Fe-catalysed oxidative C–H functionalization/C–S bond formation†
Haibo
Wang
a,
Lu
Wang
a,
Jinsai
Shang
a,
Xing
Li
a,
Haoyuan
Wang
a,
Jie
Gui
a and
Aiwen
Lei
*ab
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, P. R. China. E-mail: aiwenlei@whu.edu.cn; Fax: +86-27-68754672; Tel: +86-27-68754672
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, 730000, Lanzhou, P. R. China
First published on 8th November 2011
Abstract
Iron was used as the catalyst for the direct C–H functionalization/C–S bond formation under mild conditions. Various substrates could afford benzothiazoles in moderate to excellent yields. Preliminary mechanistic studies revealed that pyridine played a crucial role for the high yields and selectivities.
Direct C–H bond functionalization towards C–C and C–heteroatom (N, O and S etc.) bond formation promoted by transition metals has attracted extensive attention in the past decades.1 However, most efforts were focused on noble metal catalysts, such as Ru,2Rh,3Pd,4etc. Due to the wide existence and low toxicity of iron compounds, more and more attention have been paid to the iron-catalysed reaction for organic synthesis.5 Remarkable results have been reported on iron promoted “traditional” cross couplings between R1X and R2M.6 Recently, Fe-catalysed direct C–H arylation of unactivated arenes with aryl halides has also been demonstrated simultaneously by Charette et al. and our group.7 To the willing of greener and more atom-economic C–C and C–heteroatom bond formations, the Fe-catalysed direct oxidative cross-coupling between two C–H bonds or C–H and X–H bonds would be an ideal approach to realize this goal. However, few examples have been reported regarding Fe-catalysed direct oxidative C–H functionalizations.8
Benzothiazoles are a very important class of heterocycles in the pharmaceutical area.9 Generally, oxidative cyclization is an efficient approach for the synthesis of thiobenzanilides by using various oxidants,10 such as quinone,10abromine,10b and hypervalent iodine,10d or metal salt.10c,e Meanwhile, Pd-catalyzed C–S bond formation from thioamides via C–H functionalization has also been realized.11 Alternatively, cyclization of 2-halophenylthiobenzamides using Pd or Cu as a catalyst provided another approach to benzothiazoles.12 However, the pre-functionalization of the starting materials in this protocol limited its application. It is no doubt that the direct oxidative intramolecular C–S bonds formation via C–H functionalization would be an attractive approach to synthesize benzothiazole. Herein, we report our progress on iron-salts-catalysed intramolecular C(Ar)–H/X–H activation/C–S bond formation under mild conditions (Scheme 1).
 | ||
| Scheme 1 Intramolecular C–S bond formation via C–H and S–H activation. | ||
Aiming at highly efficient iron-catalysed oxidative coupling, we chose 4-chloro-N-phenylbenzothioamide 1a as the model reaction for initial investigations. Condition optimization (see details in Tables S1, S2 and S3 of ESI†) showed that using Na2S2O8 as the oxidant in DMSO with 10% FeCl3 as the catalyst at 80 °C produced 68% of 2a in 4 h (Table 1, entry 2). In the absence of Fe catalyst, only 16% of 2a was obtained (Table 1, entry 1). Additives such as HOAc or Na2CO3 showed no obvious effect on improving the reaction selectivity. The breakthrough was achieved when 2 eq. of pyridine was employed as the additive. Up to 87% of the desired product 2a was obtained with higher selectivity (2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a was 87:10) (Table 1, entry 5). Further optimization of catalyst precursors in the presence of pyridine gave no obvious improvement (Table 1, entry 6–8). Even using super pure FeCl3 (99.99%) as the catalyst, the result showed no significant difference from commercial FeCl3 (98%) (Table 1, entry 9). By elongating the reaction time, the reaction could also undergo smoothly at 40 °C (Table 1, entry 10) with the slightly decreased selectivity of 2a.
:3a was 87:10) (Table 1, entry 5). Further optimization of catalyst precursors in the presence of pyridine gave no obvious improvement (Table 1, entry 6–8). Even using super pure FeCl3 (99.99%) as the catalyst, the result showed no significant difference from commercial FeCl3 (98%) (Table 1, entry 9). By elongating the reaction time, the reaction could also undergo smoothly at 40 °C (Table 1, entry 10) with the slightly decreased selectivity of 2a.
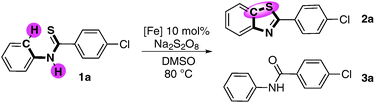
| Entry | Cat. | Additive | Conversion 1a [%] | Yield [%] | |
|---|---|---|---|---|---|
| 2a | 3a | ||||
| a Reaction conditions: 1a (0.5 mmol), Na2S2O8 (0.5 mmol) and catalyst (0.05 mmol) in 2 mL of DMSO at 80 °C for 4 h. The purity of FeCl3 is 98% unless otherwise indicated. Yields were determined by HPLC with an internal standard. b 99.99% FeCl3. c 40 °C for 10 h. | |||||
| 1 | No | — | 100 | 16 | 81 |
| 2 | FeCl3 | — | 100 | 68 | 28 |
| 3 | FeCl3 | HOAc (2 eq) | 100 | 65 | 30 |
| 4 | FeCl3 | Na2CO3 (2 eq) | 99 | 64 | 34 |
| 5 | FeCl3 | Pyridine (2 eq) | 100 | 87 | 10 |
| 6 | FeCl2 | Pyridine (2 eq) | 100 | 82 | 15 |
| 7 | FeBr2 | Pyridine (2 eq) | 100 | 46 | 33 |
| 8 | Fe(acac)3 | Pyridine (2 eq) | 90 | 30 | 32 |
| 9b | FeCl3 | Pyridine (2 eq) | 100 | 85 | 15 |
| 10c | FeCl3 | Pyridine (2 eq) | 100 | 83 | 13 |
Substrate scope of this transformation was further investigated under the optimal conditions (Table 2). The influence of the substituent on the aryl group of the thiobenzoyl part (Ar2, Table 2, substrate 1b–1f) was not the crucial factor for this transformation. Either with the electronic or steric property good to excellent yields (72–95%) can be achieved. Halo substituents (Br, I) can be well tolerated under the reaction conditions (Table 2, substrates 1e and 1f). Then, the effect of the substituents on Ar1 was examined (Table 2, substrate 1g–1m). When the substituent group was the electron-donating group, either ortho or para-position did not influence the reaction. Excellent yields (84–93%) were obtained (Table 2, substrates 1g, 1h and 1l). The reaction of N-naphthalene substituted benzothioamide gave excellent yields (97%) of the cyclization product (Table 2, substrate 1m). However, electron-withdrawing groups decreased the yields dramatically, only moderate yield was obtained when the NO2 substituent was introduced (Table 2, substrate 1k). C–Br and C–I could also be well tolerated (Table 2, substrates 1i and 1j). Interestingly, the reaction of 1n proceeded smoothly to give 2n with high selectivity.

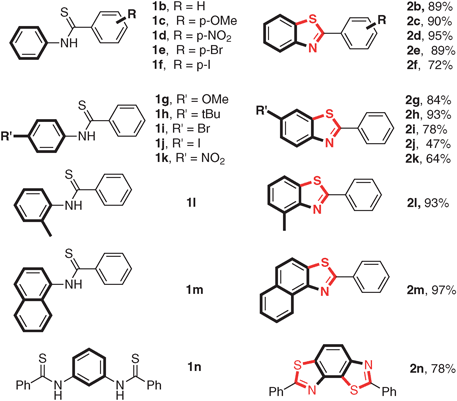
Furthermore, N-aryl alkylthioamides and thioureas were also employed as substrates (Table 3). N-Aryl tert-pentanethioamide bearing both electron-donating and withdrawing groups could undergo the C–H activation/C–S bond formation smoothly in moderate to excellent yields (71–92%) (Table 3, product 5a–5c). However, when N-phenyl alkylthioamide bore a less hindered substituted group, only low yields were obtained. For instance, only 21% desired product 5d and even no 5e were obtained. When thioureas were employed as substrates, the reaction of 1,1-dibutyl-3-phenylthiourea (4f) and 1,1-diethyl-3-phenylthiourea (4g) gave corresponding products in good (5f, 70%) and moderate (5g, 57%) yields. When R was NHPh and NHBn, 50% of 5h and 22% of 5i were obtained, respectively.
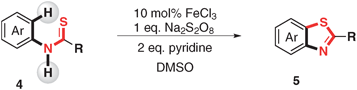

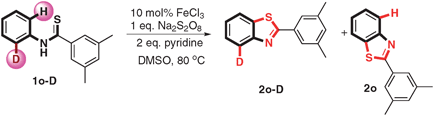
To gain preliminary mechanistic information about this transformation, isotopic effect experiments have been carried out. The reaction of deuterium-1o (1o-D) under the standard condition provided a mixture of the products deuterium-2o (2o-D) and 2o in 93% combined yields, in which the ratio of 2o-D:2o was 1.3 (eqn (1)). The KIE values revealed that the C–H bond cleavage is not the rate-determining-step. When running the reaction in the presence of a radical scavenger (TEMPO) at the same condition, only 19% product was obtained (as shown in eqn (2)) indicating that the reaction might proceed via a radical process.
 | (1) |
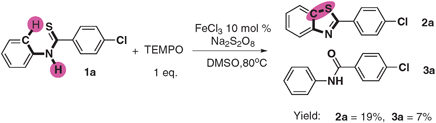 | (2) |
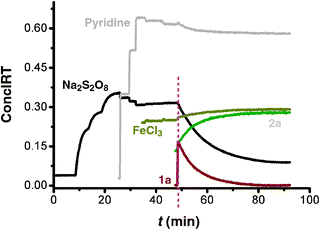 | ||
| Fig. 1 The 2D-kinetic profiles of the stoichiometric reaction of Na2S2O8 (0.2 M), pyridine (4 M), FeCl3 (0.02 M) and 1a (0.2 M) added to 5 mL DMSO at 40 °C one by one; the reaction was monitored by in situIR. | ||
Further kinetic investigation showed that the reaction was first order in [1a], and zero order in the oxidant [Na2S2O8] (see Table S4 and Fig. S10–S17 in ESI†).
Finally, in order to clarify the role of FeCl3 and pyridine in the reaction, comparison experiments were carried out and monitored by in situIR. We found that pyridine is crucial for the high selectivity of C–H activation/C–S bond formation (for detailed information see Fig. S18–S20 in ESI†).
According to all above results, we proposed a mechanism shown in Scheme 2. The substrate N-phenyl benzothioamide was oxidized by Fe(III) through path A and lost an electron and H+ to form the thioyl radical intermediate, in the meantime Fe(III) was reduced to Fe(II). Fe(II) species was re-oxidized by Na2S2O8 to regenerate Fe(III). Then, the cyclization of the thioyl radical intermediate followed by oxidation in the presence of Na2S2O8 gave the product 2-phenyl benzothiazole.
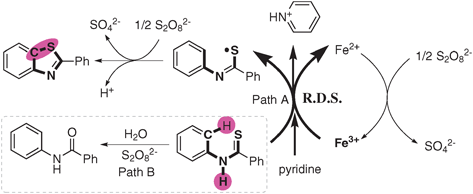 | ||
| Scheme 2 Proposed mechanism. | ||
In conclusion, we have developed an efficient iron-catalysed C–H functionalization/C–S bond formation under mild conditions. This transformation could be conveniently carried out affording various benzothiazoles in moderate to excellent yields. Preliminary mechanistic studies revealed that the reaction required the co-existence of substrate, oxidant, FeCl3 and pyridine. Kinetic studies indicated that pyridine was crucial for the high selectivity of this transformation and the reaction was first order in the substrate and zero-order in oxidant Na2S2O8.
This work was support by the National Natural Science Foundation of China (20702040, 20832003, 20972118).
Notes and references
- For recent reviews on transition metal-catalyzed C–H functionalization, see: (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS; (b) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 RSC; (c) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC; (d) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS; (e) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS.
- For selected recent examples, see: (a) F. Pozgan and P. H. Dixneuf, Adv. Synth. Catal., 2009, 351, 1737 CrossRef CAS; (b) I. Oezdemir, S. Demir, B. Cetinkaya, C. Gourlaouen, F. Maseras, C. Bruneau and P. H. Dixneuf, J. Am. Chem. Soc., 2008, 130, 1156 CrossRef CAS; (c) N. A. Foley, T. B. Gunnoe, T. R. Cundari, P. D. Boyle and J. L. Petersen, Angew. Chem., Int. Ed., 2008, 47, 726 CrossRef CAS.
- For selected recent examples, see: (a) Q. Li and Z.-X. Yu, J. Am. Chem. Soc., 2010, 132, 4542 CrossRef CAS; (b) A. S. Tsai, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6316 CrossRef CAS; (c) M. Shen, B. E. Leslie and T. G. Driver, Angew. Chem., Int. Ed., 2008, 47, 5056 CrossRef CAS.
- For selected recent examples, see: (a) D. A. Candito and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 6713 CrossRef CAS; (b) L.-C. Campeau, D. R. Stuart, J.-P. Leclerc, M. Bertrand-Laperle, E. Villemure, H.-Y. Sun, S. Lasserre, N. Guimond, M. Lecavallier and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 3291 CrossRef CAS; (c) N. Lebrasseur and I. Larrosa, J. Am. Chem. Soc., 2008, 130, 2926 CrossRef CAS; (d) H. A. Chiong, Q.-N. Pham and O. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879 CrossRef CAS.
- (a) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317 CrossRef CAS; (b) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217 CrossRef CAS.
- (a) X.-F. Wu and C. Darcel, Eur. J. Org. Chem., 2009, 4753 CrossRef CAS; (b) G. Cahiez, L. Foulgoc and A. Moyeux, Angew. Chem., Int. Ed., 2009, 48, 2969 CrossRef CAS; (c) D. Bezier and C. Darcel, Adv. Synth. Catal., 2009, 351, 1732 CrossRef CAS; (d) O. Bistri, A. Correa and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 586 CrossRef CAS.
- (a) W. Liu, H. Cao and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 2004 CAS; (b) F. Vallee, J. J. Mousseau and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 1514 CrossRef CAS.
- (a) Z. Li, L. Cao and C.-J. Li, Angew. Chem., Int. Ed., 2007, 46, 6505 CrossRef CAS; (b) Z. Li, R. Yu and H. Li, Angew. Chem., Int. Ed., 2008, 47, 7497 CrossRef CAS; (c) J. Norinder, A. Matsumoto, N. Yoshikai and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 5858 CrossRef CAS; (d) J. Wen, J. Zhang, S.-Y. Chen, J. Li and X.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 8897 CrossRef CAS; (e) Y.-Z. Li, B.-J. Li, X.-Y. Lu, S. Lin and Z.-J. Shi, Angew. Chem., Int. Ed., 2009, 48, 3817 CrossRef CAS; (f) C.-X. Song, G.-X. Cai, T. R. Farrell, Z.-P. Jiang, H. Li, L.-B. Gan and Z.-J. Shi, Chem. Commun., 2009, 6002 RSC; (g) N. Yoshikai, A. Matsumoto, J. Norinder and E. Nakamura, Angew. Chem., Int. Ed., 2009, 48, 2925 CrossRef CAS.
- For selected recent examples, see: (a) S. Massari, D. Daelemans, M. L. Barreca, A. Knezevich, S. Sabatini, V. Cecchetti, A. Marcello, C. Pannecouque and O. Tabarrini, J. Med. Chem., 2010, 53, 641 CrossRef CAS; (b) L. A. Black, M. D. Cowart, G. A. Gfesser, B. D. Wakefield, R. J. Altenbach, H. Liu, C. Zhao and G. C. Hsieh, WO 2009085945, 2009 Search PubMed; (c) S. Aiello, G. Wells, E. L. Stone, H. Kadri, R. Bazzi, D. R. Bell, M. F. G. Stevens, C. S. Matthews, T. D. Bradshaw and A. D. Westwell, J. Med. Chem., 2008, 51, 5135 CrossRef CAS.
- For selected recent examples, see: (a) D. S. Bose and M. Idrees, Tetrahedron Lett., 2007, 48, 669 CrossRef CAS; (b) D. S. Bose and M. Idrees, J. Org. Chem., 2006, 71, 8261 CrossRef CAS; (c) X.-J. Mu, J.-P. Zou, R.-S. Zeng and J.-C. Wu, Tetrahedron Lett., 2005, 46, 4345 CrossRef CAS; (d) F. M. Moghaddam and H. Z. Boeini, Synlett, 2005, 1612 CrossRef CAS; (e) D.-F. Shi, T. D. Bradshaw, S. Wrigley, C. J. McCall, P. Lelieveld, I. Fichtner and M. F. G. Stevens, J. Med. Chem., 1996, 39, 3375 CrossRef CAS; (f) F. M. Moghaddam and D. Zargarani, J. Sulfur Chem., 2009, 30, 507 CrossRef CAS.
- (a) K. Inamoto, C. Hasegawa, K. Hiroya and T. Doi, Org. Lett., 2008, 10, 5147 CrossRef CAS; (b) L. L. Joyce and R. A. Batey, Org. Lett., 2009, 11, 2792 CrossRef CAS.
- For selected recent examples, see: (a) M. D. Vera and J. C. Pelletier, J. Comb. Chem., 2007, 9, 569 CrossRef CAS; (b) G. Evindar and R. A. Batey, J. Org. Chem., 2006, 71, 1802 CrossRef CAS; (c) C. Benedi, F. Bravo, P. Uriz, E. Fernandez, C. Claver and S. Castillon, Tetrahedron Lett., 2003, 44, 6073 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, spectral data and other supplementary information. See DOI: 10.1039/c1cc16184a |
| This journal is © The Royal Society of Chemistry 2012 |