Pyridine encapsulated hyperbranched polymers as mimetic models of haeme containing proteins, that also provide interesting and unusual porphyrin-ligand geometries†
Lance J.
Twyman
*a,
Adam
Ellis
a and
Peter J.
Gittins
b
aDepartment of Chemistry, The University of Sheffield, Sheffield, S3 7HF, UK. E-mail: l.j.twyman@sheffield.ac.uk; Fax: 0114 2229346; Tel: 0114 2229560
bDepartment of Chemistry, St. Olaf College, 1520 St. Olaf Avenue, Northfield, Minnesota, MN 55057, USA
First published on 31st October 2011
Abstract
This communication describes the use of non-covalent chemistry to construct recyclable porphyrin cored HBPs. The non-covalent design allows the polymeric backbone to be rescued and reused after porphyrin degradation. The steric environment within the polymeric encapsulated ligand notably affected the porphyrin coordination geometry.
In nature, the biosynthetic pathways involved in assembling porphyrins and their associated proteins often make use of non-covalent interactions1–3 such as hydrophobic/hydrophilic interactions, metal–ligand coordination,4 electrostatic interactions,5 and hydrogen bonding.6 For example, the haeme prosthetic group in myoglobin (an iron cored porphyrin subunit) is not covalently attached to its surrounding protein structure, but is held in place non-covalently in the hydrophobic interior of the protein by coordination to a histidine group.7 As well as controlling the various interactions around any bound active, catalytic or binding group, nature also uses the surrounding area to control the electronic environment. A similar non covalent arrangement exists for other proteins, for example cytochrome-c and haemoglobin.8 There have been a number of successful attempts to mimic this ability to control the local electronic environment around a binding or catalytic site.9–11 Arguably the best of these are ordered macromolecular systems that contain active/catalytic groups within the interior of globular structure.12 As such, the area around the binding site controls the electronic environment and catalytic activity in the same way as the protein back-bone does for an enzyme. The best developed of these biomimetic macromolecules are perfectly formed and mono-disperse dendrimers.13 The architecture and interior/environment of these macromolecules can be controlled via their iterative synthesis.14–16 Although this leads to a high degree of control, it is only achieved after a lengthy and potentially difficult synthesis. To overcome this problem, we recently constructed a core functionalised hyperbranched polymer (HBP) that possessed a controlled interior electronic environment.17 Specifically, this HBP was synthesised from the AB2 branching monomer 3,5-diacetoxybenzoic acid, and the B4 core unit, tetra-acetoxyphenyl porphyrin. This was a relatively simple synthesis and the use of reversible/equilibrium chemistry ensured that every polymer molecule possessed a single porphyrin unit at its core. In addition, the molecular weight of the polymer could be controlled by selecting an appropriate core to monomer ratio.18 We were able to insert Fe(II) into the central porphyrin and show that this polymer could model the O2 binding behaviour of haemoglobin and myoglobin.17 In addition, we were also able to demonstrate that the Fe(III) derivative could perform as an efficient oxidation catalyst (a superior catalyst when compared to the non-encapsulated/free porphyrin unit). These abilities were a result of the HBP controlling both local electronic environment and any steric factors around the binding site.19 Unfortunately these HBP systems are not particularly stable and the covalently bound porphyrin was oxidised during the reaction, destroying the catalytic ability of the polymer.20 When this happens we are left with no alternative but to construct a new catalytic system. This was particularly irritating when you consider that only the central porphyrin had degraded. The bulk of the material (the polymer backbone) remained perfectly serviceable. Unfortunately, and despite the fact that only a small part of the molecule was damaged, the covalent design dictates that a completely new HBP system must be constructed. This new polymer must then be thoroughly characterised before use. Furthermore, due to the polydisperse nature of the polymer in terms of structure, architecture and size, the chances of constructing an identical polymeric back bone are almost zero. For certain applications requiring reproducibility, this can be a problem. However, if the polymer backbone and the central porphyrin were linked via non-covalent bonds, the reactive catalysts could be replaced after degradation and the HBP backbone recycled. Such systems would then require only minor characterisation to confirm porphyrin incorporation (and then reused). In addition, this non-covalent design represents a superior mimic of the natural systems they are designed to imitate. Thus, in an effort to produce a more realistic biomimetic model, work began on the design and synthesis of a non-covalent analogue of the porphyrin cored hyperbranched polymer described above.
Pyridine is able to coordinate to the metal centre of a porphyrinvia its lone pair, in the same way that histidine binds to the iron centre in haeme.21–22 In this respect the pyridine ring can be seen as analogous to the imidazole ring of the histidine group, with the zinc porphyrin behaving in a similar way to the haeme prosthetic group.23 Therefore, it was reasoned that a pyridine-cored polymer prepared using the established 3,5-diacetoxybenzoic acid polymerisation procedure should provide a credible model to test our methodology. 3-acetoxy pyridine was selected as the core unit and the polymer (PEHAcPy) was prepared using a slightly modified version of the polymerisation of 3,5-diacetoxybenzoic acid previously reported, as shown in Scheme 1. In light of the site isolation properties displayed by the hyperbranched 3,5-diacetoxybenzoic acid backbone, a lower molecular weight polymer was considered desirable. Polymers with Mn below 5000 Da are below any apparent densely packed limit (which may prevent substrate penetration) and display enhanced electronic environments with respect to binding and catalysis.19 As such, a polymer with an Mn value of around 3000–4000 was targeted. To achieve this molecular weight under standard conditions, a core to monomer ratio of around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 would usually be required (i.e. around 6–7% molar equivalents of pyridine core).18 However, 4-acetoxy pyridine is more volatile than cores previously used and when exposed to the high temperatures and vacuum required for polymerisation, it was expected that some of the pyridine core molecule would be lost. As the extent of loss is unknown, it was decided to use a relatively large amount of the pyridine core.
:15 would usually be required (i.e. around 6–7% molar equivalents of pyridine core).18 However, 4-acetoxy pyridine is more volatile than cores previously used and when exposed to the high temperatures and vacuum required for polymerisation, it was expected that some of the pyridine core molecule would be lost. As the extent of loss is unknown, it was decided to use a relatively large amount of the pyridine core.
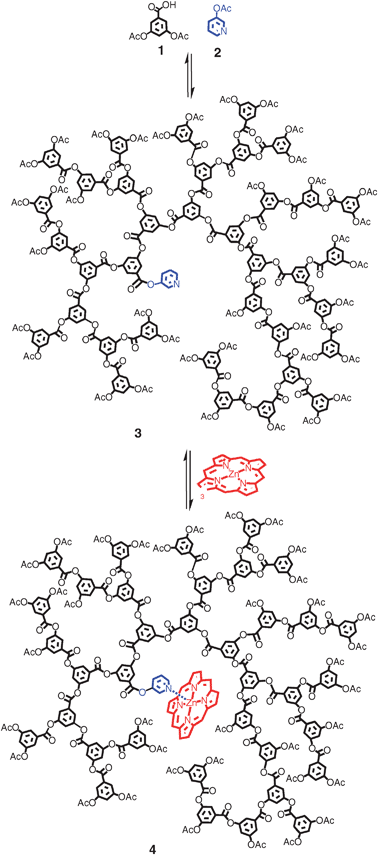 | ||
| Scheme 1 Synthesis of the pyridine cored HBP 3 and its porphyrin complex 4. The final polymers are polydisperse in molecular weight, branching and structure. Representative structures are therefore shown. | ||
Specifically, 0.5 molar equivalents of 3-acetoxy pyridine were used in the polymer feed. The monomer and core molecule were then heated to 200 °C for 45 min, before reducing the temperature to 180 °C and exposing to vacuum for one hour. The polymer was then dissolved in THF and precipitated into methanol giving a white powder in 75% yield after vacuum filtration. Solubility of the unreacted 3-acetoxy pyridine allowed pure polymer product 3 to be obtained by repeating the trituration step a number of times. 1H NMR displayed characteristic polymer peaks at 8.24–7.28 ppm from the aromatic protons, and at 2.08 ppm from the acetoxy terminal groups. In addition to these peaks, a broad doublet was clearly visible at 8.56 ppm. This was attributed to the two protons alpha to the nitrogen in the pyridyl ester group (seen at 8.45 ppm in starting material 2). On closer inspection the remaining protons from the pyridine core were seen contributing to the aromatic multiplet between 8.24 ppm and 7.28 ppm at a resonance of about 8.1 ppm. GPC analysis estimated a molecular weight/Mn value of around 3000 Da.24 The molecular weight can also be estimated from the integration ratio of the pyridyl core peaks to the bulk polymer's aromatic and acetoxy peaks in the 1H NMR. This generated an Mn value of 3450 Da and is in good agreement with that determined by GPC. Given the relatively similar values of Mn(GPC) and Mn(NMR), we can be confident that a high level of pyridine incorporation has been achieved (∼90%).18
Having constructed the polymer we were now in a position to test our hypothesis, that a potentially active core unit could be reversibly bound within the polymer. Simple binding experiments were carried out in the form of titrations to observe and evaluate any interaction between the pyridyl unit and zinc-tetraphenylporphyrin (ZnTPP), compound 4 in Scheme 1. Any binding would be compared to that recorded for a control experiment involving the same porphyrin and a much simpler and smaller ligand, 3-acetoxypyridine (AcPy) 2, Scheme 1. This is the same porphyrin unit used for the core/focal point and the pyridine is electronically very similar to the pyridine at the centre/focal point of the HBP as both have electron withdrawing ester moieties in their 3-position. The zinc metallated porphyrin was prepared according to the literature.25 After purification, a stock solution of ZnTPP was made up in dichloromethane (1 × 10−6 M). This was then used to prepare a solution of the pyridine-cored hyperbranched polymer (1 × 10−2 M), which in turn was titrated into a solution of the porphyrin stock. Performing the titration using this method ensures that a constant porphyrin concentration is maintained throughout. The titration was monitored by UV/Vis spectrophotometry, which follows changes in the porphyrin Soret band absorption. In the case of ZnTPP, the Soret band shifts from 418 nm for unbound porphyrin to 425 nm for bound porphyrin. Plotting the change in absorbance at λmax against moles of ligand added produced a binding curve that fitted for a 1:1 binding isotherm.26 From this, an association constant (Ka) of 1.09 × 103 M−1 was calculated for the porphyrin/polymer interaction using the change in absorbance at 418 nm. The control titration was conducted using 4-acetoxypyridine (AcPy 2) and a Ka value of 1.89 × 103 M−1 was calculated. Although the Ka values for the polymer and control ligand were of the same order of magnitude, the value for the smaller control ligand was roughly double that of the polymer incorporated ligand.
The reduced binding strength of the polymer is easily explained by steric effects. The bulk of the polymer results in a compact free space around the polymer incorporated pyridine site. This prevents the nitrogen lone pair orbital overlapping efficiently with the metals empty d-orbital. This was confirmed using NMR data of the resulting pyridine shifts, which showed a significantly reduced shift for the pyridine's alphaprotons when compared to a control system.27 When optimum binding occurs the preferred geometry is for pyridine to be perpendicular to the plane of the porphyrin (providing maximum orbital overlap). In this position the pyridine is perfectly orientated within the shielded region of the porphyrin, leading to a maximum effect regarding chemical shift (of the pyridine protons). However, if sterics or geometry prevent optimal binding, then the pyridine is less perfectly orientated and shifts are reduced. Specifically, the NMR spectra of a 1:1 complex of the polymer (PEHAcPy 3) and ZnTPP at 2 × 10−2 M indicated a comparatively small 4.1 ppm downfield shift for the pyridine's alphaprotons.‡ This compares to a slightly larger 5.7ppm downfield shift for a control system (a 2 × 10−2 M 1:1 solution of ZnTPP and AcPy 2).
In conclusion; the use of 3-acetoxy pyridine as a core/focal point within hyperbranched 3,5-diacetoxybenzoic acid has successfully been shown to allow reversible non-covalent incorporation of a porphyrin within the bulk structure. As such, this method provides a more robust system that would allow easy recycling of the polymer backbone (after porphyrin degradation). We are currently attempting to exploit this methodology and investigate these and related systems as potential biomimetic models for use in oxygen storage and catalysis. As well as porphyrin incorporation, the methodology described could also allow more exotic and sensitive molecules to be incorporated post synthetically (and non-covalently). Finally, although sterics prevented optimum orbital overlap, this should not necessarily be viewed as a disadvantage For example, the ideas developed in this work could be used to modify ligand binding or to encourage unusual coordination properties and geometries.
References
- C. J. Reedy and B. R. Gibney, Chem. Rev., 2004, 104, 617–649 CrossRef CAS.
- F. Arnesano, L. Banci, I. Bertini, F. Capozzi, S. Ciofi-Baffoni, S. Ciurli, C. Luchinat, S. Mangani, A. Rosato, P. Turano and M. S. Viezzoli, Coord. Chem. Rev., 2006, 250, 1419–1450 CrossRef CAS.
- F. A. Walker, Chem. Rev., 2004, 104, 589–615 CrossRef CAS.
- A. S. Galstyan, S. D. Zaric and E.-W. Knapp, JBIC, J. Biol. Inorg. Chem., 2005, 10, 343–354 CrossRef CAS.
- A. Warshel and R. M. Weiss, J. Am. Chem. Soc., 1981, 103, 446–451 CrossRef CAS.
- S. D. Stojanovic and S. D. Zaric, Open. Struct. Biol. J., 2009, 3, 34–41 CrossRef CAS.
- J. C. Kendrew, G. Bodo, H. M. Dintzis, R. G. Parrish, H. Wyckoff and D. C. Phillips, Nature, 1958, 181, 662–666 CrossRef CAS.
- I. Bertini, G. Cavallaro and A. Rosato, Chem. Rev., 2006, 106, 90–115 CrossRef CAS.
- C. W. Wharton, Int. J. Biol. Macromol., 1979, 1, 3–16 CrossRef CAS.
- P. J. Dandliker, F. Diederich, M. Gross, C. B. Knobler, A. Louati and E. M. Sanford, Angew. Chem., 1994, 106, 1821–1824 CrossRef CAS (see also Angew. Chem., Int. Ed. Engl., 1994, 1833(1817), 1739–1842).
- J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS.
- P. J. Dandliker, F. Diederich, A. Zingg, J. P. Gisselbrecht, M. Gross, A. Louati and E. Sanford, Helv. Chim. Acta, 1997, 80, 1773–1801 CrossRef CAS.
- R. Breslow, Chem. Soc. Rev., 1972, 1, 553–580 RSC.
- G. R. Newkome, Z. Yao, G. R. Baker and V. K. Gupta, J. Org. Chem., 1985, 50, 2003–2004 CrossRef CAS.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Macromolecules, 1986, 19, 2466–2468 CrossRef CAS.
- C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1990, 112, 7638–7647 CrossRef CAS.
- L. J. Twyman and Y. Ge, Chem. Commun., 2006, 1658–1660 RSC.
- P. J. Gittins, J. Alston, Y. Ge and L. J. Twyman, Macromolecules, 2004, 37, 7428–7431 CrossRef CAS.
- X. Zheng, I. R. Oviedo and L. J. Twyman, Macromolecules, 2008, 41, 7776–7779 CrossRef CAS.
- Ref. 19 above and X. Zheng, PhD Thesis, University of Sheffield, 2008 Search PubMed.
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press: San Diego, 2000, vol. 3 Search PubMed.
- N. Datta-Gupta, D. Malakar and R. G. Ramcharan, J. Inorg. Nucl. Chem., 1981, 43, 2079–2080 CrossRef CAS.
- M. Ushiyama, H. Nishikawa, Y. Morio and T. Yamamura, Peptide Sci., 1999, 35, 401–404 Search PubMed.
- L. J. Twyman, A. E. Beezer and J. C. Mitchell, J. Chem. Soc., Perkin Trans. 1, 1994, 407–411 RSC.
- W. Su, K. Singh, J. Rogers, J. Slagle and P. Fleitz, Mater. Sci. Eng., B, 2006, 132, 12–15 CrossRef CAS.
- H.-J. S. A. Yatsimirsky, Principles and Methods in Supramolecular Chemistry, Wiley-Blackwell, 1999 Search PubMed.
- R. J. Abraham, G. R. Bedford and B. Wright, Org. Magn. Reson., 1982, 18, 45–52 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1cc14396d |
| ‡ No uncomplexed pyridine could be detected at these concentrations. |
| This journal is © The Royal Society of Chemistry 2012 |