A 2,2′,6,6′-tetraphosphinobiphenyl†
Holm
Petzold
* and
Albara I. S.
Alrawashdeh
Inst. für Chemie, Strasse der Nationen 62, D-09111 Chemnitz, Germany. E-mail: holm.petzold@chemie.tu-chemnitz.de; Fax: +49 (0)371 531-21219
First published on 31st October 2011
Abstract
The first example of a 2,2′,6,6′-tetraphosphinobiphenyl was prepared and used as a ligand in a dinuclear palladium(II) complex. The close proximity of the two complex fragments induces a coupling pathway allowing the allosteric communication between the two palladium complex fragments.
Bidentate chelating ligands are key building blocks in modern transition metal catalysts. For enantioselective catalysis especially 2,2′-diphosphinobiphenyls have attracted much interest. The most prominent derivative is 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) first synthesized by Noyori and co-workers (Scheme 1).1 Noyori was not only encouraged to the synthesis of BINAP by the fascinating applications in asymmetric chemistry2 but also inspired by “its molecular beauty” due to the high symmetry.3 Nowadays a high number of derivatives have been synthesized4 and numerous publications appear every year applying 2,2′-diphosphinobiphenyl derivatives as ligands for catalytic active transition metal complexes,5 interestingly only few examples of biaryl based tri-6 and tetraphosphanes7 have been reported. In this communication we would like to report the first synthesis of a highly symmetric 2,2′,6,6′-tetraphosphinobiphenyl and its palladium(II) complex (Scheme 2).
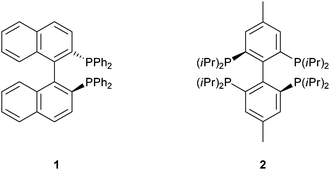 | ||
| Scheme 1 BINAP 1 and the 2,2′,6,6′-tetraphosphinobiphenyl 2. | ||
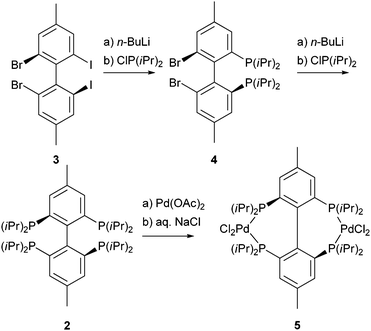 | ||
| Scheme 2 Synthesis of (4,4′-dimethylbiphenyl-2,2′,6,6′-tetrayl) tetrakis(diisopropylphosphane) (2) and its related complex 5. | ||
Employing well known synthesis strategies,6b,8 we gained access to the isopropyl9 decorated tetraphosphane 2 by a “one-pot-two-step” synthesis starting from the easily available 2,2′-dibromo-6,6′-diiodo-4,4′-dimethylbiphenyl 3.10 Upon treatment of 3 with two equivalents of n-BuLi and subsequent reaction with excess of chlorodiisopropylphosphane molecule 4 is formed. Diphosphane 4 gives upon reaction with another two equivalents of n-BuLi and subsequently with chlorodiisopropylphosphane crude product 2. Flash chromatography and crystallization from ethanol yield pure 2. The 31P{1H} NMR spectrum shows the high symmetry of 2, only one signal at δ = −4.1 ppm (C6D6) is found. Similarly the 1H NMR spectrum of 2 shows one set of signals for the eight equivalent isopropyl groups, one signal for the four equivalent aromatic protons as well as one resonance which can be assigned to the two equivalent methyl groups in the 4,4′ position at the biphenyl moiety. The number of signals is consistent with the expected D2h symmetry of 2 with three C2-axes (C2(z)-axes in line with the central C–C bond of the biphenyl moiety and the two symmetry equivalent axes C2(x) and C2(y) perpendicular to the central C–C bond) as well as an S4-axis and two mirror planes.
Due to the high proximity of the phosphane moieties in 2 the lone pairs of the phosphorus atoms are effectively shielded which is also reflected by the low reactivity of 2 towards Pd(cod)Cl2 (cod = cycloocta-1,5-diene). Therefore, molecule 2 was reacted with the more reactive palladium acetate followed by addition of sodium chloride solution to yield the desired dinuclear palladium complex 5 (Scheme 2, Fig. 1).‡ Complex 5 shows a fluxional behaviour and a site exchange is observed indicated by highly temperature dependent NMR spectra. Coordination of 2 to palladium caused large conformational changes in the ligand and led to a hindrance of the free rotation of the isopropyl groups in 5. This becomes apparent in very broad lines in all NMR spectra recorded at 298 K. For example, one signal with line widths about 600 Hz is found at 51 ppm in the 31P{1H} NMR spectrum. Recording the spectra at 473 K in d7-DMF decreases the broadening and the averaged signal over all conformers at δ = 53.9 again showed the high symmetry of the molecule (Fig. 2). Due to the coordination of phosphane 2 to palladium the eight isopropyl groups split into two sets of four equivalent isopropyl groups. A selective NOE NMR experiment (Fig. S1, ESI†) with excitation on the aromatic proton showed contact to the axial isopropyl groups that allowed the assignment to the axial array and the equatorial side. The number of signals found at 373 K is consistent with the lower D2 symmetry of complex 5 compared to the free ligand 2. Decreasing the symmetry from D2h to D2 leads to the loss of the S4-axes and the mirror planes. As a consequence, 5 is chiral with three perpendicular C2 axes. The axes C2(x) (through the Pd-atoms and middle of the central C–C bond) and C2(y) (perpendicular to C2(x) and C2(z)) are not symmetry equivalent. Similarly, cooling the solution results in a splitting of the broad signal in the 31P{1H} NMR spectrum into 14 detected signals (Fig. 2). Based on the intensities the signals found at 223 K can be assigned to four conformers: three C1 symmetric conformers A, B, C giving rise to 4 signals each: A1−4 (δ = 54.2; 49.9; 46.0; 42.3 ppm), B1−4 (δ = 61.0; 40.5; 64.4; 45.2 ppm), C1−4 (δ = 53.5; 40.4; 49.7; 46.2 ppm) and one D1−2 (δ = 51.9; 48.3 ppm) with two signals indicating that one C2 axis is still appropriate (Fig. 2 and Fig. S2, ESI†). Conformers A–D are found in the ratio of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :21:17:49 at 223 K; at lower temperature signals for conformers A and B, respectively, are weaker and the signals for C and D become dominant (at 198 K: 11:17:20:51 and at 178 K: 11:8:24:57). That means the energy minimum conformer of complex 5 is either C or D. Down to the lowest accessible temperature for solution studies the signals sharpen which indicates that rotation of the isopropyl groups is frozen at the 31P NMR time scale (Fig. S4, ESI†). P–P exchange in conformer D must occur through at least one conformer higher in energy and lower in symmetry, which might be one assigned to the signal groups A, B, or C, respectively. To deepen the knowledge of this process, we recorded 2D 31P{1H} EXSY NMR spectra at 208 K in a d7-DMF/CH2Cl2 mixture (ratio 1:2) (Fig. S3, ESI†). The observed exchange signals for the direct P–P exchange are very small, although they should be the strongest as conformer D is the dominant species. Exchange cross peaks of signal D1 with B1/2 and C1/2 as well as D2 with B3/4 and C3/4, respectively, are found. However, this alone does not explain the P–P exchange. Due to limitation in the solubility of complex 5 and the necessary low temperatures we have not been able to obtain a signal to noise ratio allowing detection of cross peaks between other conformers. However, a good guess of the reaction rate is the coalescence point that gives k = 0.5 kHz at 298 K for the P–P exchange.
:21:17:49 at 223 K; at lower temperature signals for conformers A and B, respectively, are weaker and the signals for C and D become dominant (at 198 K: 11:17:20:51 and at 178 K: 11:8:24:57). That means the energy minimum conformer of complex 5 is either C or D. Down to the lowest accessible temperature for solution studies the signals sharpen which indicates that rotation of the isopropyl groups is frozen at the 31P NMR time scale (Fig. S4, ESI†). P–P exchange in conformer D must occur through at least one conformer higher in energy and lower in symmetry, which might be one assigned to the signal groups A, B, or C, respectively. To deepen the knowledge of this process, we recorded 2D 31P{1H} EXSY NMR spectra at 208 K in a d7-DMF/CH2Cl2 mixture (ratio 1:2) (Fig. S3, ESI†). The observed exchange signals for the direct P–P exchange are very small, although they should be the strongest as conformer D is the dominant species. Exchange cross peaks of signal D1 with B1/2 and C1/2 as well as D2 with B3/4 and C3/4, respectively, are found. However, this alone does not explain the P–P exchange. Due to limitation in the solubility of complex 5 and the necessary low temperatures we have not been able to obtain a signal to noise ratio allowing detection of cross peaks between other conformers. However, a good guess of the reaction rate is the coalescence point that gives k = 0.5 kHz at 298 K for the P–P exchange.
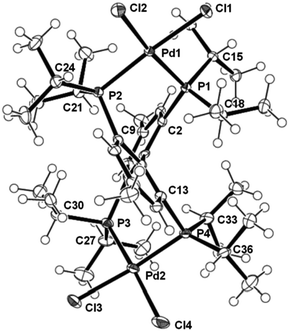 | ||
| Fig. 1 Molecular structure of complex 5 in solid state, ellipsoid probability level is 50%, second molecule and solvent molecules have been omitted for clarity. Bond lengths (Å): Pd1–Pd2 7.7430(4), Cl1–Pd1 2.3738(9), Cl2–Pd1 2.3628(9), Cl3–Pd2 2.3640(9), Cl4–Pd2 2.3327(12), P1–Pd1 2.2744(9), P2–Pd1 2.2782(9), P3–Pd2 2.2993(11), P4–Pd2 2.2492(9), and bond angle (°): P1–Pd1–P2 91.46(3), P1–Pd1–Cl2 170.51(4), P2–Pd1–Cl2 95.37(3), P1–Pd1–Cl1 86.78(3), P2–Pd1–Cl1 176.01(4), Cl2–Pd1–Cl1 85.98(3), P4–Pd2–P3 92.80(4). | ||
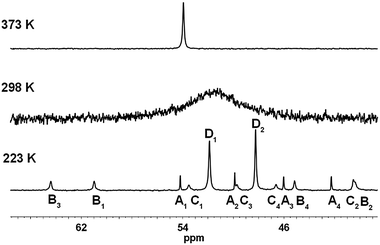 | ||
| Fig. 2 Temperature dependent 31P{1H} NMR spectra of complex 5 in d7-DMF solution at 223, 298, and 373 K, respectively (from bottom to top). The spectra have been scaled in order to make the broad signals visible. See also Fig. S4, ESI.† | ||
By diffusion of n-pentane vapour into a solution of complex 5 in CH2Cl2/MeOH crystals suitable for single crystal X-ray structure determination could be obtained. Complex 5 crystallized in the monoclinic crystal system, space groupP21/c. The asymmetric unit includes two molecules of 5 and five molecules of CH2Cl2 and one of methanol. In contrast to the results of the low temperature NMR spectra, in the solid state none of the molecules of complex 5 has crystallographic symmetry. The isopropyl groups of the ligand are tightly packed and show short contacts to neighboring methyl groups as well as to the aromatic protons at the biphenyl moiety.
Not unexpectedly one of the two molecules of 5 in the asymmetric unit shows disorder that could not be fully resolved; however, the other has been refined without disordered parts (Fig. 1). The most striking feature is the almost square planar coordination of the two palladium complex fragments. Contrary to the expectation for a 2,2′-diphosphane biphenyl ligated palladium complex but in accordance with NMR spectroscopy the palladium atoms are bent away from the expected C2(x) axes through the midpoints of the phosphorus atoms in 2,2′- and 6,6′-positions, respectively, and the centroid of the central C–C bond in the biphenyl moiety. Therefore, the two palladium complex fragments are C1 symmetric, similar observations have been found in mononuclear transition metal complexes with related structure.11 The isopropyl groups do not show a clear preference for edge or face orientation, however, in nearly all PdP-CH(Me)2 moieties stacked conformation is found. The found multitude of orientations of the isopropyl groups in the solid state raises the question which conformer is the dominant one assigned to signal group D observed in the low temperature 31P NMR spectrum. As the crystals have been grown at ambient temperature different conformers are thermally accessible. Moreover, the one with the highest entropy is more dominant in solution at higher temperature and also the one fitting best in the crystal lattice will be depicted from the mixture and subsequently found in the solid state structure. For those reasons it is not wise to draw too many correlations between conformers observed in solution and in the solid state. But if one omits the orientation of the isopropyl groups, the molecular structure of all molecules found in the solid state points to the assumption that the former C2(z)-axes remain valid in D. Hence, most likely conformer D is C2-symmetric with the C2-axis in line with the central C–C bond. However, clearly the spatial arrangement of the isopropyl groups is highly interdependent as the observation of only four conformers in the 31P NMR spectrum and only one C2 symmetric conformer D as the dominant species demonstrates. The two complex fragments combined in 5 cannot simply be treated as a combination of two fluxional fragments but are cross linked and sense each other.
In summary, we have for the first time synthesized a highly symmetric 2,2′,6,6′-tetraphosphinobiphenyl 2 and a related dinuclear palladium complex 5 employing 2 as a ligand. The complex shows complicated temperature dependent NMR spectra that have been rationalized by intramolecular interaction of the isopropyl groups (gear effect).
As the spatial arrangement of the ligands around a metal center often controls the reactivity and properties (e.g. colour, spin state) the found interaction can be recognized as a kind of communication or coupling pathway between the palladium complex fragments through the ligand skeleton similar to the allosteric interaction in enzymes. Related highly symmetric tetraphosphanes like 1,2,4,5-tetrakis(diphenylphosphino)benzene12 or 2,2′,6,6′-tetrakis((diphenylphosphino)methyl)biphenyl7 can also serve as bridging ligands but the larger P⋯P distances between the complex fragments does not allow allosteric interactions of a comparable magnitude.
This work was financially supported by Fonds der Chemischen Industrie and Tafila Technical University. We would also like to thank Prof. H. Lang and Dr T. Rüffer for valuable discussions.
Notes and references
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345–350 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- (a) H. Shimizu, I. Nagasaki and T. Saito, Tetrahedron, 2005, 61, 5405–5432 CrossRef CAS; (b) N. Kurono, M. Uemura and T. Ohkuma, Eur. J. Org. Chem., 2010, 1455–1459 CrossRef CAS.
- (a) P. Wang, X. Liu, J. Yang, Y. Yang, L. Zhang, Q. Yang and C. Li, J. Mater. Chem., 2009, 19, 8009–8014 RSC; (b) S.-Y. Wang and T.-P. Loh, Chem. Commun., 2010, 46, 8694–8703 RSC; (c) H. Aramoto, Y. Obora and Y. Ishii, J. Org. Chem., 2009, 74, 628–633 CrossRef CAS; (d) K. Aikawa, Y. Hioki and K. Mikami, Org. Lett., 2010, 12, 5716–5719 CrossRef CAS; (e) M. McCarthy and P. J. Guiry, Org. Lett., 2007, 9, 4025–4028 CrossRef CAS; (f) U. K. Singh, E. R. Strieter, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14104–14114 CrossRef CAS; (g) M. Kitamura, M. Tsukamoto, Y. Bessho, M. Yoshimura, U. Kobs, M. Widhalm and R. Noyori, J. Am. Chem. Soc., 2002, 124, 6649–6667 CrossRef CAS; (h) K. Mikami and M. Hatano, J. Am. Chem. Soc., 2003, 125, 4704–4705 CrossRef CAS; (i) K. Tanaka and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 8078–8079 CrossRef CAS; (j) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5360–5361 CrossRef; (k) D. A. Evans and R. J. Thomson, J. Am. Chem. Soc., 2005, 127, 10506–10507 CrossRef CAS; (l) D. A. Evans, R. J. Thomson and F. Franco, J. Am. Chem. Soc., 2005, 127, 10816–10817 CrossRef CAS; (m) S. Shekhar, P. Ryberg, J. F. Hartwig, E. R. Strieter and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 3584–3591 CrossRef CAS; (n) A. Kina, H. Iwamura and T. Hayashi, J. Am. Chem. Soc., 2006, 128, 3904–3905 CrossRef CAS; (o) S.-Y. Wang, S.-J. Ji and T.-P. Loh, J. Am. Chem. Soc., 2007, 129, 276–277 CrossRef CAS; (p) S. Maeda and K. Ohno, J. Am. Chem. Soc., 2008, 130, 17228–17229 CrossRef CAS; (q) Y. Hsiao, N. R. Rivera, T. Rosner, S. W. Krska, E. Njolito, F. Wang, Y. Sun, J. D. Armstrong, E. J. j. Grabowski, R. D. Tillyer, F. Spindler and C. Malan, J. Am. Chem. Soc., 2004, 126, 9918–9919 CrossRef CAS; (r) M. McCarthy and P. J. Guiry, Tetrahedron, 2001, 57, 3809–3844 CrossRef CAS; (s) B. H. Lipshutz and H. Shimizu, Angew. Chem., Int. Ed., 2004, 43, 2228–2230 CrossRef CAS; (t) A. M. Johns, J. W. Tye and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 16010–16011 CrossRef CAS; (u) S. Doherty, C. H. Smyth, R. W. Harrington and W. Clegg, Organometallics, 2009, 28, 5273–5276 CrossRef CAS.
- (a) K. Aikawa and K. Mikami, Angew. Chem., Int. Ed., 2003, 42, 5455–5458 CrossRef CAS; (b) L. Bonnafoux, R. Gramage-Doria, F. Colobert and F. R. Leroux, Chem.–Eur. J., 2011, 17, 11008–11016 CrossRef CAS.
- S. Yu, X. Zhang, y. Yan, C. Cai, L. Dai and X. Zhang, Chem.–Eur. J., 2010, 16, 4938–4943 CAS.
- (a) R. Schmid, M. Cereghetti, B. Heiser, P. Schoenholzer and H.-J. Hansen, Helv. Chim. Acta, 1988, 71, 897–929 CAS; (b) F. R. Leroux and H. Mettler, Synlett, 2006, 766–770 CrossRef CAS; (c) F. R. Leroux and H. Mettler, Adv. Synth. Catal., 2007, 349, 323–336 CrossRef CAS.
- So far only the diisopropylphosphane 4 allowed the second lithium/bromide exchange reaction. Diphenylphosphanes and di(n-butyl)phosphanes akin to 4 undergo P–C bond cleavage6a,8b.
- A. Larkem, H. Larkem and J. W. Barton, Abhath Al-Yarmad Basic Sci. Eng., 2003, 12(2B), 491–501 Search PubMed.
- K. Mikami, T. Miyamoto and M. Hatano, Chem. Commun., 2004, 2082–2083 RSC.
- (a) P. Rosa, A. Debay, L. Capes, G. Chastanet, A. Bousseksou, P. L. Floch and J.-F. Letard, Eur. J. Inorg. Chem., 2004, 3017–3019; (b) P.-W. Wang and M. A. Fox, Inorg. Chem., 1994, 33, 2938–2945 CrossRef CAS; (c) G. Hogarth and T. Norman, Inorg. Chim. Acta, 1996, 248, 167–174 CrossRef CAS; (d) V. W.-W. Yam, S. W.-K. Choi and K.-K. Cheung, Organometallics, 1996, 15, 1734–1739 CrossRef CAS; (e) H. C. E. McFarlane and W. McFarlane, Polyhedron, 1988, 7, 1875–1879 CrossRef CAS; (f) H. C. E. McFarlane and W. McFarlane, Polyhedron, 1999, 18, 2117–2127 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the synthesis of compounds 2, 4, and 5 as well as details concerning structure refinement and NMR spectra (Fig. S1–S4). CCDC 832393 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1cc15917h |
| ‡ Synthesis of complex5: One equivalent of ligand 2 (46 mg, 0.0712 mmol) was dissolved in CH2Cl2 (5 mL), and 2 equivalents of palladium acetate (32 mg, 0.1424 mmol) in CH2Cl2 (5 mL) were added dropwise at ambient temperature. The yellow brown suspension was stirred for 6 h at ambient temperature. The reaction mixture was washed several times with brine and extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were dried over MgSO4 and concentrated under reduced pressure. Crystallization from chloroform yielded a yellow solid of 5 (60 mg, 84% based on 2). Mp: 209 °C. 1H NMR (500 MHz, d7-dmf, 100 °C) δ 8.25 (d, 3J(P,H) = 10 Hz, Ar-H, 4H), 4.04 (dsep; 2J(P,H) = 10.4, 3J(H,H) = 7.1 Hz, CH(Me)2, 4H), 2.85 (s, biphen-CH3, 6H), 2.15 (dsep; 2J(P,H) 8.7, 3J(H,H) 7.1 Hz, CH(Me)2, 4H), 1.97 (dd, 3J(P,H) = 17.0, 3J(H,H) = 7.2 Hz, Me, 12H), 1.81 (dd, 3J(P,H) = 15.0, 3J(H,H) = 7.1 Hz, Me, 12H), 1.47 (dd, 3J(P,H) = 20.6, 3J(H,H) = 7.2 Hz, Me, 12H), 1.40 (dd, 3J(P,H)=14.0, 3J(H,H) = 7.2 Hz, Me, 12H). 31P{1H} NMR (202.5 MHz, d7-dmf, 100 °C) δ 53.9 ppm. Elemental analysis calcd (%) for C38H66P4Pd2Cl21/2 CHCl3 (1001.477 g mol−1): C 43.58, H 6.32; found C 43.39, H 6.35. |
| This journal is © The Royal Society of Chemistry 2012 |