Electrophoretic mobility measurement by laser Doppler velocimetry and capillary electrophoresis of micrometric fluorescent polystyrene beads
Bo
Xiong
a,
Antoine
Pallandre
*a,
Isabelle
le Potier
a,
Pierre
Audebert
b,
Elias
Fattal
a,
Nicolas
Tsapis
a,
Gillian
Barratt
a and
Myriam
Taverna
a
aUniv Paris Sud 11, Faculté de Pharmacie, CNRS UMR 8612, LabEx LERMIT, 5 rue Jean-Baptiste Clément, Châtenay-Malabry, 92290, France. E-mail: antoine.pallandre@u-psud.fr
bEcole Normale Supérieure de Cachan, Laboratory of Photophysics and Photochemistry Supramolecular and Macromolecular, CNRS UMR 8531, 61 avenue du P Wilson, Cachan, 94235, France
First published on 30th November 2011
Abstract
Many studies have been made and techniques developed to measure the mobility of particles and molecules by laser Doppler velocimetry and capillary electrophoresis. We propose here to evaluate and compare these two measurement techniques for their ability to characterize various fluorescent polystyrene beads as a function of the buffer pH. The repeatability of electrophoretic mobility determination by the two techniques in buffer at different pHs (neutral to alkaline) was first examined and compared. The accuracy of the determination was then evaluated. A wide range of beads which varied in their size (diameters ranging from 270 to 1000 nm), surface functional groups (NH2, COOH, and neutral), and the presence or absence of surfactants or incorporated dye molecules were investigated in order to perform a comprehensive study. The results indicated that apart from large amino beads (with a diameter over 800 nm), capillary electrophoresis generally gave better or similar relative standard deviations for most polystyrene beads, which could be attributed to a stronger adsorption of these beads onto the silica capillary surface in CE. Beads with neutral pH were more difficult to measure accurately with both methods. We also concluded that capillary electrophoresis measurements are not accurate for amino beads in the pH range of this study. However, both methods were capable of distinguishing polystyrene beads with different sizes or surface groups. We found that dye molecules introduced in beads did not alter their electrophoretic mobility values. Taken together, the data and discussion provide a guide to choose the right technique to characterize any given set of functional particles precisely and with the highest accuracy.
Introduction
The last decade has witnessed considerable development of various types of particles and of their potential applications in life science. These particles include light-emitting semiconductor quantum dots, magnetic metal oxide particles, fluorescent polymer beads, and nanoparticles for drug delivery.1–3 Improvements have been made in colloidal stability, photostability, various possibilities of chemical modifications, superparamagnetism and control of specific excitation wavelength.4,5 However, all these properties are determined by their chemical composition, shape, size distribution, charge, surface-attached functional groups, and the fluorescence excitation/emission spectra of incorporated fluorescent probes.6–9 In this context, reliable strategies for characterizing functional beads are required both for monitoring synthesis and for controlling fluorescent particles to be used as probes.Electrophoretic mobility is one of the most important particle properties affecting their movement in electrical fields, and thus their possible sorting.10–12 The zeta potential, directly related to the electrophoretic mobility, is another key parameter giving an insight into their surface state.13–15 For micrometric and submicrometric functional particles, the electrophoretic mobility is no longer proportional to the charge divided by the shearing coefficient, and may become independent of the size of the bead. In this case, the electrophoretic mobility can be calculated from the following equation:16
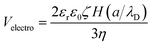 | (1) |
The electrophoretic mobility is conventionally calculated from experiments based on corresponding theoretical approximate models. There are several reported models, among which Smoluchowski and Huckel are the most frequently used.17,18 In short, the Henry function is equal to 1 in the Huckel model where the thickness of the ionic layer immobilized on the bead surface is large with respect to the diameter of the bead itself. In the Smoluchowski model, this value attains 1.5, and this model is employed when the particle velocity is similar to electroosmotic mobility.18
Laser Doppler velocimetry (LDV) is an established method for determining the flow mobility of particles used in life science,19–21 and has been integrated in several commercial instruments.22,23 It uses dynamic laser light scattering in a microtube under electrophoresis conditions to determine the electrophoretic mobility of the particles. On the other hand, capillary electrophoresis (CE) also offers the possibility of measuring the electrophoretic mobility of various particles.18,24–26 McIntire and coworkers carried out size-based separation by this technique. They evaluated the repeatability of electrophoretic mobility values for PBs,27 and also determined the electrophoretic mobility for cationic beads.26,28 In addition, Jones and Ballou optimized the CE conditions and characterized the electrophoretic mobility of chemically different PBs.29 Several years later, Gilman and coworkers reported the use of CE coupled with light scattering technology to characterize PBs.30
There are some obvious differences between LDV and CE. LDV determination is conventionally performed on a microtube of several millimetres internal diameter, and depending on the commercial instruments, these vessels may be composed of PEEK, polycarbonate or polystyrene. The CE determination is carried out on a capillary with an internal diameter of several tens of micrometres generally made of fused silica. These dimensional and chemical differences may lead to different results. For example, beads may interact with the capillary surface in CE due to the high surface energy of the silica inner wall. To overcome this phenomenon several strategies have been developed, including silica coatings and surface treatments.31,32 However, the adsorption/desorption phenomena of the surfactants and additives, that may be present in the particle suspensions, can also lead to some interaction with the polymeric coatings and EOF mobilities may evolve in an unpredictable manner during CE measurements. Moreover, the stability of these coatings may be a problem at extreme pHs. For these reasons, and as the pH was varied over quite a large range in our study, an uncoated capillary was preferred in the present study. Furthermore, LDV measurements are usually made by detecting the phase or frequency shift of an incident laser beam, while the CE is conventionally coupled with UV or laser-induced fluorescence detection, which is based on optical absorbance or emission mechanisms, respectively. These differences in detection as well as in instrumental aspects between the two methods can lead to different accuracies for the determination of electrophoretic mobility. In this context, the present paper aims at comparing electrophoretic mobility determinations made using LDV and CE methods. Up to now, only Guillaumont et al. have reported brief comparisons between LDV and CE,25,33 and these reports did not focus on beads with different functional groups or carrying different dye molecules. A more extensive study presenting a comparison between LDV and CE characterizations of nanolatexes was recently published by Oukacine et al.28 The size range and the chemical functionalities of their beads are different from the micrometric beads investigated in the present study. However, their study benefited from more detailed information concerning the chemical nature of the beads and the surfactant, which helps to draw conclusions from their results.
The synthesis of fluorescent polystyrene beads (PBs) is now well mastered and the control of their size distribution is comparably accurate.34,35 PBs are available commercially and are widely used in various types of research.36,37
In this comprehensive study, the electrophoretic mobilities of PBs with different surface functional groups, sizes and incorporated fluorescent dyes were carefully determined by both LDV and CE. The PBs, provided by the supplier with several characterization data and described in Table 1, were chosen to cover a wide range of bead properties in order to highlight differences between LDV and CE determinations. In particular, we focused on the accuracy and relative standard deviation (RSD) of these determinations, in order to draw conclusions about the suitability of the two strategies for determining electrophoretic mobilities depending on the properties of the investigated beads.
| Sample no. | Supplier | Reference | Diameter/nm | Functional group | λ ex/nm | λ em/nm |
|---|---|---|---|---|---|---|
| a Merck Chimie SAS-Estapor (Paris, France). b Sigma-Aldrich Chemie (Steinheim, Germany). | ||||||
| PB-NH2-268-A | Mercka | F2-XC025 | 268 | Amino | 471 | 523 |
| PB-NH2-268-B | Mercka | F2-Y025 | 268 | Amino | 520 | 569 |
| PB-NH2-1000-C | Sigmab | L0905-1 mL | 1000 | Amino | 418 | 470 |
| PB-alk-NH2-871-A | Mercka | F3-XC080 | 871 | Alkyl chain-amino | 483 | 525 |
| PB-alk-NH2-871-B | Mercka | F3-Y080 | 871 | Alkyl chain-amino | 516 | 566 |
| PB-COOH-328-B | Mercka | F1-Y030 | 328 | Carboxyl | 513 | 566 |
| PB-COOH-392-A | Mercka | F1-XC030 | 392 | Carboxyl | 474 | 523 |
| PB-COOH-1002-A | Mercka | F1-XC100 | 1002 | Carboxyl | 469 | 523 |
| PB-COOH-1009-B | Mercka | F1-Y100 | 1009 | Carboxyl | 519 | 563 |
| PB-plain-319-A | Mercka | FXC-030 | 319 | — | 471 | 526 |
| PB-plain-930-A | Mercka | FXC-100 | 930 | — | 475 | 529 |
Experimental
Chemicals and reagents
The PBs with different sizes, bearing various functional decoration groups and containing different dye molecules, were purchased from either Merck Chimie SAS-Estapor (Paris, France) or SIGMA-ALDRICH CHEMIE (Steinheim, Germany), and all sample references as well as size, functional groups and maximum excitation/emission wavelength in fluorescence spectroscopy are given in Table 1. According to dynamic light scattering determinations, all PBs showed a size variation of less than 5% compared with the data sheet of the suppliers, and the variability of their zeta potentials was less than 10%, as measured by the Nano ZS (Malvern Instruments, Malvern, UK) in zeta potential mode. The electroosmotic flow (EOF) marker thiourea and other chemicals were purchased from SIGMA-ALDRICH CHEMIE (Steinheim, Germany). Throughout the experiments, deionized water from a Milli-Q system (Millipore Corporation, Bedford, Massachusetts, USA) was used to prepare the different solutions. Apart from filtration when necessary and dilution as mentioned, all chemicals were used without any further treatment or purification.Sodium dihydrogen phosphate was obtained from Carlo Erba Reagents (Val de Reuil, France). 10 mM phosphate buffers at defined pH values were prepared from stock solutions of 500 mM sodium dihydrogen phosphate and 1 M sodium hydroxide solution. The pH of the buffer was carefully adjusted to the desired value with a pH-meter (Inolab pH730, WTW, Weilheim, Germany).
Electrophoretic mobility determination by CE
All PB samples were diluted in deionized water and prepared at a concentration of 0.01% (w/w) before CE determinations, and 0.1 mg mL−1thiourea solution was used to determine the EOF under the same CE conditions.The CE determination conditions employed in this study essentially followed those optimized by Jones and Ballou.29 CE experiments were performed on a PACE 5500 (Beckman Coulter Inc., Brea, CA, USA) with an UV detector and 75 μm I.D. capillary. The 0.1 mg mL−1 neutral marker thiourea sample and all PB samples were analysed in 10 mM phosphate buffers at 5 different pH values. The ionic strength of the phosphate buffer was kept low in order to avoid agglomeration of all studied PS particles in suspension in the pH range used for these characterizations. When determining the electroosmotic mobility with thiourea, the UV detector was set at 190 nm, while it was changed to 254 nm when PB samples were determined. A 47 cm capillary, with a 40 cm effective length, was used. A 30 kV voltage was applied for the separation. Between each sample the capillary was flushed with 0.1 M sodium hydroxide solution for 5 minutes, followed by deionized water for 10 minutes and finally with the separation buffer for 15 minutes. The temperature for the CE separation was fixed at 25 °C. CE determination was repeated at least five times.
LDV determination
LDV determinations for the electrophoretic mobility of PBs were performed in a dynamic light scattering instrument Nano ZS (Malvern Instruments, Malvern, UK) in zeta potential mode. A U-shape disposable LDV determination cell (DTS1060, Malvern Instruments, Malvern, UK), with a 750 μL volume, was used. Before each determination, 0.01% (w/w) PB samples in phosphate buffer at the desired pH were freshly prepared by mixing PB samples and 10 mM of the appropriate phosphate buffers, and then were injected into the cell. After inserting the cell into the instrument, a two minute automatic optimization was performed to select the conditions giving the best sensitivity and repeatability of the LDV determination. Thereafter, at least five replicate measurements of the electrophoretic mobility of the PBs were made. The cell was maintained at 25 °C during the measurements. The refractive index and absorption values for polystyrene latex incorporated into the Malvern DTS software were used. The “general purpose” method and a 150 V voltage were used for all measurements. The use of a low molarity buffer ensured that the electric current flowing through the sample remained low, thus avoiding heating and artifacts due to convection.Results and discussion
To compare the electrophoretic mobility values of various PB samples determined by CE or LDV, 10 mM phosphate buffer was chosen as the electrophoresis buffer for CE29 and the dispersion medium for LDV determinations. This condition was selected, based on literature data, to avoid any sedimentation or agglomeration of PBs. This low concentration also afforded a limited Joule heating during electrophoretic processes. To evaluate the influence of pH on CE and LDV determinations, we investigated a pH range from 6.6 to 10.7, as many previous studies have demonstrated good stabilities of beads at these pH values. To measure the electrophoretic mobility of beads by CE, we first evaluated the electroosmotic mobility using thiourea as an EOF marker. Although several studies have reported the usefulness of coating the capillary to limit the bead adsorption and to allow a more accurate estimation of electrophoretic mobility, we did not employ this strategy, as up to now, no coating has demonstrated sufficient stability at all these pHs. In addition, it was not our aim to improve CE performance for this determination but rather to compare the performances of CE and LDV in determining the electrophoretic mobility. Therefore, the polycarbonate cell used for LDV and the CE silica capillaries were not modified for this study. A wide range of PBs varying by their size, surface charge, type of functional groups and incorporated fluorescent dyes were investigated to display a comprehensive comparison of the two techniques.For CE determinations, the electrophoretic mobility of the investigated PBs was estimated by the following equation:
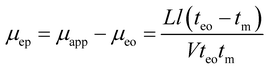 | (2) |
The electrophoretic mobilities of all PBs were determined at five different pH values (6.6, 7.4, 8.4, 9.6 and 10.7) by both CE and LDV. Some of the results are summarized in Table 2. The negative electrophoretic mobilities of amino and plain beads from Merck were unexpected. However, according to the supplier, a negatively charged surfactant was used to stabilize these beads, leading to these values at alkaline pH. An overall inspection of the relative standard deviations (RSD) obtained at different pHs for different PBs generally indicated higher RSDs with LDV compared with CE, except for some specific PBs.
| Sample no. | Electrophoretic mobility/×10−4 cm2 V−1 S−1 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH 6.6 | pH 8.4 | pH 10.7 | ||||||||||
| CE | RSD (%, n = 5) | LDV | RSD (%, n = 5) | CE | RSD (%, n = 5) | LDV | RSD (%, n = 5) | CE | RSD (%, n = 5) | LDV | RSD (%, n = 5) | |
| a All samples. b All samples except PB-NH2-1000-C, PB-alk-NH2-871-A and PB-alk-NH2-871-B. | ||||||||||||
| PB-NH2-268-A | −4.28 | 0.5 | −4.48 | 3.2 | −5.74 | 4.5 | −5.48 | 3.0 | −5.94 | 0.5 | −6.15 | 2.8 |
| PB-NH2-268-B | −4.49 | 5.2 | −5.10 | 2.1 | −5.40 | 3.6 | −6.00 | 1.6 | −6.22 | 0.5 | −6.02 | 3.2 |
| PB-NH2-1000-C | 1.61 | 18.0 | 2.29 | 0.5 | 2.26 | 7.6 | 1.72 | 5.7 | 1.52 | 12.0 | 1.42 | 4.1 |
| PB-alk-NH2-871-A | −2.01 | 23.4 | −2.69 | 1.8 | −3.45 | 1.7 | −3.73 | 1.4 | −4.77 | 13.5 | −5.39 | 2.9 |
| PB-alk-NH2-871-B | −1.48 | 61.0 | −2.25 | 15.1 | −3.11 | 6.0 | −3.88 | 3.3 | −4.70 | 5.1 | −5.37 | 1.5 |
| PB-COOH-328-B | −2.38 | 7.6 | −4.14 | 2.0 | −2.16 | 1.7 | −4.02 | 3.5 | −4.18 | 0.5 | −4.12 | 3.0 |
| PB-COOH-392-A | −2.53 | 7.3 | −3.56 | 3.1 | −2.55 | 0.9 | −3.87 | 0.7 | −3.25 | 1.9 | −3.72 | 4.6 |
| PB-COOH-1002-A | −4.00 | 5.9 | −2.10 | 28.0 | −6.44 | 3.5 | −5.46 | 14.7 | −6.75 | 3.9 | −6.90 | 3.7 |
| PB-COOH-1009-B | −4.45 | 1.3 | −3.09 | 15.2 | −5.58 | 5.9 | −5.30 | 12.1 | −7.49 | 1.4 | −6.55 | 8.0 |
| PB-plain-319-A | −3.20 | 10.2 | −3.08 | 2.2 | −4.63 | 16.5 | −3.99 | 0.8 | −6.24 | 0.6 | −4.73 | 0.6 |
| PB-plain-930-A | −5.80 | 1.8 | −5.70 | 3.9 | −6.25 | 2.9 | −6.31 | 7.8 | −7.55 | 2.0 | −7.42 | 1.9 |
| Mean RSDa | 12.9 | 7.0 | 5.0 | 5.0 | 3.8 | 3.3 | ||||||
| Mean RSDb | 5.0 | 7.5 | 4.9 | 5.5 | 1.4 | 3.5 | ||||||
Indeed, for amino PBs with a diameter larger than 800 nm, such as the samples PB-NH2-1000-C, PB-alk-NH2-871-A and PB-alk-NH2-871-B, the LDV method yielded a much lower relative standard deviation (RSD). This may be related to the difference in the surface nature of PBs and the capillaries employed in the two methods. Some interactions were indeed expected between the amino groups from those PBs and the ionized silanols of the inner surface of the silica capillary used in CE. This kind of interaction would influence the migration behaviour of amino PBs and decrease their apparent velocity. The polycarbonate capillaries used in LDV determination are probably less prone to interact with the cationic beads. With CE, RSDs also seem to be correlated with the size of the PB, and this is more pronounced for certain pH values. For example, at pH 6.6, the RSD increases from 0.5% to 18–61% as the particle size increases from 268 nm to 900/1000 nm. The two largest cationic PBs have comparable diameters, and according to colloid theories, those particles present weaker mutual repulsion, which may make them more unstable with a tendency to self-aggregate; this feature may intensify the adsorption problem already mentioned for CE. This would be expected to lead to a large variation in electrophoretic mobility determinations and may explain the high RSD observed for the largest beads, especially at pH 6.6 and 10.7. In addition, LDV measurements are performed on a larger sample volume and the cooperative effect of particle association onto the inner wall of the polycarbonate cell is probably much less pronounced. So, CE determination in uncoated capillaries is not acceptable in the case of cationic beads with large diameters. Garell et al. have proposed smart strategies to characterize amino nanoparticles and propose a procedure to successfully characterize such functionalized microparticles by CE.38,39 In contrast, cationic PBs with smaller diameters yield, at most pH values, lower RSDs than those obtained from LDV.
In contrast, anionic PBs with smaller diameter beads exhibited lower RSDs with CE than those observed with LDV when the pH is higher than 7.5. This is illustrated in Fig. 1, where the RSD with CE decreases to less than 4% above pH 7.5 and tends to be stable. This is probably related again to the surface chemistry of the capillary. When the pH is lower than 8, the silanols of the capillary are not fully charged, which renders the surface slightly unstable with regard to the EOF. This variation is probably responsible for the electrophoretic mobility variation and the consequently high RSD.
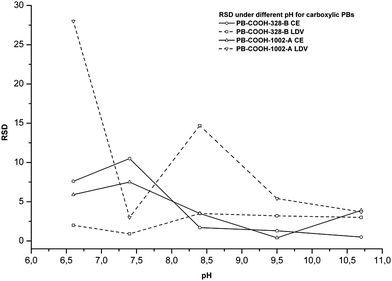 | ||
| Fig. 1 Relative standard deviations at different pHs for some carboxylic PBs by CE and LDV (n = 5). | ||
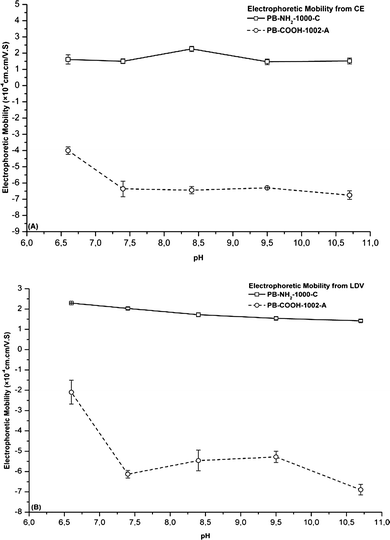
From Table 2, it appears that the electrophoretic mobility values are not always identical when determined with CE and LDV. However, a similar decrease of electrophoretic mobilities with increasing pH is observed with both methods, whatever the beads. As an illustration, Fig. 2 compares the electrophoretic mobility for four fluorescent PBs evaluated in 10 mM phosphate buffer at different pH values. We observed that the electrophoretic mobilities determined for PB-plain-930-A by CE and LDV are very similar. At neutral pH, obvious differences are observed between LDV and CE for most of the beads investigated. Under neutral conditions, the surface charge of all PBs tends to be more neutral, and as a result sedimentation and agglomeration of PBs would be more marked. Therefore, PBs are probably more unstable under neutral conditions. In general, neutral pH is not a favourable medium for either CE or LDV determination.
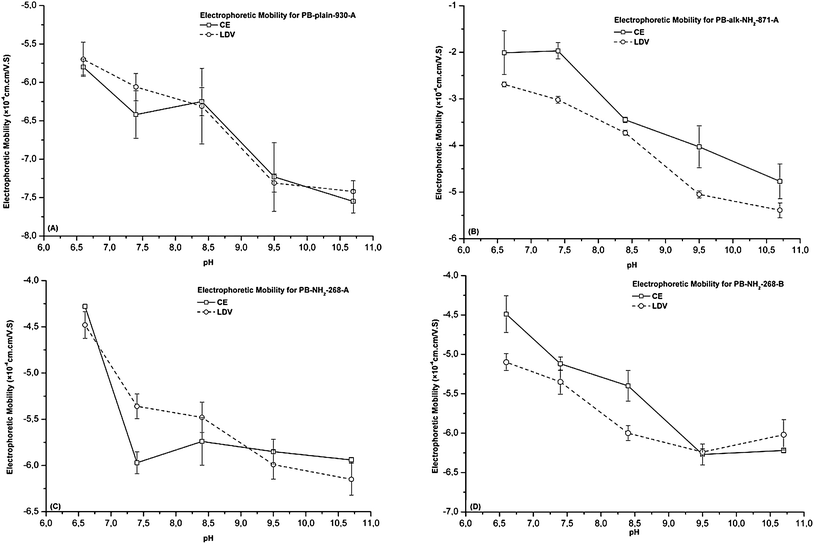 | ||
| Fig. 2 Comparison of the electrophoretic mobility from CE and LDV for various PBs in 10 mM phosphate buffer at different pHs (n = 5). (A) PB-plain-930-A; (B) PB-alk-NH2-871-A; (C) PB-NH2-268-A; and (D) PB-NH2-268-B. | ||
The size distribution is an important factor that influences the electrophoretic mobility of PBs. For the same concentration (expressed as w/v), the PB samples with smaller diameters contain more beads and therefore have a larger total surface area than those containing bigger particles. Therefore, it is interesting to compare CE and LDV for their capacity to discriminate PBs of different diameters by determining their electrophoretic mobility. This would be an indirect way to test the reliability of each technique. For two similar beads only differing in their diameter, we evaluated the ratio between their electrophoretic mobility differences to their diameter differences (Δμep/ΔD). The results are illustrated in Fig. 3 for carboxylic, plain and amino beads. For carboxylic PBs, CE performed better in discriminating the size of the beads based on their electrophoretic mobility, while LDV was generally better for plain PBs. For amino beads, CE led to a better size discrimination for pH values above 8.5, and was comparable to LDV when the pH was between 6 and 9. We can conclude that both CE and LDV are able to differentiate PB samples with different sizes, but CE may be preferable for carboxylic and amino PBs (but only under alkaline conditions) while LDV is better for plain beads.
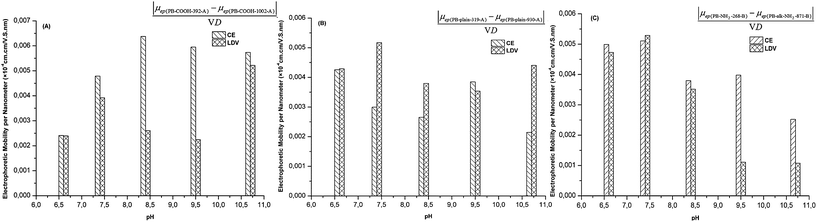 | ||
| Fig. 3 The electrophoretic mobility difference per nanometre in PB diameters determined by CE and LDV at different pHs. (A) Between PB–COOH-392-A and PB–COOH-1002-A; (B) between PB-plain-319-A and PB-plain-930-A; (C) between PB-NH2-268-B and PB-alk-NH2-871-B. | ||
Grafted functional groups should strongly influence the electrophoretic mobility of PBs because they modify the surface charge of PBs. In order to evaluate the capacity of the two methods to distinguish two similar beads bearing different functional groups, the electrophoretic mobility of carboxyl and amino PBs with similar sizes (∼1000 nm) was compared by CE and LDV, as a function of the pH. Fig. 4 shows that similar and consistent results were obtained by CE and LDV. Amino PBs had a positive electrophoretic mobility which was constant over the whole pH range, while PB-COOH-1002-A samples exhibited a slight decrease of their negative electrophoretic mobility from pH 6.6 to pH 7.4, which was observed by both CE and LDV. This could be attributed to the ionization of the carboxylic groups on the surface when the pH rises from 6.6 to 7.4.
 | ||
| Fig. 4 The influence of different functional groups on the electrophoretic mobility of PBs determined by CE and LDV (n = 5). (A and B) Electrophoretic mobility determined by CE and LDV for 1002 nm and 1000 nm PBs with carboxyl groups and amino groups. | ||
The influence of the nature of the dyes incorporated into the PBs on the electrophoretic mobility was also evaluated. The different dyes did not lead to any significant changes in the electrophoretic mobility for alkylamino PBs (Fig. 5A and B) and alkylamino PBs (Fig. 5C and D) when measured by either CE or LDV. The dyes are probably located deep inside the core of the polystyrene nanoparticle and have probably no influence on the electrophoretic mobility of PBs. This was particularly obvious for the large amino beads, when the electrophoretic mobility was estimated by CE.
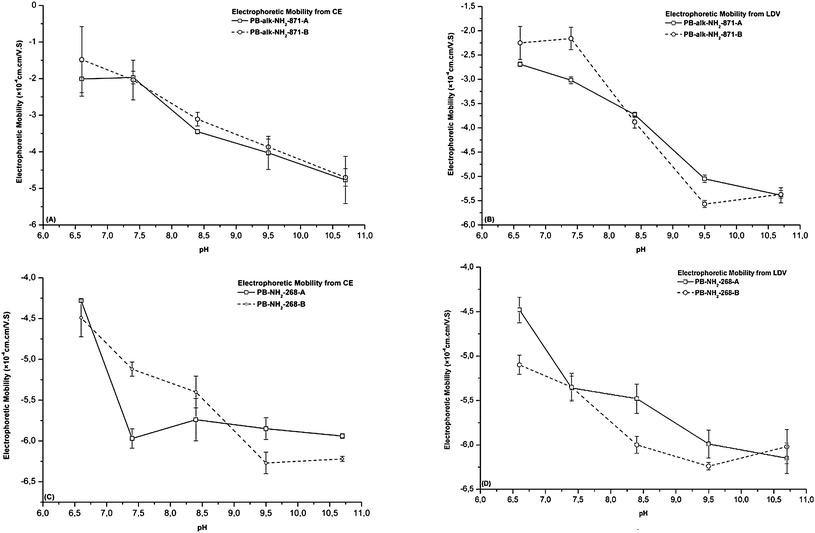 | ||
| Fig. 5 The influence of different dyes in PBs for their electrophoretic mobilities determined by CE and LDV (n = 5). (A and B) Electrophoretic mobility determined by CE and LDV for PB-alk-NH2-871-(A, B); (C and D) electrophoretic mobility determined by CE and LDV for PB-NH2-268-(A, B). | ||
Conclusion
In this study, the electrophoretic mobility of various fluorescent PBs has been determined by CE and LDV and the results compared. The results indicated that, for amino PBs with diameters larger than 800 nm, LDV exhibits lower or comparable RSDs compared with CE, and this is probably related to the interaction of PBs between them or the unstable character of these samples. On the other hand, for other PBs such as smaller amino PBs, carboxylic PBs and plain PBs, CE generally yields a lower or similar RSD when compared with LDV, which is probably due to the better stability of those PBs. In addition, when the pH is above 8, the RSDs for CE are much better, since the surface of the fused silica capillary is fully ionized at these pH values. This is logical since amino groups and the inner wall of the capillary may interact together at alkaline pHs. Neutral polymeric coating would be an interesting strategy to improve CE measurement of amino-functionalized particles by reducing these interactions. Another route to improve this manner of characterizing PBs would be to carry out more experiments to optimize the choice of the buffer. However, most of the time the higher RSD of CE results is related to the methodology of the measurement since each measurement starts with a new injection whereas for LDV characterization the PB solution remains in the cell. Consequently, CE is the most reliable way to measure electrophoretic mobilities when the interactions between PBs and the capillary are low by tuning the pH and the ionic strength of the BGE. It is noteworthy that CE and LDV are both capable of distinguishing PBs with different sizes or surface functional groups. Regarding the influence of different dye probes in PBs on their electrophoretic mobility, the results suggest that there are no significant changes in the electrophoretic mobilities due to the presence of dyes in PB. This result is an experimental proof of the core location of dye molecules in fluorescent PBs. Another point highlighted in this article is that an amino end-terminated PB with a negatively charged surfactant could not easily be analyzed by CE.Abbreviations
| CE | capillary electrophoresis |
| LDV | laser Doppler velocimetry |
| PBs | polystyrene beads |
| EOF | electroosmotic flow |
| RSD | relative standard deviation |
| PEEK | polyether ether ketone |
| BGE | background electrolyte |
Acknowledgements
This research and Dr Bo Xiong were financially supported by Pôle de Recherche et d'Enseignement Supérieur (PRES) foundation, and Agence Nationale de la Recherche (P3N DIMIPOLE).Notes and references
- R. Moller and W. Fritzsche, Curr. Pharm. Biotechnol., 2007, 8, 274 Search PubMed.
- A. Gomez-Hens, J. M. Fernandez-Romero and M. P. Aguilar-Caballos, Mini-Rev. Med. Chem., 2009, 9, 1064 CrossRef CAS.
- Y. Maitani, J. Drug Delivery Sci. Technol., 2011, 21, 27 CAS.
- M. Schaferling and S. Nagl, Anal. Bioanal. Chem., 2006, 385, 500 CrossRef.
- M. J. Choi, A. M. McDonagh, P. Maynard and C. Roux, Forensic Sci. Int., 2008, 179, 87 CrossRef CAS.
- Y. Y. Li, H. Q. Dong, K. Wang, D. L. Shi, X. Z. Zhang and R. X. Zhuo, Sci. China Chem., 2010, 53, 447 CrossRef CAS.
- W. Liu, W. Zhong and Y. W. Du, J. Nanosci. Nanotechnol., 2008, 8, 2781 CAS.
- L. S. Nair and C. T. Laurencin, J. Biomed. Nanotechnol., 2007, 3, 301 CrossRef CAS.
- S. G. Sun, Acta Physico-Chimica Sinica, 2004, 20, 1017 CAS.
- S. S. Ho, K. Critchley, G. D. Lilly, B. Shim and N. A. Kotov, J. Mater. Chem., 2009, 19, 1390 RSC.
- X. Y. Xu, K. K. Caswell, E. Tucker, S. Kabisatpathy, K. L. Brodhacker and W. A. Scrivens, J. Chromatogr., A, 2007, 1167, 35 CrossRef CAS.
- A. I. Lopez-Lorente, B. M. Simonet and M. Valcarcel, TrAC, Trends Anal. Chem., 2011, 30, 58 CrossRef CAS.
- M. H. Chang, D. Dosev and I. M. Kennedy, Sens. Actuators, B, 2007, 124, 172 CrossRef.
- A. Helle, S. Hirsjadrvi, L. Peltonen, J. Hirvonen and S. K. Wiedmer, J. Chromatogr., A, 2008, 1178, 248 CrossRef CAS.
- M. Kosmulski, J. Colloid Interface Sci., 2009, 337, 439 CrossRef CAS.
- D. C. Henry, Proc. R. Soc. London, Ser. A, 1931, 133, 106 CrossRef CAS.
- U. Pyell, Electrophoresis, 2008, 29, 576 CrossRef CAS.
- U. Pyell, Electrophoresis, 2010, 31, 814 CrossRef CAS.
- N. J. Lawson, Proc. Inst. Mech. Eng., Part G, 2004, 218, 33 Search PubMed.
- E. P. Hassel and S. Linow, Meas. Sci. Technol., 2000, 11, R37 CrossRef CAS.
- G. S. Elliott and T. J. Beutner, Prog. Aerosp. Sci., 1999, 35, 799 CrossRef.
- W. Bucking, S. Massadeh, A. Merkulov, S. Xu and T. Nann, Anal. Bioanal. Chem., 2010, 396, 1087 CrossRef.
- C. Sestier, M. F. Da-Silva, D. Sabolovic, J. Roger and J. N. Pons, Electrophoresis, 1998, 19, 1220 CrossRef CAS.
- N. Surugau and P. L. Urban, J. Sep. Sci., 2009, 32, 1889 CrossRef CAS.
- B. Fourest, N. Hakem and R. Guillaumont, Radiochim. Acta, 1994, 66–67, 173 Search PubMed.
- B. V. Huff and G. L. McIntire, J. Microcolumn Sep., 1994, 6, 591 CrossRef CAS.
- B. B. Vanorman and G. L. McIntire, Am. Lab., 1990, 22, 66 CAS.
- F. Oukacine, A. Morel and H. Cottet, Langmuir, 2011, 27, 4040 CrossRef CAS.
- H. K. Jones and N. E. Ballou, Anal. Chem., 1990, 62, 2484 CrossRef CAS.
- Y. H. Rezenom, A. D. Wellman, L. Tilstra, C. D. Medley and S. D. Gilman, Analyst, 2007, 132, 1215 RSC.
- B. B. VanOman and G. L. McIntire, J. Microcolumn Sep., 1989, 1, 289 CrossRef.
- Z. Kucerova, M. Szumski, B. Buszewski and P. Jandera, J. Sep. Sci., 2007, 30, 3018 CrossRef CAS.
- B. Fourest, N. Hakem, J. Perrone and R. Guillaumont, J. Radioanal. Nucl. Chem., 1996, 208, 309 CrossRef CAS.
- C. T. Adkins and E. Harth, Macromolecules, 2008, 41, 3472 CrossRef CAS.
- S. Santra, D. Dutta, G. A. Walter and B. M. Moudgil, Technol. Cancer Res. Treat., 2005, 4, 593 CAS.
- S. Ookawara, N. Oozeki, K. Ogawa, P. Lob and V. Hessel, Chem. Eng. Process., 2010, 49, 697 CrossRef CAS.
- H. S. Cho, Z. Y. Dong, G. M. Pauletti, J. M. Zhang, H. Xu, H. C. Gu, L. M. Wang, R. C. Ewing, C. Huth, F. Wang and D. L. Shi, ACS Nano, 2010, 4, 5398 CrossRef CAS.
- J. Petr, B. Teste, S. Descroix, J.-M. Slaugue, P. Garell and A. Varenne, Electrophoresis, 2010, 31, 2754 CrossRef CAS.
- F. d'Orlyé, A. Varenne, T. Georgelin, J.-M. Slaugue, B. Teste, S. Descroix and P. Gareil, Electrophoresis, 2009, 30, 2572 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |