Corannulene and its penta-tert-butyl derivative co-crystallize 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :1 with pristine C60-fullerene†
:1 with pristine C60-fullerene†
Louise N.
Dawe
*a,
Tayel A.
AlHujran
a,
Huu-Anh
Tran
a,
Jason I.
Mercer
b,
Edward A.
Jackson
c,
Lawrence T.
Scott
c and
Paris E.
Georghiou
*a
aDepartment of Chemistry, Memorial University of Newfoundland, St. John's, Newfoundland and Labrador, Canada A1B3X7. E-mail: louise.dawe@mun.ca; parisg@mun.ca; Tel: +1-709-864-8904 Tel: +1-709-864-8517
bDepartment of Computer Science, Memorial University of Newfoundland, St. John's, Newfoundland and Labrador, Canada A1B3X5. E-mail: jason.mercer@mun.ca; Tel: +1-709-864-4315
cDepartment of Chemistry, Merkert Chemistry Centre, Boston College, Chestnut Hill, Massachusetts 02467-3860, USA. E-mail: lawrence.scott@BC.edu; Fax: +1-617-552-6454; Tel: +1-617-552-8024
First published on 14th March 2012
Abstract
The first X-ray structural determinations of pristine fullerene C60, cocrystallized 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with corannulene and with its pentaalkyl-substituted derivative, 1,3,5,7,9-penta-tert-butyl-corannulene, have now been achieved.
:1 with corannulene and with its pentaalkyl-substituted derivative, 1,3,5,7,9-penta-tert-butyl-corannulene, have now been achieved.
The chemistry and properties of corannulene (1) have been of long-standing interest to many research groups, but particularly so in recent years due to its structural resemblance to segments of the C60-fullerene. Corannulene represents the smallest C60-fullerene fragment which is built around a five-membered ring and is also the first “geodesic” polyarene to have been synthesized.1 Barth and Lawton first reported its synthesis via a 16-step procedure in 1966.2 Since then more practical syntheses of corannulene have been developed, some being variants3 of flash vacuum pyrolysis approaches,4 and more recently, variants of the solution-phase methods originally developed by the Siegel group.5 As a result of these advances, controlled substitution at corannulene's rim has been possible to achieve in synthetically-useful amounts which has resulted in diverse derivatives whose properties have been studied.6 Recently, Petrukhina's group showed that the “hub” of corannulene could also be functionalized via Friedel–Crafts alkylation to form stable salts which could be further functionalized at the adjacent carbon on the rim to produce neutral bifunctionalized derivatives.7
The curved concave surface of corannulene has long been postulated as being ideally suited for supramolecular binding to the electron-deficient convex C60. There are many computational studies which have shown that, at least in the gas phase, complexation from the concave face of corannulene to a C60 molecule should be energetically favourable;8 however, direct experimental proof has not been demonstrated until now.
Corannulene has indeed been shown to form a complex with the cation of C60 in the gas phase.9 More recently, Fasel and coworkers reported a Cu(110)-surface supported corannulene:C60 host–guest system.10 Both of these findings are consistent with the hypothesis of a preferred concave-corannulene convex-C60 complementary π–π interaction. We have previously shown11 that corannulenes 2 and 3, which are more electron-rich than corannulene itself, exhibit modest binding constant values of 280–475 M−1 as measured by 1H-NMR studies in toluene-d8. Later, a “Venus fly-trap”-resembling corannulene derivative,12pentakis(1,4-benzodithiino)corannulene (4), proved to be a better host for C60, having a binding constant of 1420 M−1. In probably the best previously-demonstrated evidence of a corannulene concave-face binding to C60, Sygula et al. reported a double concave “buckycatcher” containing two opposing concave corannulene subunits linked via a central motif. In addition to showing a much higher binding constant of ∼8600 M−1 by 1H-NMR studies in toluene-d8, these authors obtained an X-ray crystal structure of a 1:1 C60:buckycatcher complex.13
Despite the studies described above, no solution-phase or solid-phase complexation of “pristine” or un-modified, corannulene with “pristine” C6014 has been reported to date. We now report that we have succeeded in obtaining the first X-ray structures of both a stable pristine 1:1 corannulene:C60 co-crystal (Fig. 1) and also of a C60:penta-tert-butylated corannulene, 5 (Fig. 2). Compound 5 was the first alkyl-substituted corannulene derivative for which an X-ray structure was previously determined15 and it, like corannulene itself, shows no evidence for complexation with C60 in solution studies.
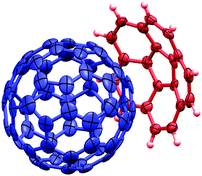 | ||
| Fig. 1 50% probability ellipsoid representation of the asymmetric unit of C60:1, with the minor disorder component of C60 omitted for clarity. | ||
 | ||
| Fig. 2 30% probability ellipsoid representation of C60:5, with the minor disorder component of C60 omitted for clarity. | ||
A solution of corannulene containing an approximately equimolar amount of C60 in toluene was allowed to slowly evaporate over a period of several weeks during which time red prisms formed from which a single crystal suitable for X-ray diffraction measurements was obtained. In the case of C60:5 suitable crystals for X-ray diffraction measurements could only be obtained from the slow evaporation of a solution containing equimolar amounts of 5 and C60 in 1,2-dichlorobenzene.
For C60:1, the C60 molecule was disordered and was modeled with four different orientations. The major component was located completely from difference maps, and subsequent atom positions for smaller components were determined and refined by first locating the highest peaks from difference maps for the next major disorder component, using Monte Carlo methods to calculate the remaining atom positions and then refining these positions with a central dummy atom that acted as the pivot. This was repeated for all orientations. Refinement values are still high,16 and reflect the fact that the C60 crystallized with many different orientations. An investigation into modeling this as a spherical shell of diffuse electron density is ongoing and will be reported elsewhere. For C60:5, the C60 molecule was disordered and modeled with two different orientations that were refined.17 Two disordered lattice solvent 1,2-dichlorobenzene molecules were present in the asymmetric unit.
The C60 molecules in the structure of C60:1 pack in a zigzag manner (coincident with the two-fold screw axes), with centroid–centroid separations of 10.04 Å (close to the van der Waals limit of fullerene, 10.0 Å18) and an angle for three successive C60 molecule centroids of 135.9° (Fig. 3). This packing motif is common for close contacts between C60 molecules in host–guest crystals,18,19 and, in particular, the 1:1 C60:calix[5]arene complex exhibits this packing motif.20 The depth of penetration of the C60 into the corannulene is 6.94 Å, measured from the centroid of the corannulene five-membered ring to the centroid of C60, and 3.75 Å, measured from the shortest distance from the concave surface of 1 to a C60 surface (“A” in Fig. 4). Other short contacts of 6.64 Å and 3.21 Å can be seen between the centroid of a six-membered ring of the convex face of a second corannulene to the centroid of C60, and to the C60 surface, respectively (see “B” in Fig. 4). Each C60 is in close contact (less than 10.3 Å18) with six other C60 molecules. For uncomplexed 1 the average bowl depth at −100 °C is 0.875 Å,15 compared to 0.89 Å in the current study. This small difference does not indicate any significant change in geometry upon co-crystallization with C60. By way of contrast, an average depth of 0.72 Å was reported for uncomplexed 515 whereas 0.59 Å was found for its C60 complex in this study.
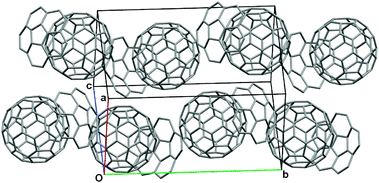 | ||
| Fig. 3 Capped stick representation of the extended packed unit cell (0 ≤ x ≤ 1, −0.5 ≤ y ≤ 1.5, 0 ≤ z ≤ 1) for C60:1. Minor disorder components of C60 and H-atoms omitted for clarity. | ||
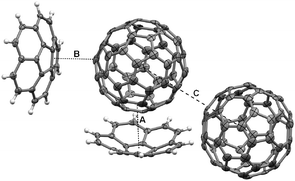 | ||
| Fig. 4 30% probability ellipsoid representation of C60:1 (minor disorder components of C60 omitted for clarity). Dashed lines indicate: A: concave surface of 1 to C60 short contact 3.75 Å; B: convex surface of 1 (symmetry operator 1.5 − x, −½ + y, ½ − z) to C60 short contact of 3.21 Å; and C: C60 centroid–centroid (symmetry operator ½ − x, ½ + y, ½ − z) contact of 10.04 Å. | ||
The packing motif of C60:5 is similar to that of C60:1; the C60 molecules pack in a zigzag manner (coincident with the two-fold screw axes), with centroid–centroid separations of 10.16 Å, and an angle of 97.4° for three successive C60 molecule centroids (Fig. 5). By analogy with the close contacts seen in 1:C60, the depth of penetration of the C60 into 5 is 6.78 Å and 3.30 Å respectively. Multiple methyl C–H (5)–C60 interactions are present, with contacts ranging from 2.78–2.90 Å, while lattice solvent dichlorobenzene molecules also interact with C60via π(solvent)–π(C60) (3.35 Å) and C–Cl(solvent)–C60 (3.02 Å) associations. Finally, unlike the C60:1 complex, significant host–host interactions are also present; the host molecules are staggered with respect to their five-membered rings, with centroid–centroid distances of 3.30 Å (Fig. 2). The relatively greater ordering of the “guest” C60 in this structure is consistent with observations made by us previously in which the tetra-tert-butyl-alkylated calix[4]naphthalene21 proved to be a better host for C60 than the corresponding unsubstituted calix[4]naphthalene, presumably due to the extra methyl–π interactions that can take place between the host and guest.22a–c
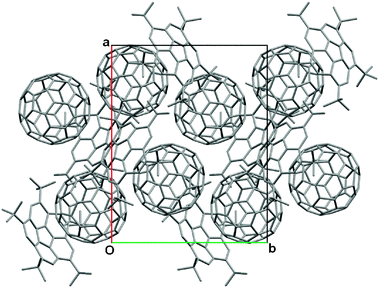 | ||
| Fig. 5 Capped stick representation of the extended packed unit cell (0 ≤ x ≤ 1, −0.5 ≤ y ≤ 1.5, 0 ≤ z ≤ 1) for C60:5, showing the zigzag motif with respect to C60. Minor disorder components of C60, H-atoms and lattice solvent C6H4Cl2 omitted for clarity. | ||
In general, it would be expected that strong host–guest interactions should lead to reduced disorder for C60. In the structures reported here, C60 was disordered for both C60:1 and C60:5. This is particularly problematic for C60:1, where the fullerene showed many different orientations and would be better described as a spherical shell of electron density. While there is shape complementarity between C60 and corannulene, the disorder we have found in the C60:1 structure points to the absence of a strong host–guest interaction for any particular orientation of C60 with respect to the corannulene.
The authors thank NSERC, Memorial University of Newfoundland and the U.S. Dept. of Energy for financial support. The following individuals are acknowledged: Dr Khalil Abboud (U. of Florida) for helpful discussions on the refinement of C60:1; Dr Hilary Jenkins (McMaster Analytical X-ray Diffraction Facility) for the data collection for C60:5; Dr Christine Beavers (Advanced Light Source, LBL. Berkeley) and Dr Lee Daniels (Rigaku Americas Corporation) for helpful discussions on the solution and refinement of C60:5.
Notes and references
- Recent reviews: (a) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868–4884 CrossRef CAS; (b) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS; (c) A. Sygula, Eur. J. Org. Chem., 2011, 1611–1625 CrossRef CAS.
- (a) W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1966, 88, 380 CrossRef CAS; (b) R. G. Lawton and W. E. Barth, J. Am. Chem. Soc., 1971, 93, 1730 CrossRef.
- (a) G. Mehta and G. Panda, Tetrahedron Lett., 1997, 38, 2145–2148 CrossRef CAS; (b) L. T. Scott, P.-C. Cheng, M. M. Hashemi, M. S. Bratcher, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1997, 119, 10963–10968 CrossRef CAS.
- L. T. Scott, M. M. Hashemi, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1991, 113, 7082–7084 CrossRef CAS.
- (a) T. J. Seiders, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1996, 118, 2754–2755 CrossRef CAS; (b) T. J. Seiders, E. L. Elliott, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 1999, 121, 7804–7813 CrossRef CAS; (c) A. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 1999, 121, 7800–7803 CrossRef CAS; (d) A. Sygula, G. X. Xu, Z. Marcinow and P. W. Rabideau, Tetrahedron, 2001, 57, 3637–3644 CrossRef CAS; (e) A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012 DOI:10.1021/op200387s.
- (a) D. Bandera, K. K. Baldridge, A. Linden, R. Dorta and J. S. Siegel, Angew. Chem., Int. Ed., 2011, 50, 865–867 CrossRef CAS; (b) R. Maag, B. H. Northrop, A. Butterfield, A. Linden, A. Zerbe, Y. M. Lee, K.-W. Chi, P. J. Stang and J. S. Siegel, Org. Biomol. Chem., 2009, 7, 4881–4885 RSC.
- A. V. Zabula, C. Dubceac, A. S. Filatov and M. A. Petrukhina, J. Org. Chem., 2011, 76, 9572–9576 CrossRef CAS.
- (a) P. A. Denis, Chem. Phys. Lett., 2011, 516, 82–87 CrossRef CAS; (b) C. Muck-Lichtenfeld, S. Grimme, L. Kobryn and A. Sygula, Phys. Chem. Chem. Phys., 2010, 12, 7091–7097 RSC; (c) E. A. Jackson, B. D. Steinberg, M. Bancu, A. Wakamiya and L. T. Scott, J. Am. Chem. Soc., 2006, 129, 484–485 Search PubMed; (d) Y. Zhao and D. G. Truhlar, Phys. Chem. Chem. Phys., 2008, 10, 2813–2818 RSC.
- H. Becker, G. Javahery, S. Petrie, P. C. Cheng, H. Schwarz, L. T. Scott and D. K. Bohme, J. Am. Chem. Soc., 1993, 115, 11636–11637 CrossRef CAS.
- W. Xiao, D. Passerone, P. Ruffieux, K. Ait-Mansour, O. Groning, E. Tosatti, J. S. Siegel and R. Fasel, J. Am. Chem. Soc., 2008, 130, 4767–4771 CrossRef CAS.
- S. Mizyed, P. E. Georghiou, M. Bancu, B. Cuadra, A. K. Rai, P. Cheng and L. T. Scott, J. Am. Chem. Soc., 2001, 123, 12770–12774 CrossRef CAS.
- P. E. Georghiou, A. H. Tran, S. Mizyed, M. Bancu and L. T. Scott, J. Org. Chem., 2005, 70, 6158–6163 CrossRef CAS.
- A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842–3843 CrossRef CAS.
- For a recent review of various hosts for “pristine” C60 see P. E. Georghiou, in Handbook of Carbon NanoMaterials Vol 1. Synthesis and Supramolecular Systems, ed. F. D'Souza and K. M. Kadish, World Scientific Publishing Co. Pvt. Ltd., Singapore, 2011 Search PubMed.
- Y. Sevryugina, A. Y. Rogachev, E. A. Jackson, L. T. Scott and M. A. Petrukhina, J. Org. Chem., 2006, 71, 6615–6618 Search PubMed.
- Crystal data for C60:1: (C60)·C20H10, M = 970.96, monoclinic, a = 13.0695(10) Å, b = 18.6034(15) Å, c = 17.3068(13) Å, α = 90.00°, β = 110.768(8)°, γ = 90.00°, V = 3934.5(5) Å3, T = 163(2) K, space group P21/n, Z = 4, μ(MoKα) = 0.094 mm−1, 35719 reflections measured, 7297 independent reflections (Rint = 0.0537). Final R1 = 0.1551 (I > 2σ(I)) and wR(F2) = 0.4859 (all data). The goodness of fit on F2 was 2.112. C60 was disordered and modeled with four orientations that were refined using a SHELXL SUMP command with total occupancy = 1. SIMU restraints were applied to all C–C bonds in C60. CCDC 864755.
- Crystal data for C60:5: C60·C40H50·1.5(C6H4Cl2), M = 1472.00, monoclinic, a = 21.975(6) Å, b = 15.264(4) Å, c = 23.000(7) Å, α = 90.00°, β = 117.783(5)°, γ = 90.00°, V = 6825(3) Å3, T = 100(2) K, space group P21/n, Z = 4, μ(CuKα) = 1.674 mm−1, 35649 reflections measured, 14175 independent reflections (Rmerge = 0.0989). Final R1 = 0.1184 (I > 2σ(I)) and wR(F2) = 0.3355 (all data). The goodness of fit on F2 was 1.050. The selected crystal was a non-merohedral twin (hklf5). C60 was disordered with two occupancy-refined orientations. SHELXL SADI and SIMU restraints were applied to all C–C bonds in C60. CCDC 864756.
- M. Makha, A. Purich, C. L. Raston and A. N. Sobolev, Eur. J. Inorg. Chem., 2006, 507–517 CrossRef CAS.
- J. U. Franco, J. C. Hammons, D. Rios and M. M. Olmstead, Inorg. Chem., 2010, 49, 5120–5125 CrossRef CAS.
- J. L. Atwood, L. J. Barbour, M. W. Heaven and C. L. Raston, Angew. Chem., Int. Ed., 2003, 42, 3254–3257 CrossRef.
- P. E. Georghiou, A. H. Tran, S. S. Stroud and D. W. Thompson, Tetrahedron, 2006, 62, 2036–2044 CrossRef CAS.
- For some other examples see (a) S. Mizyed, M. Ashram, D. O. Miller and P. E. Georghiou, J. Chem. Soc., Perkin Trans. 2, 2001, 1916–1919 RSC; (b) A. Hosseini, S. Taylor, G. Accorsi, N. Armaroli, C. A. Reed and P. D. W. Boyd, J. Am. Chem. Soc., 2006, 128, 15903–15913 CrossRef CAS; (c) W. Van Rossom, O. Kundrat, T. H. Ngo, P. Lhotak and W. Dehaen, Tetrahedron Lett., 2010, 51, 2423–2426 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional crystallographic details and figures. CCDC 864755 (1) and 864756 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2cc30652b |
| This journal is © The Royal Society of Chemistry 2012 |