Metal to ligand charge transfer induced DNA photobinding in a Ru(II)–Pt(II) supramolecule using red light in the therapeutic window: a new mechanism for DNA modification†
Samantha L. H.
Higgins
a,
Allison J.
Tucker
a,
Brenda S. J.
Winkel
b and
Karen J.
Brewer
*a
aVirginia Polytechnic Institute and State University, Department of Chemistry, Blacksburg, VA 24061-0212, USA. E-mail: k.brewer@vt.edu; Tel: 540-231-6579
bVirginia Polytechnic Institute and State University, Department of Biological Sciences, Blacksburg, VA 24061-0406, USA
First published on 10th November 2011
Abstract
The Ru(II)–Pt(II) supramolecular complex, [(Ph2phen)2Ru(dpp)PtCl2]2+, displays a new mechanism for DNA modification: photobinding through a 3MLCT excited state. Gel shift analysis, selective DNA precipitation, and DNA melting point experiments support efficient covalent DNA binding following visible light excitation.
The interaction of metal-based complexes with DNA is of great interest for the development of anticancer drug therapies. DNA offers several potential binding sites for transition metals, including the anionic phosphate backbone, electron-rich bases, and the major or minor grooves.1 Transition metal complexes can associate with DNA using a variety of binding motifs: ionic binding, covalent modification, intercalation, and major/minor groove binding. The well-studied first generation anticancer complex, cisplatin [cis-Pt(NH3)2Cl2], covalently binds predominantly to the N7-guanine in DNA, causing a distortion in the structure of the DNA and ultimately cell death.2Cisplatin undergoes sequential thermal chloride substitution to form [cis-Pt(NH3)2(OH2)2]2+, important in nuclear membrane permeability and DNA binding.3,4 Several Ru(II)–Pt(II) bimetallic complexes with varied Ru molecular architecture, but similar cis-PtCl2 moieties show thermal chloride ligand substitution with enhanced association and rapid covalent binding compared to cisplatin. Few photocleavage studies and no photobinding are reported.5
Photodynamic therapy (PDT) is a cancer therapy, which utilizes a photosensitizer, such as Photofrin®, molecular oxygen and light that are individually non-toxic, but when combined provide an effective and targeted therapy.6 The prototypical [Ru(bpy)3]2+, bpy = 2,2′-bipyridine, photosensitizer in the presence of visible light and oxygen can cause oxidative DNA damage, through the production of reactive oxygen species (ROS), however the chromophore only weakly associates with DNA.7 Recently, [(bpy)2Os(dppn)]2+, dppn = benzo[i]dipyrido-[3,2-a:2,3-c]phenazine, was reported to photomodify DNA in the therapeutic window utilizing the low energy 3MLCT absorption.8 Generally, successful PDT agents have strong absorption in the therapeutic window (600 to 900 nm) and/or can strongly associate with a specific target in the cell.9
Light activation of DNA binding has been reported for transition metal complexes, which undergo photo-ligand exchange to form covalent bonds with various biological substrates typically through UV excitation and ligand field (LF) excited states.10,11 Morrison and co-workers developed [cis-Rh(phen)2Cl2]+, where phen = 1,10-phenanthroline. Dark ionic binding of the Rh(III)-based complex is dependent upon ionic solution strength. Irradiation (λirr > 330 nm) of the metal complex with DNA results in photo-ligand exchange to form covalent bonds to polynucleic acids. Pt(IV)-based metal complexes using iodide or azide as the reducing ligands have been reported to form reactive Pt(II) species, which can then covalently bind to DNA. Singh and Turro reported the photoinitiated binding of cis-[Ru(bpy)2(NH3)2]2+ to DNA with irradiation >345 nm providing photo-ligand exchange of the ammines to form covalent bonds to DNA. The Turro group also reported the cis-[Rh2(μ-O2CCH3)2(CH3CN)6]2+ complex that undergoes facile thermal and photo hydrolysis. In the presence of light (λirr > 455 nm) and linear pUC18 DNA, the Rh–Rh bimetallic photobinds to DNA. Typically, LF or less commonly LMCT states are responsible for all observed DNA photobinding. The molar absorptivity of MLCT transitions and their ability to be tuned into the low energy visible region make MLCT facilitated DNA photobinding highly desirable as a new DNA photo-modification mechanism applicable for PDT drug development.
Reported herein is the first demonstrated photobinding of a Pt bioactive site to DNA occurring via a new mechanism, MLCT excitation of the coupled Ru chromophore. This new mode of DNA modification by [(Ph2phen)2Ru(dpp)PtCl2]2+ (Ph2phen = 4,7-diphenyl-1,10-phenanthroline and dpp = 2,3-bis(2-pyridyl)pyrazine)) was assayed using electrophoretic gel mobility shift analysis, selective calf thymus DNA (CT-DNA) precipitation, and DNA melting point assays.
Recently, we reported the synthesis, redox, spectroscopic, and photophysical properties of [(Ph2phen)2Ru(dpp)PtCl2]2+, where Ph2phen is the terminal ligand (TL) and dpp is the bridging ligand (BL).12 The Ru(II)–Pt(II) bimetallic uses a polyazine BL to couple these components and impart the light absorbing properties onto the Pt bioactive site. The coupling affords a supramolecule that binds to and photocleaves DNA. The complex was synthesized by a building block method, which allowed for ease of purification and characterization at each step†. The electronic absorption spectroscopy shows strong ligand π → π* transitions in the UV region and Ru(dπ) to TL(π*) or BL(π*) charge transfer (1MLCT) transitions in the visible region, Fig. 1. The electrochemistry supports a Ru(dπ)-based highest occupied molecular orbital (HOMO) and a dpp(π*)-based lowest unoccupied molecular orbital (LUMO), which correlates with the electronic absorption spectroscopy as the lowest energy 1MLCT transition or specifically the Ru(dπ) → dpp(π*) charge transfer transition (ε525 = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 M−1 cm−1, H2O). Excitation of [(Ph2phen)2Ru(dpp)PtCl2]2+ in the UV or visible region results in the population of the lowest lying 3MLCT emissive excited state. Emission of the bimetallic complex occurs at 740 nm with an excited state lifetime of 40 ns. The Ph2phen TL has been shown to enhance the 3MLCT excited state lifetime and reactivity, although the nature of the state is formally Ru(dπ) → dpp(π*) CT.12,13 The Ru(dπ) → dpp(π*) MLCT nature of the lowest lying excited state affords enhanced electron density at the dpp bound to Pt, decreasing the Lewis acidity of the Pt, providing the opportunity for photolabilization, hydrolysis, and DNA photobinding.
100 M−1 cm−1, H2O). Excitation of [(Ph2phen)2Ru(dpp)PtCl2]2+ in the UV or visible region results in the population of the lowest lying 3MLCT emissive excited state. Emission of the bimetallic complex occurs at 740 nm with an excited state lifetime of 40 ns. The Ph2phen TL has been shown to enhance the 3MLCT excited state lifetime and reactivity, although the nature of the state is formally Ru(dπ) → dpp(π*) CT.12,13 The Ru(dπ) → dpp(π*) MLCT nature of the lowest lying excited state affords enhanced electron density at the dpp bound to Pt, decreasing the Lewis acidity of the Pt, providing the opportunity for photolabilization, hydrolysis, and DNA photobinding.
![Structure of [(Ph2phen)2Ru(dpp)PtCl2]2+ and electronic absorption spectrum in H2O at RT.](/image/article/2012/CC/c1cc15780a/c1cc15780a-f1.gif) | ||
| Fig. 1 Structure of [(Ph2phen)2Ru(dpp)PtCl2]2+ and electronic absorption spectrum in H2O at RT. | ||
All photobinding studies were completed using a 5 W LED array (λirr = 455 nm) or a 1000 W Xe arc lamp fitted with 455 and 590 nm cutoff filters (λirr ≥ 590 nm) as the light source (absorption profiles shown in supplementary Fig. 1) and were carried out in phosphate buffer (pH = 7.4).14 Photobinding to pUC18 plasmid DNA by [(Ph2phen)2Ru(dpp)PtCl2]2+ was analyzed using a gel mobility shift assay. The Ru(II)–Pt(II) bimetallic complex was photolyzed (λirr = 455 nm or ≥590 nm) in the presence of pUC18 plasmid DNA for 0, 2.5, 5, 10, 20, 30, 45, and 60 min at a 5:1 BP:MC (base pair to metal complex) ratio. Binding was assayed as a change in DNA migration using gel electrophoresis after irradiation for 0, 2.5, 5, 10, 20, 30, 45, or 60 min at 455 nm or ≥590 nm, shown in Fig. 2A and B, respectively. Both treatments resulted in reduced migration of Form I DNA over time and the formation of a more intense Form II band. The slowing of Form I DNA migration is indicative of metal complex binding, which alters the tertiary structure15 and demonstrates that this complex photobinds DNAvia a unique MLCT excitation mechanism. The enhancement of Form II is indicative of photocleavage of pUC18 DNA facilitated by the generation of 1O2 from the Ru-based 3MLCT state via energy transfer quenching.16 Binding of the complex to DNA is also facilitated by photolysis of the [(Ph2phen)2Ru(dpp)PtCl2]2+ system which presumably produces the aqua complex followed by DNA addition although to a lesser extent, see supplemental information.
![Gel electrophoresis mobility shift assay showing the photobinding of [(Ph2phen)2Ru(dpp)PtCl2]2+ to pUC18 plasmid DNA following 455 nm (A) or ≥ 590 nm (B) irradiation. The lanes correspond to: (λ) lambda molecular weight marker, (C) pUC18 DNA plasmid control, and 0, 2.5, 5, 10, 20, 30, 45, and 60) 5 : 1 BP:MC solutions photolyzed for 0, 2.5, 5, 10, 20, 30, 45, and 60 min, respectively.](/image/article/2012/CC/c1cc15780a/c1cc15780a-f2.gif) | ||
| Fig. 2
Gel electrophoresis mobility shift assay showing the photobinding of [(Ph2phen)2Ru(dpp)PtCl2]2+ to pUC18 plasmid DNA following 455 nm (A) or ≥ 590 nm (B) irradiation. The lanes correspond to: (λ) lambda molecular weight marker, (C) pUC18 DNA plasmid control, and 0, 2.5, 5, 10, 20, 30, 45, and 60) 5:1 BP:MC solutions photolyzed for 0, 2.5, 5, 10, 20, 30, 45, and 60 min, respectively. | ||
Selective precipitation of CT-DNA was used to further probe the photobinding of the metal complex to DNA. Metal complex-DNA solutions were either incubated in the dark or photolyzed and the CT-DNA was selectively precipitated using a previously reported method.17 The amount of unbound metal complex, was determined using electronic absorption spectroscopy of the supernatant. The control samples were incubated at room temperature and at 37 °C in the dark, and the photolysis samples were photolyzed at 455 nm or ≥590 nm for 0, 2.5, 5, 10, 20, 30, 45, and 60 min. The change in the 1MLCT (λabsmax = 525 nm) absorbance of the supernatant over time (in triplicate) was presented as At/A0vs. time, shown in Fig. 3. The dark control shows minimal change in the metal complex absorbance over time, suggesting that the complex binds slowly (not shown within this time frame) to CT-DNA in the dark at RT. Samples that were incubated in the dark at 37 °C only showed ca. 15% of the metal complex bound to the DNA at 60 min. Under photolysis conditions (λirr = 455 nm), the bimetallic complex showed a rapid change in absorbance up to 10 min and reached a steady state by 30 min. The [(Ph2phen)2Ru(dpp)PtCl2]2+-treated samples reached a similar saturation level using lower energy, λirr ≥ 590 nm, red light to induce photobinding, but with slower initial kinetics, consistent with the results of the mobility shift assays. These selective precipitation studies show slow thermal binding and rapid photobinding of the complex to DNAvia MLCT excitation. To the best of our knowledge, this is the first example of an efficient low energy light induced photobinding agent that utilizes a MLCT excitation to generate a unique DNA photobinding event. The experimental binding site size determined based on metal saturation for the Ru(II)–Pt(II) bimetallic complex was 32 Å, which correlates to the minimum of 20 Å.
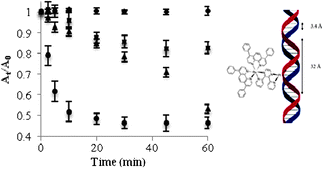 | ||
| Fig. 3 Selective CT-DNA precipitation experiment following incubation at RT in the dark (◆), incubation at 37 °C in the dark (■), photolysis, λirr ≥ 590 nm (▲) and photolysis, λirr = 455 nm (●). The binding site was determined to be approximately 32 Å. | ||
The third method used to analyze the photobinding of [(Ph2phen)2Ru(dpp)PtCl2]2+ to CT-DNA was to examine effects on melting point, or the temperature at which the DNA denatures. The solutions of CT-DNA, 5:1 BP:MC incubated at 37 °C in the dark, and photolyzed at 455 nm for 60 min were heated from 60 to 100 °C by 2.5 °C increments and the absorbance at 260 nm was recorded. CT-DNA controls showed a melting point reported and confirmed by experiment of 85 °C. The solutions incubated at 37 °C, showed no change in melting point. Upon photolysis of the 5:1 BP:MC ratio sample, the Tm decreased by 5 °C, indicative of photobinding of the complex to DNA.
The [(Ph2phen)2Ru(dpp)PtCl2]2+ complex has been shown to photobind to DNA, via MLCT excitation using visible light. The coupling of a Ru-based MLCT light absorber with the Ph2phen TL provides for a Ru → dpp3MLCT excited state with a lifetime of 40 ns. This state moves electron density to the dpp BL, which is bound to the Pt bioactive site. The increased electron density at dpp and therefore decreased Lewis acidity of the attached Pt, may facilitate photolabilization of the chlorides and hydrolysis, leading to efficient DNA binding.4,18 The pUC18 photobinding and photocleavage studies show that photolysis of the metal complex in the presence of DNA results in photobinding at early times†, enhancing DNA photocleavage via ROS localization at the DNA target at later times. The selective DNA precipitation experiments show rapid Ru(II)–Pt(II) photobinding to DNA. Moreover, the metal complex-DNA solutions photolyzed with visible light exhibited a 5 °C decrease in Tm, further indicating DNA modification. The [(Ph2phen)2Ru(dpp)PtCl2]2+-DNA interactions indicate photobinding activity in the therapeutic window, a result of the broad MLCT transitions and large molar absorptivity.
The gel electrophoresis, selective DNA precipitation, and DNA melting point studies demonstrate that the [(Ph2phen)2Ru(dpp)PtCl2]2+ complex photobinds to DNA. This is the first example of a Ru(II)–Pt(II) complex reported to photobind to DNA, which occurs by a new mechanism, ligand labilization via MLCT excitation. The use of visible light, where typical Pt(II) complexes do not absorb displays the importance of the supramolecular architecture, where the spectroscopic properties of the Ru(II) chromophore are imparted onto the cis-PtCl2 bioactive site. Red therapeutic light excitation is possible in this system providing for one of only a handful of metal complexes that can be activated in the therapeutic window and the only Ru(II)–Pt(II) bimetallic complex.8,11,19 This work shows the application of the [(Ph2phen)2Ru(dpp)PtCl2]2+ supramolecular complex as a MLCT activated DNA photobinding agent with enhanced spectroscopic properties allowing for the use of visible light excitation. Studies are underway to explore this new reactivity in further detail.
Notes and references
- L. J. Boerner and J. M. Zaleski, Curr. Opin. Chem. Biol., 2005, 9, 135–144 CrossRef CAS.
- Y. W. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS; S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153–1181 CrossRef CAS; A. S. Abu-Surrah and M. Kettunen, Curr. Med. Chem., 2006, 13, 1337–1357 CrossRef CAS; E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS.
- W. H. Ang, M. Nyint and S. J. Lippard, J. Am. Chem. Soc., 2010, 132, 7249–7435; S. R. Rajski and R. M. Williams, Chem. Rev., 1998, 98, 2723–2796 CrossRef CAS.
- K. B. Garbutcheon-Singh, M. P. Grant, B. W. Harper, A. M. Krause-Heuer, M. Manohar, N. Orkey and J. R. Aldrich-Wright, Curr. Top. Med. Chem., 2011, 11, 521–542 CrossRef CAS.
- M. Milkevitch, H. Storrie, E. Brauns, K. J. Brewer and B. W. Shirley, Inorg. Chem., 1997, 36, 4534–4538 CrossRef CAS; Z. Fang, S. Swavey, A. Holder, B. Winkel and K. J. Brewer, Inorg. Chem. Commun., 2002, 5, 1078–1081 CrossRef CAS; R. L. Williams, H. N. Toft, B. Winkel and K. J. Brewer, Inorg. Chem., 2003, 42, 4394–4400 CrossRef CAS; A. Herman, J. M. Tanski, M. F. Tibbetts and C. M. Anderson, Inorg. Chem., 2008, 47, 274–280 CrossRef CAS; A. Jain, J. Wang, E. R. Mashack, B. S. J. Winkel and K. J. Brewer, Inorg. Chem., 2009, 48, 9077–9084 CrossRef CAS; K. Sakai, H. Ozawa, H. Yamada, T. Tsubomura, M. Hara, A. Higuchi and M. A. Hagac, Dalton Trans., 2006, 3300–3305 RSC.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS.
- C. V. Kumar, J. K. Barton and N. J. Turro, J. Am. Chem. Soc., 1985, 107, 5518–5523 CrossRef CAS.
- Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 6759–6761 RSC.
- R. Bonnett, Chemical Aspects of Photodynamic Therapy, Gordon and Breach Science Publishers, London, 2000 Search PubMed; P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 Search PubMed.
- R. E. Mahnken, M. A. Billadeau, E. P. Nikonowicz and H. Morrison, J. Am. Chem. Soc., 1992, 114, 9253–9265 CrossRef CAS; H. C. Fry, C. Deal, E. Barr and S. D. Cummings, J. Photochem. Photobiol., A, 2002, 150 Search PubMed; T. N. Singh and C. Turro, Inorg. Chem., 2004, 43, 7260–7262 CrossRef CAS; D. A. Lutterman, P. K.-L. Fu and C. Turro, J. Am. Chem. Soc., 2006, 128, 738–739 CrossRef CAS.
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Anti-Cancer Agents Med. Chem., 2007, 7, 75–93 CrossRef CAS.
- S. L. H. Higgins, T. A. White, B. S. J. Winkel and K. J. Brewer, Inorg. Chem., 2011, 50, 463–470 CrossRef CAS.
- M. T. Mongelli and K. J. Brewer, Inorg. Chem. Commun., 2006, 9, 877–881 CrossRef CAS.
- A. J. Prussin, S. Zhao, A. Jain, B. S. J. Winkel and K. J. Brewer, J. Inorg. Biochem., 2009, 103, 427–431 CrossRef.
- B. Zhang, S. Seki, K. Akiyama, K. Tsutsui, T. Li and K. Nagao, Acta Medica Okayama, 1992, 46, 427–434 Search PubMed.
- B. Armitage, Chem. Rev., 1998, 98, 1171–1200 CrossRef CAS.
- R. L. Williams, H. N. Toft, B. Winkel and K. J. Brewer, Inorg. Chem., 2003, 42, 4394–4400 CrossRef CAS.
- D. P. Bancroft, C. A. Lepre and S. J. Lippard, J. Am. Chem. Soc., 1990, 112, 6860–6781 CrossRef CAS; T. W. Hambley, J. Chem. Soc., Dalton Trans., 2001, 2711–2718 RSC.
- J. Wang, S. L. H. Higgins, B. S. J. Winkel and K. J. Brewer, Chem. Commun., 2011, 47, 9786–9788 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, characterization of [(Ph2phen)2Ru(dpp)PtCl2]2+ and photobinding methods. See DOI: 10.1039/c1cc15780a |
| This journal is © The Royal Society of Chemistry 2012 |