Mapping local electric fields in proteins at biomimetic interfaces†
Gal
Schkolnik
a,
Tillmann
Utesch
a,
Johannes
Salewski
a,
Katalin
Tenger
b,
Diego
Millo
a,
Anja
Kranich
a,
Ingo
Zebger
a,
Claudia
Schulz
a,
László
Zimányi
b,
Gábor
Rákhely
c,
Maria Andrea
Mroginski
a and
Peter
Hildebrandt
*a
aTechnische Universität Berlin, Institut für Chemie, Sekr. PC14, Straße des 17. Juni 135, D-10623 Berlin, Germany. E-mail: hildebrandt@chem.tu-berlin.de; Fax: +49 30 31421122; Tel: +49 30 31421419
bInstitute of Biophysics, Biological Research Centre, H-6726 Szeged Temesv, á, ri krt. 62., Hungary
cDepartment of Biotechnology, University of Szeged, Közép fasor 52, H-6726 Szeged, Hungary
First published on 11th November 2011
Abstract
We present a novel approach for determining the strength of the electric field experienced by proteins immobilised on membrane models. It is based on the vibrational Stark effect of a nitrile label introduced at different positions on engineered proteins and monitored by surface enhanced infrared absorption spectroscopy.
Most biochemical and biophysical processes of proteins take place at and in membranes and thus under the influence of electrostatic fields. Particularly strong local electric fields of the order of 109 V m−1 prevail at the membrane/solution interface and in the boundary region between the hydrophobic core and the polar or charged headgroups of the membrane.1 Such high external electric fields may modulate the structure of integral membrane and membrane-attached proteins as well as the conformational and reaction dynamics of proteins, such as in enzymatic processes, ion transport, and electron transfer.1–7 Despite a large body of experimental data, no concept has yet been established to describe the electric-field dependence of protein functions on a molecular level. As an essential part of such a concept, the local electric field strengths at the protein/membrane interface need to be quantified, which requires novel methodological strategies.
Electric field strengths may be determined on the basis of the vibrational Stark effect (VSE), which refers to the electric field induced frequency shift Δν of a vibrational mode according to
hcΔν = −Δ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) ![[F with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0046_20d1.gif) | (1) |
is the change in the dipole moment between the vibrational ground and excited states, is the electric field strength, and h and c denote the Planck constant and the speed of light, respectively. A particularly sensitive VSE reporter group is the nitrile function. In their pioneering work, Boxer and co-workers8–10 have introduced a nitrile label at different sites on the protein surface and at the active site of the enzyme human aldose reductase, and probed the respective C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching modes of the proteins in solution. In the present work, we have, for the first time, extended this strategy to proteins immobilised on simple membrane models monitoring the CN stretchings using surface enhanced infrared absorption (SEIRA) spectroscopy.
N stretching modes of the proteins in solution. In the present work, we have, for the first time, extended this strategy to proteins immobilised on simple membrane models monitoring the CN stretchings using surface enhanced infrared absorption (SEIRA) spectroscopy.
We have chosen the heme protein cytochrome c (Cyt-c) as a convenient test protein that can be readily electrostatically bound to Au electrodes coated with self-assembled monolayers (SAMs) of carboxyl-terminated alkylthiols. Such devices may be considered as a simple model for biological membranes, specifically with respect to the electrostatics at the SAM/solution (membrane/solution) interface.7 Furthermore, the structure and reaction dynamics of Cyt-c have been shown to depend sensitively on the interfacial electric field upon binding to SAM-coated electrodes and liposomes. It has been proposed that electric field variations at the mitochondrial membrane may contribute to the switching of the protein function from electron transport in the respiratory chain to lipid peroxidation as one of the first steps in apoptosis.7 Thus, this work constitutes the starting point for a comprehensive analysis of the electric field control of Cyt-c's function.
4-Mercaptobenzonitrile (MBN) has been used as a nitrile label, covalently attached to a Cys side chain of site-directed engineered horse heart Cyt-c variants. In this work, we have introduced a Cys at two different positions, one in the vicinity of the redox centre (Lys8) and one at a relatively remote position (Lys39). The two variants, K8C and K39C, were then treated with MBN such that the aromatic nitrile was selectively bound to the thiol function of the cysteine (see ESI† for experimental details).
IR spectra of the labelled Cyt-c variants in solution display weak but clearly identifiable peaks in an optical window between 2200 and 2300 cm−1 that is free of any interference by IR bands of the protein, but which includes a strong background absorption (Fig. S1, ESI†). To determine the frequencies of the nitrile stretching modes with higher precision, we have thus used the second derivatives of the spectra in which the minima correspond to the peak maxima of the IR absorption bands (Fig. 1 and 2; for details of the spectra analysis, see ESI†). For the labelled K39C variant in solution (Fig. 1, top), the nitrile stretching mode is observed at 2235.1 cm−1. This frequency, which is distinctly higher than that of solid MBN (2225.7 cm−1) or MBN dissolved in DMSO (2228.3 cm−1, Fig. S2, ESI†), is indicative of a hydrogen bonded nitrile group.11,12 This observation is in line with the results of molecular dynamics (MD) simulations which predict that the nitrile group is solvent-exposed and, therefore, involved in hydrogen bonding interactions with water molecules (Fig. 3). For the labelled K8C variant in solution (Fig. 2, top), the CN stretching mode is found at 2234.5 cm−1, in the same region as that measured for the K39C protein variant, consistent with a solvent-exposed nitrile group. Also in this case, MD simulations predict a solution exposed nitrile group.
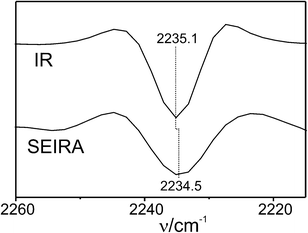 | ||
| Fig. 1 Second derivatives of the IR (top) and SEIRA (bottom, measured at open circuit) spectra of the MBN-labelled K39C variant of Cyt-c. The root-mean standard deviation was ±0.15 (IR) and ±0.4 cm−1 (SEIRA). | ||
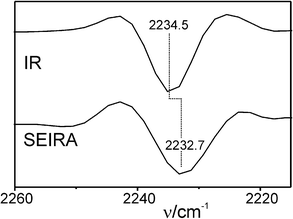 | ||
| Fig. 2 Second derivatives of the IR (top) and SEIRA (bottom; measured at open circuit) spectra of the MBN-labelled K8C variant of Cyt-c. The root-mean standard deviation was ±0.15 (IR) and ±0.9 cm−1 (SEIRA). | ||
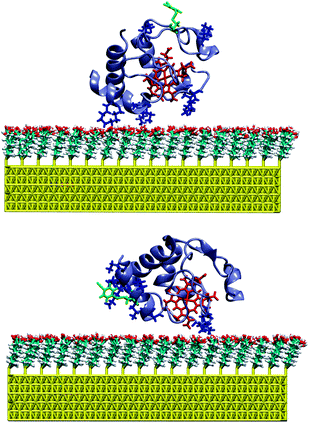 | ||
| Fig. 3 MD snapshot of the labelled K39C (top) and K8C (bottom) variants of Cyt-c bound to the SAM-coated Au surface. The MBN group and the cationic lysine residues are indicated in green and blue, respectively. | ||
In addition to hydrogen bonding interactions, the stretching mode of a nitrile group attached to a protein is affected by the electrostatic field caused by charged and polar residues in its immediate environment. Since in both protein variants the nitrile label is solvent exposed and thus subject to similar hydrogen bonding interactions, the frequency difference ΔνK39C–K8C of the respective bands at 2235.1 cm−1 (K39C) and 2234.5 cm−1 (K8C) is likely to arise mainly from the difference in the component of the protein's intrinsic electric field ΔFK39C–K8C along the CN bond, corresponding to the direction of Δ.11 Thus, one obtains
| ΔνK39C–K8C = νK39C − νK8C = −ΔμSTRΔFK39C–K8C | (2) |
N stretching of MBN has been previously determined to be Δμ = −7.4 × 10−7 cm−1/V cm−1.9 According to eqn (2), one obtains ΔFK39C–K8C = 8.1 × 107 V m−1. This difference in the local electric field may be mainly related to the larger number of positively charged lysine residues in the vicinity of position 8.
In the next step, the labelled proteins were immobilized on an Au film functionalised with a SAM of 6-mercaptohexanoic acid. The Au film, deposited on a silicon prism, serves as a working electrode as well as for enhancing the IR signal in an attenuated total reflection (ATR) SEIRA setup13 (see ESI† for further details). The negatively charged SAM allows for the electrostatic immobilization of Cyt-cvia its positively charged lysine-rich binding domain surrounding the exposed heme crevice.7,13,14 Under these immobilization conditions the native protein structure and function are preserved as demonstrated by cyclic voltammetry and SEIRA spectroscopy13 (ESI†, Fig. S3). In the SEIRA spectrum of the labelled K39C variant, measured at open circuit, the CN stretching mode is observed at 2234.5 cm−1, i.e. at a frequency slightly lower than for the protein in solution (Fig. 1), whereas the SEIRA spectrum of the labelled K8C displays a band at a distinctly lower frequency, i.e. 2232.7 cm−1 (Fig. 2).
MD simulations were carried out for the labelled K39C and K8C variants immobilized on a SAM-coated Au surface, starting with an initial configuration in which the proteins' dipole moments were oriented perpendicularly to the surface. This set-up is expected to afford the energetically most favourable orientation of the electrostatically bound Cyt-c on the SAM-coated Au electrode.15,16 The results indicate that, as for the proteins in solution, the nitrile function remains solvent exposed in both Cyt-c variants. Furthermore, the MD simulations show no indication for immobilization-induced structural changes of the protein in the vicinity of the label (ESI†). In addition, for K8C the simulations rule out a direct interaction of the nitrile function with the carboxyl groups of the SAM, even though the CN label is located in the vicinity of the protein binding domain (Fig. 3). Thus, one may assume that the frequency changes of the nitrile stretching observed upon binding of the proteins to the SAM surface largely result from the modulation of the local electrostatic field by the potential drop across the electrode/SAM/solution interface.
Correspondingly, one may relate the difference between the IR and SEIRA frequencies Δν to the external electric field (x) at a distance x from the SAM surface according to
| Δν = νSEIRA − νIR = |ΔμSTR||(x)|cos(α) | (3) |
(x) and Δ, corresponding to the angle between the CN bond and the surface normal. Thus, the frequency differences Δν of −0.6 cm−1 (K39C) and −1.8 cm−1 (K8C) allow determining the quantity |(x)|·cos(α) according to eqn (3), yielding −8.1 × 107 V m−1 and −2.4 × 108 V m−1 for K39C and K8C, respectively.
As a first approximation, one may assume that the distance-dependent decay of the electric field strength from the SAM surface to the solution follows an exponential function according to
| |(x)| = −ESAMκexp(−κx) | (4) |
The MD simulations allow determining the distance from the centre of the CN bond to the SAM surface as well as the angle of the CN bond with respect to the surface normal, i.e. the vector of the interfacial electric field. Averaging the respective parameters over the last 5 ns of a 10 ns production run, these quantities are evaluated to be 4.4 nm and 127° for K39C, and 2.2 nm and 105° for K8C. Combining eqn (3) and (4), one may then evaluate the ratio of the frequency difference Δν(K39C)/Δν(K8C). The quantity ESAM cancels out. The resultant value is 0.78 and thus larger than the experimental ratio of 0.33, implying that the theoretical approach overestimates the distance-dependent decay of the local electric field. The discrepancy may be mainly due to the underlying assumptions of eqn (4) that neglect spatially fixed charges in the diffuse double layer which are introduced by the immobilisation of Cyt-c at the SAM/solution interface. The consequences are particularly severe for the label at K8C since it is in the vicinity of several positively charged lysine residues. These residues may reorient towards the negatively charged SAM surface, thereby modulating the electric field. Such effects are likely to be less severe for the more remote label in K39C, both due to the longer distance from the SAM surface and the lower number of nearby charged residues.
In conclusion, we have presented an approach that allows mapping the local electric field experienced by proteins immobilized at charged interfaces. Based on the comparative spectroscopic analysis of the protein in solution and in the immobilized state, it is possible to deconvolute contributions arising from the internal electrostatic field of the protein |protein| from the external field |(x)| originating from the potential drop across the SAM/solution interface. Differences in |protein|, determined for two solvent-exposed label positions, are attributed to the local arrangement of charged and polar residues which may in turn also modulate the interfacial electric field experienced by the protein immobilized on a charged surface. As a consequence, an exponential distance dependence of |(x)| at the SAM/solution interface is insufficient for describing the local field strength for labels exposed to the diffuse double layer. The value of |(x)| for the label position of K8C is determined to be 9.4 × 108 V m−1, according to eqn (3) (α = 105°). This result agrees in order of magnitude with previous estimates.17
The present study demonstrates a proof-of-principle and thus constitutes a promising starting point for a future comprehensive investigation of the electric field distribution at the SAM/protein interface, which is required for a quantitative analysis of the electric field effects on the structure and electron transfer dynamics of Cyt-c and other proteins at biomimetic interfaces.
Notes and references
- R. J. Clarke, Adv. Colloid Interface Sci., 2001, 89, 263 CrossRef.
- S. Geibel, T. Friedrich, P. Ormos, P. G. Wood, G. Nagel and E. Bamberg, Biophys. J., 2001, 81, 2059 CrossRef CAS.
- F. Bezanilla, Nat. Rev. Mol. Cell Biol., 2008, 9, 323 CrossRef CAS ; and references therein.
- (a) A. Pilotelle-Bunner, P. Beaunier, J. Tandori, P. Maroti, R. J. Clarke and P. Sebban, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 1039 CrossRef CAS; (b) Z. D. Popovic, G. J. Kovacs, P. S. Vincett, G. Alegria and P. L. Dutton, Chem. Phys., 1986, 110, 227 CrossRef CAS; (c) S. Franzen, R. F. Goldstein and S. G. Boxer, J. Phys. Chem., 1990, 94, 5135 CrossRef CAS.
- M. C. Koag and D. M. Papazian, Channels, 2009, 3, 356 CrossRef CAS.
- C. Weidemüller and K. Hauser, Biochim. Biophys. Acta, Bioenerg., 2009, 1787, 721 CrossRef , and references therein.
- K. K. Ly, M. Sezer, N. Wisitruangsakul, J. J. Feng, A. Kranich, D. Millo, I. M. Weidinger, I. Zebger, D. H. Murgida and P. Hildebrandt, FEBS J., 2011, 278, 1382 CrossRef ; and references therein.
- I. T. Suydam, C. D. Snow, V. S. Pande and S. G. Boxer, Science, 2006, 313, 200 CrossRef CAS.
- I. T. Suydam and S. G. Boxer, Biochemistry, 2003, 42, 12050 CrossRef CAS.
- A. T. Fafarman, L. J. Webb, J. I. Chuang and S. G. Boxer, J. Am. Chem. Soc., 2006, 128, 13356 CrossRef CAS.
- A. T. Fafarman, P. A. Sigala, D. Herschlag and S. G. Boxer, J. Am. Chem. Soc., 2010, 132, 12811 CrossRef CAS.
- D. J. Aschaffenburg and R. S. Moog, J. Phys. Chem. B, 2009, 113, 12736 CrossRef CAS.
- N. Wisitruangsakul, I. Zebger, K. H. Ly, D. H. Murgida, S. Egkasit and P. Hildebrandt, Phys. Chem. Chem. Phys., 2008, 10, 5276 RSC.
- D. H. Murgida and P. Hildebrandt, Phys. Chem. Chem. Phys., 2005, 7, 3773 RSC.
- O. Rüdiger, J. M. Abad, E. C. Hatchikian, V. M. Fernandez and A. L. De Lacey, J. Am. Chem. Soc., 2005, 127, 16008 CrossRef.
- D. Alvarez-Paggi, D. F. Martín, P. M. De Biase, P. Hildebrandt, M. A. Martí and D. H. Murgida, J. Am. Chem. Soc., 2010, 132, 5769 CrossRef CAS.
- D. H. Murgida and P. Hildebrandt, J. Phys. Chem. B, 2001, 105, 1578 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Description of experimental procedures and the MD simulation as well as further spectroscopic data on MBN. See DOI: 10.1039/c1cc13186a |
| This journal is © The Royal Society of Chemistry 2012 |