | 8.0222(7) |
| c/Å | 13.4912(12) |
| β/° | 106.581(1) |
| Unit cell volume/Å3 | 2640.2(4) |
| Calculated density/g cm−3 | 1.402 |
| Space group | C2 (no. 5) |
| No. of formula per unit cell, Z, Z′ | 2, 0.5 |
| Absorption coefficient/μ mm−1 | 0.481 |
| No. of reflections measured | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 636 636 |
| No. of independent reflections | 5364 |
| R int | 0.0202 |
| No. of reflections with I > 2σI | 4681 |
| Final R1 value (I > 2σI) | 0.0657 |
| Final wR2 value (I > 2σI) | 0.1713 |
| Final R1 value (all data) | 0.0739 |
| Final wR2 value (all data) | 0.1802 |
| Goodness on fit | 1.029 |
The asymmetric unit is composed of one molecule of acid (AH), one molecule of deprotonated acid (A−) and one half molecule of base H2B2+ (Fig. 2). Therefore, the 2-fold axis regenerates a building unit (Fig. 3) composed of a single H2B2+ and two pairs of AH and A− that are tightly connected through strong ionic interactions (Table 2).
 | ||
| Fig. 2 Asymmetric unit with ellipsoidal contribution at room temperature (50% of probability). Molecule of the dibase is depicted in grey and white (to illustrate the symmetry generated part), the molecule of deprotonated acid is presented in purple and the full protonated acid is featured in yellow. Symmetry used to generate the equivalent atom: * (−x, y, 2 − z). | ||
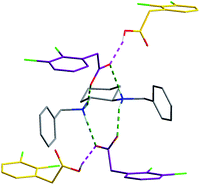 | ||
| Fig. 3 The building unit formed by one molecule of H2B2+ (grey), two molecules of A− (purple) and two molecules of AH (yellow). The ionic bonds are depicted in dashed green lines or pink dashed lines regarding molecules involved. | ||
The oxygen atoms, O(1A) and O(2A), from the carboxylate function of A− establish strong ionic bonds with the two hydrogen atoms of the protonated amine moiety, N(1B)H2. Molecule AH is involved in a second ionic interaction through the hydroxyl group, O(2)H, of the carboxylic moiety with the oxygen atom O(2A) of A−.
For molecule A−, the carbon–oxygen bond lengths are similar whereas for molecule AH these lengths exhibit an unambiguous difference (Table 3). Furthermore, the IRFT spectrum confirms the presence of protonated and deprotonated carboxylic moieties. Indeed, the representative vibrational band of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 1750–1700 cm−1 is split, with equal intensity, into two bands, one at 1700 cm−1 and the other at 1630 cm−1. This latter can be attributed to the carboxylate moiety.
O at 1750–1700 cm−1 is split, with equal intensity, into two bands, one at 1700 cm−1 and the other at 1630 cm−1. This latter can be attributed to the carboxylate moiety.
| Molecule AH | Molecule A− | ||
|---|---|---|---|
| C(8)–O(1) | 1.195(5) Å | C(8A)–O(1A) | 1.233(4) Å |
| C(8)–O(2) | 1.289(6) Å | C(8A)–O(2A) | 1.255(4) Å |
According to these results, a proton transfer occurs between the two acids A− and the base H2B2+, whereas no proton exchange occurs within the other acid molecules AH. Therefore, the title compound is at the same time a salt because of the proton transfer and a cocrystal because the carboxylic moiety is not altered and remains neutral.22,23
The building units are aligned along the b direction and thus formed rows in this direction (Fig. 4). Every building unit interacts with equivalent elementary blocks from adjacent rows along the c axis. These interactions are established between aromatic rings of molecules AH from one row to molecules A− of adjacent rows; the distance between the centroids of these phenyl groups is circa 3.6 Å.
 | ||
| Fig. 4 Representation of the building unit stacking. The π–π interactions are represented in dashed blue lines. The 2-fold axes along the b axis are represented in black. | ||
Therefore, the building units are stacked through π–π interactions and give rise to slices parallel to the (bc) plane with d200 thickness. Moreover, two types of subslices exist along the c axis: the first composed of molecules AH with d003 (=4.31 Å) thickness and the second containing chiral ions: H2B2+ and 2A− with 2d003 (=8.62 Å) thickness (Fig. 5).
 | ||
| Fig. 5 Projection along the b axis, (200) layers are stacked along the a axis with d200 thickness and (003) layers are stacked along the c axis. The symmetry elements are depicted in black. | ||
The comparison between the experimental and the calculated morphologies by using the geometrical BFDH method24 clearly shows that the main growth axis is [010] (Fig. 6). The fibrous aspect corresponds well to the rows spreading along b (Fig. 7).
 | ||
| Fig. 6 Comparison between the experimental and the calculated morphologies. | ||
 | ||
| Fig. 7 SEM picture of a single crystal of a hybrid salt–cocrystal. A fibrous aspect can be observed on the single crystals. | ||
In order to see if AH exhibits special structural features, its crystal structure was also investigated. The crystallographic data and details of the resolution are summarized in Table 4.
| Chemical formula | [C6H3Cl2–CH2–COOH] |
| CSD number | CCDC 818798 |
| Molecular weight/g mol−1 | 205.03 |
| Crystal system | Monoclinic |
| a/Å | 4.8955(5) |
| b/Å | 12.3245(12) |
| c/Å | 14.5030(14) |
| β/° | 94.270(2) |
| Unit cell volume/Å3 | 872.6(15) |
| Calculated density/g cm−3 | 1.561 |
| Space group | P21/c (no. 14) |
| No. of formula per unit cell, Z, Z′ | 4, 1 |
| Absorption coefficient/μ mm−1 | 0.695 |
| No. of reflections measured | 6796 |
| No. of independent reflections | 1774 |
| R int | 0.0186 |
| No. of reflections with I > 2σI | 1523 |
| Final R1 value (I > 2σI) | 0.0477 |
| Final wR2 value (I > 2σI) | 0.1307 |
| Final R1 value (all data) | 0.0542 |
| Final wR2 value (all data) | 0.1362 |
| Goodness on fit | 1.058 |
The asymmetric unit (Fig. 8) is composed of a single molecule of 2,3-dichlorophenylacetic acid. The molecules are tightly linked by the usual centrosymmetric hydrogen bonds between their acidic moieties O(2)⋯O(1), d = 2.67(1) Å (Fig. 9). During the last steps of the structure refinement, high electronic density was located at almost 1 Å to the oxygen atom O(2); therefore it was attributed to the hydrogen atom of the carboxylic moiety. However, the bond lengths observed within the carboxylic moiety of the 2,3-dichlorophenyl acetic acid are similar, C–O(1) 1.231(3)Å and C–O(2) 1.267(3)Å. Therefore the proton of the carboxylic moiety probably exhibits a disordered behaviour.
 | ||
| Fig. 8 Asymmetric unit with thermal ellipsoid representation (50% probability) and with atom labels. | ||
 | ||
| Fig. 9 Centrosymmetric dimers interacting through π–π stacking along the a axis. These interactions are represented in dashed pink lines. The hydrogen bonds are represented in dashed blue lines. | ||
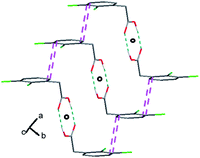
These bonds give rise to centrosymmetric dimers that is a common feature when protonated carboxylic functions are involved. The dimers are interconnected by π-stacking spreading along the [100] direction (Fig. 9 and 10).
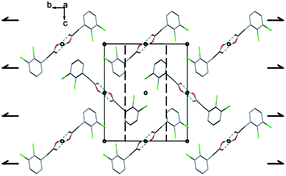 | ||
| Fig. 10 Projection along the a axis. Between the pseudo-chains the cohesion is ensured by van der Waals interactions. The symmetry elements are depicted in black. | ||
Two consecutive aromatic rings are at a distance of 3.7 Å from carbon atoms C(4)–C(3) to carbon atoms C(6)–C(7) from the adjacent ring, respectively. These interactions lead to pseudo-chains, along the a axis. The cohesion between these pseudo-chains is ensured by van der Waals interactions.
The crystal structure of the 2,3-dichlorophenylacetic acid is thus totally different with regard to the AH subslice of the phase between B and AH.
Preferential crystallization (AS3PC)
The main data of four consecutive batches in methanol are summarized in Table 5. The productivity is defined as the mass of pure enantiomer harvested per hour and per litre of solvent. This preferential crystallization exhibits a good reproducibility and a mean optical purity of ∼87% for the crude product obtained without washing or recrystallization. Moreover, the final enantiomeric excess of the mother liquors (e.e.final) appeared reproducibly greater than 20%.| Batch | Raw mass/g | Optical purity (%) | Pure enantiomer mass/g | e.e.final (%) | Duration |
|---|---|---|---|---|---|
| 1 | 4.77 | 86 | 4.10 | −20.20 | 55 min |
| 2 | 5.49 | −83 | 4.56 | 22.80 | 45 min |
| 3 | 5.31 | 90 | 4.78 | −20.64 | 50 min |
| 4 | 5.20 | −88 | 4.58 | 20.73 | 55 min |
| Average | 5.19 | 87 | 4.51 | 21.10 | 51.25 min |
| Productivity/g L−1 h−1 | 52.80 |
The main data of three consecutive batches in THF are summarized in Table 6. Compared with methanol, the productivity and the optical purity are similar; however e.e.final is halved.
| Batch | Raw mass/g | Optical purity (%) | Pure enantiomer mass/g | e.e.final (%) | Duration |
|---|---|---|---|---|---|
| a The values of the optical purity correspond to the enantiomeric excess of the harvested solid without further purification. e.e.final stands for the enantiomeric excess in the final mother liquor. | |||||
| 1 | 6.16 | 84 | 5.17 | −8.42 | 70 min |
| 2 | 6.80 | −81 | 5.51 | 9.55 | 70 min |
| 3 | 6.79 | 89 | 6.04 | −11.53 | 55 min |
| Average | 6.58 | 84.7 | 5.57 | 9.83 | 65 min |
| Productivity/g L−1 h−1 | 51.42 |
Ideally, Meyerhoffer's rule25 gives αmol = Smol(±)/Smol(+) = 2 for covalent compounds, and αmol = √2 for (1–1) stoichiometric salts. For this hybrid salt–cocrystal, even if the concentration in THF is circa twice as that in methanol, the preferential crystallization performances remained roughly the same in both solvents. These results come from the molar α ratio which is more favourable in methanol (αmol = 1.20, Table 7) than in dry THF (αmol = 1.52, Table 8). This is consistent with the ability of methanol to be a better solvent for ionic species than pure THF. Furthermore, the metastable zone width (MSZW assessed by β = S(±) at TB/S(±) at TF) is much wider in methanol (β = 1.55) than in dry THF (β = 1.23) confirming the important role of the solvent in the performances of preferential crystallization.
| Solubilities in methanol | |||
|---|---|---|---|
| Temperature/°C | 25 | 30 | 40 |
| Solubilities (±) (% mass) | 7.5 | 8.9 | 11.6 |
| Solubilities (−) (% mass) | 6.3 | 7.4 | 9.8 |
| Solubilities (±) (molar fraction) × 102 | 0.23 | 0.28 | 0.37 |
| Solubilities (−) (molar fraction) × 102 | 0.19 | 0.23 | 0.31 |
| α mol | 1.20 | 1.22 | 1.19 |
| Solubilities in THF | |||
|---|---|---|---|
| Temperature/°C | 30 | 35 | 40 |
| Solubilities (±) (% mass) | 22.5 | 25 | 27.7 |
| Solubilities (−) (% mass) | 15.8 | 17.9 | 20.2 |
| Solubilities (±) (molar fraction) × 102 | 1.84 | 2.11 | 2.42 |
| Solubilities (−) (molar fraction) × 102 | 1.2 | 1.39 | 1.61 |
| α mol | 1.53 | 1.52 | 1.50 |
Fig. 11 and 12 illustrate the projection of the solution point trajectory during an entrainment (AS3PC mode) in methanol and THF, respectively. Based on experimental results, it seems that the stereoselective crystallizations persist up to F points which means up to circa 23% in methanol and circa 10% in THF. These F points are located on the tie-lines which connect the antipode 〈−〉 to the racemic liquid at TF. They correspond to the boundaries between the three phase domain: 〈+〉, 〈−〉 and racemic solution and the biphasic domain: 〈−〉 and saturated solution.
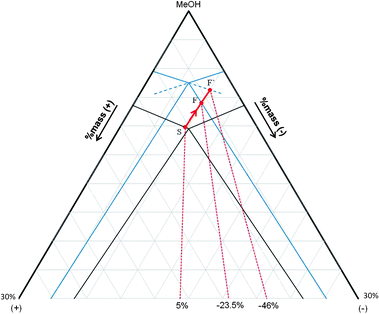 | ||
| Fig. 11 Projection of the solution point trajectory (red line) during entrainment in methanol. The initial and final isothermal sections of the ternary system (+)-(−)-MeOH are drawn in black and in blue, respectively. | ||
 | ||
| Fig. 12 Projection of the solution point trajectory (red line) during the entrainment in dry THF. The initial and final isothermal sections of the ternary system (+)-(−)-THF are drawn in black and in blue, respectively. | ||
Prior to testing PC, the crystal structure features raised some concerns about the possibility to keep on with stereoselective crystallization while the two antipodes are supersaturated.
Considering the (001) orientation and assuming that AH could be the protruding units in solution, the non-chiral AH layers might induce a loss of transmission of chirality from underneath (H2B2+–A2− layers) depending on the growing mode of the crystal (Fig. 13 and 14). This alternation of subslices forms a hybrid salt–cocrystal. This dual character has, even more surprisingly, been observed in the crystal structure of a (1–1) phase with Z′ = 3/2 containing phthalazine and fumaric acid.26
 | ||
| Fig. 13 Growth by two-dimensional nucleation mode. Alternate achiral subslices of molecule AH thickness d003 and chiral subslices of thickness 2d003 of ions: H2B2+ and A−. | ||
 | ||
| Fig. 14 Illustration of the crystal growth by assemblage of solvated chiral building units. | ||
One possible hypothesis consistent with all the results is that this crystal growth does not proceed by 2D nucleation on the (001) surface27 but by assemblage of chiral building units (Fig. 14).
In this case, a full pre-assemblage in the vicinity of the growing surfaces might prevail and therefore allow the high efficiency in chiral discrimination in the solid state. That is to say, 2AH and 2A− and H2B2+ could be linked (building unit) prior to docking onto the growing surface. Thus, the necessary transmission of chirality throughout the crystal growth could be kept without any epitaxy of the antipode. The fibrous aspect along the b axis of single crystals together with large defects at the tips of the crystals obtained in methanol under smooth conditions seems consistent with this hypothesis. Another hypothesis could also involve an extensive reconstruction of this surface.
Experimental section
Analytical techniques
NMR spectra were recorded on a Bruker Advance-300 instrument (300 MHz for 1H NMR and 75 MHz for 13C NMR) using CDCl3 or DMSO-d6 as solvent; δ values are quoted in ppm downfield from TMS using the residual proton signal of the solvent as reference.X-Ray powder diffraction measurements were carried out on a SIEMENS D5000 Matic diffractometer (Bruker analytical X-ray Systems, D-76187 Karlsruhe, Germany) with a Bragg Brentano geometry, in theta/2-theta reflection mode. The instrument is equipped with a copper anticathode (40 kV, 40 mA, Kα1 radiation: 1.540 Å, Kα2 radiation: 1.544 Å) and a scintillation detector. The diffraction patterns were collected by steps of 0.04° (in 2-theta) over the angular range 3–30°, with a counting time of 4 s per step.
Polarimetric measurements were performed with a Perkin-Elmer® 341 (λ = 436 nm, T = 25 °C, l = 10 cm).
Scanning electron microscopy (SEM) pictures were obtained with a JEOL JCM-5000 NeoScope instrument (secondary scattering electron) at an accelerated voltage of 15 kV. Samples were stuck on an SEM stub with gloss carbon and coated with gold to reduce electric charges induced during analysis with a NeoCoater MP-19020NCTR.
Infrared spectra were measured on a Bruker Alpha-P FT-IR instrument in the ATR geometry with a diamond ATR unit.
Syntheses
All reagents and solvents were purchased from Acros Organics and were used without further purification.trans-N,N′-Dibenzyldiaminocyclohexane (compound B) was prepared according to Jones and Mahon,28 which was applied with modifications (Fig. 15).
 | ||
| Fig. 15 Reaction of formation of trans-N,N′-dibenzyldiaminocyclohexane. | ||
A solution of benzaldehyde (2.05 eq., 75.02 g, 707 mmol) in methanol (400 mL) was added rapidly dropwise to a solution of trans-1,2-diaminocyclohexane ((±) DACH) (39.38 g, 345 mmol) in methanol (1 L) at room temperature. The reaction mixture was stirred for 6 hours at room temperature and then cooled down to 0 °C, followed by a slow addition of NaBH4 (1.5 eq., 19.5 g, 515 mmol). After stirring the reaction mixture overnight at ambient temperature, the solvent was removed under vacuum. After addition of dichloromethane, the organic layer (300 mL) was washed with water (2 × 200 mL) and dried (MgSO4). Dichloromethane was evaporated and the pure harvested product was a yellow oil (88.2 g, 299 mmol). The total yield was 87%. δH (300 MHz, CDCl3), 1.10 (2H, t, J = 10.56 Hz), 1.25 (2H, d, J = 7.02 Hz), 1.74 (2H, d, J = 7.02 Hz), 2.17 (2H, d, J = 10.56 Hz), 2.30 (2H, br s), 3.68 (2H, d, J = 13.2 Hz), 3.92 (2H, d, J = 13.2 Hz), 7.23–7.28 (10H, m). δC (75 MHz, CDCl3) 25.1, 31.5, 50.8, 60.9, 127.0, 128.2, 128.5, 140.8.
The enantiomeric product was synthesized from the starting chiral material (+ or –) DACH citrate monohydrate obtained from a preferential crystallization previously carried out in the laboratory.20
A suspension of K2CO3 (2.5 eq., 532 mg, 3.85 mmol) and DACH citrate monohydrate (500 mg, 1.54 mmol) was prepared in methanol (20 mL) then a solution of benzaldehyde (2.11 eq., 344 mg, 3.25 mmol) in methanol was added dropwise. After stirring the reaction mixture overnight at ambient temperature, the solvent was removed under vacuum. After addition of dichloromethane (15 mL), the organic layer was washed with water (2 × 5 mL) and dried (MgSO4). Dichloromethane was evaporated under vacuum. The crude product was dissolved in methanol (20 mL) then the solution was cooled at 0 °C, and NaBH4 (1.6 eq., 93 mg, 2.46 mmol) was added slowly. After stirring the reaction mixture overnight at ambient temperature, the solvent was removed under vacuum. Dichloromethane (15 mL) was added and then the organic layer was washed with water (2 × 5 mL) and dried (MgSO4). Dichloromethane was evaporated and the pure got back was a yellow oil (372 mg, 1.26 mmol). The results of NMR analyses were the same as that of the racemic oil. The final yield was 82%.
Screening of conglomerate
A screening of salts was carried out in order to spot stable conglomerates with compound B. All salts were prepared by using the same protocol; only the nature of the acid introduced varied. This screening allows access to all kinds of stoichiometries that lead to the possible conglomerate.29A methanol solution of pure acid was added slowly to a methanol solution of compound B at room temperature. Solutions were prepared so that the ratio of acidic/basic functions was respected. After a few minutes, the spontaneous crystallization of salt occurred at room temperature. The resulting suspension was filtered on the glass filter (porosity 4). The recovered salts were analyzed by SHG and in the case of positive results, pure enantiomeric salts were also crystallized. The X-ray powder patterns of these two salts were compared to confirm, or not, the existence of a conglomerate forming system.
Only the 2,3-dichlorophenylacetic acid (Fig. 16) in methanol gave rise to a conglomerate with compound B (Fig. 17).
 | ||
| Fig. 16 Structure of 2,3-dichlorophenylacetic acid. | ||
 | ||
| Fig. 17 X-Ray powder patterns of the racemic salt (bottom), the enantiomer salt (middle) and the calculated salt (top) from the single crystal diffraction data. | ||
As the main condition for preferential crystallization was fulfilled, AS3PC was tested.
Resolution by preferential crystallization
Preferential crystallizations using the Auto-Seeded variant (AS3PC)12 were performed in a flask equipped with a magnetic stirrer. Temperature was accurately controlled by a cryo-thermostat (Huber_polystat CC240). The course of the entrainment was monitored by off-line polarimetric measurements of the mother liquor (λ = 436 nm). The presence of very small particles requires a filtration prior to measurement with disposable Gelman® Acrodiscs media (0.45 μm). Simultaneously, the clear mother liquor was sampled to determine its global composition (mass concentration) by refractometry (Fig. 18). | ||
| Fig. 18 Evolution of the refractive index of the solution versus the mass concentration of the hybrid salt–cocrystal (in methanol). | ||
The AS3PC process is briefly recalled below (Fig. 19).
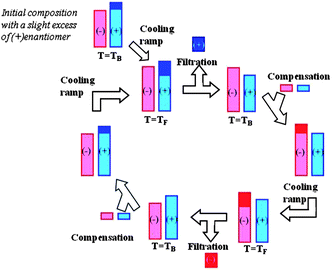 | ||
| Fig. 19 Principle of the AS3PC. Two consecutive temperature cycles are represented: the enantiomeric excess at TB engenders a preferential secondary nucleation and crystal growth during the cooling ramp. At TF, the suspension is filtered and the crop obtained is pure enantiomer, whereas the solution is enriched in the counter-enantiomer (solid parts and liquid parts of the system are depicted in dark and in light colours, respectively). | ||
A small amount of pure (+) enantiomer was added to a methanol saturated solution of racemic mixture at the initial temperature, TB, so that the initial enantiomeric excess (e.e.) remained at circa 10%. A cooling ramp was applied to the system with a controlled rate, down to TF in order to induce a dual enantioselective effect: secondary nucleation and crystalline growth of the (+) enantiomer. The crystallization of this enantiomer was maintained beyond the thermodynamic limit (i.e. e.e. = 0) due to the entrainment effect. Therefore the mother liquor became supersaturated in the counter-enantiomer (i.e. no spontaneous nucleation of the (−) enantiomer occurred for a given period of time). This slurry had to be filtered at TF before the primary nucleation of the (−) enantiomer began. At this time, the system was a solution enriched in (−) enantiomer, but depleted in matter, so a compensation in the racemic mixture and solvent was made, and this prior to each new cycle. By means of continuous recycling of the mother liquor, every odd operation gave the (¬) enantiomer, and every even operation gave the (+) enantiomer.
The filtration cakes were neither washed nor recrystallized. The enantiomeric purities of the crops were determined by polarimetry. For each batch, the final composition of the mother liquor was measured (mass concentration and e.e.final = enantiomeric excess of the mother liquor at the end of a run).
In our case, the conglomerate exhibits good solubility in methanol (Table 7) and in THF (Table 8), so that these solvents were chosen for the preferential crystallization by the AS3PC method.
The first entrainments were performed with 10.4 g of racemic mixture in 100 mL of methanol and with 34 g in 100 mL of THF and led to the main AS3PC parameters, listed in Table 9.
During these first tests, the αD of the mother liquor was measured by off-line polarimetry. The filtration time was determined according to the monitoring of the αD of the mother liquor.
Single crystal X-ray structural determination
A suitable single crystal of the title compound, (trans-N,N′-dibenzyldiaminocyclohexane)1(2,3-dichlorophenylacetic acid)4, was obtained by slow evaporation of a saturated solution in methanol at room temperature. A single crystal of 2,3-dichlorophenylacetic acid was obtained by slow evaporation of a saturated solution in dichloromethane at room temperature.The crystal structures were determined by single crystal diffraction on a SMART APEX diffractometer (with MoKα1 radiation: λ = 0.71073 Å). The structures were solved by direct methods (SHEL-XS30). Anisotropic displacement parameters were refined for all non-hydrogen atoms using SHEL-XL31 available with the WinGX32 package. All hydrogen atoms were included in the models in calculated positions and were refined as contained to bonding atoms.
For (trans-N,N′-dibenzyldiaminocyclohexane)1(2,3-dichloro-phenylacetic acid)4, the final cycle of full-matrix least-square refinement on F2 was based on 5300 observed reflections and 318 variable parameters and converged with unweighted and weighted agreement factors of R1 = 0.0698, wR2 = 0.1649 for 3171 reflections with I > 2σI and R1 = 0.1165, wR2 = 0.1888 for all data (see Table 1). Simulated morphology, calculated from the structural data with the geometric method published by Bravais, Friedel, Donnay and Harker (BFDH),24 was determined, thanks to the Material Studio molecular modelling software.33
For 2,3-dichlorophenylacetic acid, the final cycle of full-matrix least-square refinement on F2 was based on 1774 observed reflections and 110 variable parameters and converged with unweighted and weighted agreement factors of R1 = 0.0476, wR2 = 0.1334 for 1523 reflections with I > 2σI and R1 = 0.0537, wR2 = 0.1334 for all data (see Table 3).
Conclusion
An unexpected hybrid salt–cocrystal between the dibase (H2B2+) and four monoacids (2AH and 2A−) has been characterized. It crystallizes as a stable conglomerate. The stoichiometry of this compound emphasizes the importance of enlarging the “classic” stoichiometries when screening conglomerates.The resolutions in methanol and in THF reveal that the entrainment effect, at its best, persists up to 22% e.e.final in the counter-enantiomer for a global concentration of 13% in methanol. Furthermore these results prove that the non-chiral protonated acid subslices do not induce any loss in the chiral selectivity of the self-assembling process (i.e. the stereoselective crystallization). The morphology of crystals and the high performance of AS3PC suggest that the crystal growth proceeds by direct incorporation of building units composed of H2B2+, 2A−, 2AH or reconstruction of some surfaces of the crystal might occur.
Acknowledgements
We are grateful for financial support to the European collaborative project: “IntEnant” FP7-NMP2-SL-2008-214219.Notes and references
- T. E. Beesley and R. P. W. Scott, Chiral Chromatography, J., Wiley, Chichester, England, New York, 1998 Search PubMed.
- R. A. Sheldon, Chirotechnology: Industrial Synthesis of Optical Active Compounds, Marcel Dekker, New York, 1993 Search PubMed.
- J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates and Resolutions, Krieger, Malabar, Fla, 1994 Search PubMed.
- D. Kozma, CRC Handbook of Optical Resolutions via Diastereomeric Salt Formation, CRC Press, Hoboken, 2001 Search PubMed.
- P. Newman, Optical Resolutions Procedures for Chemical Compounds, Optical Resolution Information Center, Manhattan College, Riverdale, NY, 1978 Search PubMed.
- K. Harata, Chem. Rev., 1998, 98, 1803–1828 CrossRef CAS.
- L. Addadi, J. Van Mil and M. Lahav, J. Am. Chem. Soc., 1981, 103, 1249–1251 CrossRef CAS.
- N. Doki, M. Yokota, S. Sasaki and N. Kubota, Cryst. Growth Des., 2004, 4, 1359–1363 CAS.
- C. Viedma, J. Cryst. Growth, 2004, 261, 118–121 CrossRef CAS.
- W. L. Noorduin, E. Vlieg, R. M. Kellogg and B. Kaptein, Angew. Chem., Int. Ed., 2009, 48, 9600–9606 CAS.
- G. Coquerel, in Novel Optical Resolution Technologies, Springer, Heidelberg, 2007, pp. 1–51 Search PubMed.
- G. Coquerel, M.-N. Petit and R. Bouaziz, PCT Pat., WO 95/08522, 1995.
- A. N. Collins, G. N. Sheldrake and J. Crosby, Chirality in Industry: the Commercial Manufacture and Applications of Optically Active Compounds, Wiley, Chichester, 1992 Search PubMed.
- R. M. Secor, Chem. Rev., 1963, 63, 297–309 CrossRef CAS.
- R. Duschinsky, J. Soc. Chem. Ind., London, 1934, 53, 10–20 CrossRef.
- M. Helmreich, C.-P. Niesert, D. Cravo, G. Coquerel, G. Levilain, S. Wacharine-Antar and P. Cardinaël, Process for Enantiomeric Separation of Racemic Dihydro-1,3,5-Triazines via Preferential Crystallization, WO Pat., 2010109015, 2010 Search PubMed.
- E. Aubin, M. N. Petit and G. Coquerel, J. Phys. IV France, 2004, 122, 157–162 CrossRef CAS.
- G. Tauvel, M. Sanselme, S. Coste-Leconte, S. Petit and G. Coquerel, J. Mol. Struct., 2009, 936, 60–66 CrossRef CAS.
- A. Galland, V. Dupray, B. Berton, S. Morin-Grognet, M. Sanselme, H. Atmani and G. Coquerel, Cryst. Growth Des., 2009, 9(6), 2713–2718 CAS.
- A. Galland, V. Dupray, A. Lafontaine, B. Berton, M. Sanselme, H. Atmani and G. Coquerel, Tetrahedron: Asymmetry, 2010, 21(18), 2212–2217 CrossRef CAS.
- J. F. Larrow and E. N. Jacobsen, Organic Syntheses, Coll., 2004, 10, 96 CAS; J. F. Larrow and E. N. Jacobsen, Organic Syntheses, Coll., 1998, 75, 1 Search PubMed.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef.
- S. L. Childs, G. P. Stahly and A. Park, Mol. Pharmaceutics, 2007, 4, 323–338 CrossRef CAS.
- J. D. H. Donnay and D. Harker, Am. Mineral., 1937, 22, 446–447 CAS.
- W. Meyerhoffer, Ber Dtsch. Chem. Ges., 1904, 37, 2604–2610 CrossRef CAS.
- B. Olenik, T. Smolka, R. Boese and R. Sustmann, Cryst. Growth Des., 2003, 3(2), 183–188 CAS.
- O. Söhnel and J. Garside, Precipitation: Basic Principles and Industrial Applications, Butterworth–Heinemann, 1992 Search PubMed.
- M. D. Jones and M. F. Mahon, J. Organomet. Chem., 2008, 693(13), 2377–2382 CrossRef CAS.
- S. Gonella, J. Mahieux, M. Sanselme and G. Coquerel, Org. Process Res. Dev., 2011 DOI:10.1021/op200092f , articles ASAP (As Soon As Publishable).
- Include in WinGX suite: SHELXS-97, G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, Include in WinGX: SHELXL-97—a Program for Crystal Structure Refinement, University of Goettingen, Germany, 1997, release 97-2 Search PubMed.
- L. J. Farrugia, WinGX: Version 1.70.01: An integrated system of Windows Programs for the solution, refinement and analysis of Single Crystal X-Ray Diffraction Data, Dept. of chemistry, University of Glasgow Search PubMed.
- Materials Studio v. 5.5.0.0, Accelrys Software Inc., 2010 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: FT-IR spectra of hybrid salt–cocrystal and 2,3-dichlorophenylacetic acid. CIF-file including crystallographic data (excluding structure factors) of hybrid salt–cocrystal (CCDC 818797) and 2,3-dichlorophenylacetic acid (CCDC 818798). See DOI: 10.1039/c1ce05484h |
| ‡ http://www.intenant.com/ |
| This journal is © The Royal Society of Chemistry 2012 |