An O2 smart plastic film for packaging
Andrew
Mills
*a,
Katherine
Lawrie
a,
Julie
Bardin
b,
Alistair
Apedaile
b,
Graham A.
Skinner
b and
Christopher
O'Rourke
a
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, Stranmillis Road, Belfast, BT9 5AG, UK
bWestCHEM, Department of Pure & Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Thomas Graham Building, Glasgow, G1 1XL, UK
First published on 10th November 2011
Abstract
The preparation and characterisation of a novel, water-proof, irreversible, reusable, UV-activated, O2 sensitive, smart plastic film is described. A pigment, consisting of a redox dye, methylene blue (MB), and a sacrificial electron donor, DL-threitol, coated onto an inorganic support with semiconductor functionality, TiO2, has been extruded in low-density polyethylene (LDPE). The blue-coloured indicator is readily photobleached in <90 s using UVA light (4 mWcm−2), whereby MB is converted to its colourless, leuco form, leuco-methylene blue (LMB). This form persists in the absence of oxygen, but is re-oxidised to MB in ∼2.5 days in air under ambient conditions (∼21°C, ∼65% RH) within the O2 smart plastic film. The rate of recovery is linearly dependent upon the ambient level of O2. At the lower temperature of 5°C, the kinetics of the photobleaching activation step is largely unchanged, whereas that of recovery is markedly reduced to t1/2 = 36 h at 5°C (cf. 9 h at 21°C); the activation energy for the recovery step was calculated as 28 kJmol−1. The O2-sensitive recovery step was found to be moderately dependent upon humidity at 21°C, but not significantly dependent upon humidity at 5°C. The possible application of this type of indicator in food packaging is illustrated and discussed briefly.
1. Introduction
Detection of oxygen is prominent within many industrial processes, in particular where an absence of oxygen, or the accurate control of its concentration, is essential. One area of particular interest1–4 is in the monitoring of headspace gas within food packages, since oxygen is responsible for a variety of food-spoiling processes, including promotion of microbial growth, lipid oxidation, protein decomposition and discolouration. Importantly, in order to extend shelf-life, many food packages utilise modified atmosphere packaging (MAP), whereby the package is flushed with an inert gas, such as N2, or an active gas, such as CO2, in order to reduce the O2 concentration from ambient 21% to ∼0.5–2%. There are many established analytical techniques, such as gas chromatography (GC), capable of headspace oxygen detection, which are used to confirm both the efficient removal of O2 during MAP and that effective sealing of the package has occurred. However, most of these techniques are expensive, need a trained operator and require sampling of the packages (and so make a high level of quality control in MAP impossible).As a consequence of the above, much research has therefore been conducted on optical sensors as an alternative analytical technique for O2 measurement;3,4 in particular, with regard to luminescence-based sensors5–7 which are reversible, highly sensitive, with fast response times. However, although commercial luminescence-based O2 sensor systems8–10 have been developed, and shown to be as reliable as GC at O2 headspace analysis in food packages,11 they are still a relatively expensive analytical tool and so are used mainly for MAP R&D. The recent emergence of luminescence-based traffic light O2indicators12–14 offer an exciting alternative to the usual luminescence-based indicators, but in their present form they are unlikely to find application in MAP, since most require specific lighting conditions (typically a UV or coloured LED in an otherwise dark environment) to highlight the effect sufficient for detection by eye.
As a consequence there remains a need for an inexpensive, easy to read and store, irreversible O2indicator, which can provide assurance of package integrity in situ, both on the packaging line and once it reaches the consumer. Furthermore in order to be of use to the consumer, an ideal indicator should produce a discernable colour change, detectable by the untrained eye, i.e. not require any specialist analytical equipment in order to detect the presence of unacceptable levels of oxygen within the package.
Many patents exist15–22 for colorimetric oxygen indicators, as well as some commercial systems, such as the Ageless Eye™,23 but these indicators suffer from high retail costs and a lack of reliability due to their reversible nature. Therefore, many have tried to develop effective, irreversible, O2indicators for MAP. For example, Wilhelm and Wolfbeis recently reported two irreversible O2indicators24,25 that utilise the dyes: indigo (blue) and thioindigo (red), respectively, the reduced forms of which are colourless. Dye reduction was achieved through the addition of sodium dithionite under nitrogen. Since, the preparation of the indicator film and its subsequent storage also requires oxygen-free conditions, its direct application in MAP appears unlikely. Its use in micro-capillary clocks in food packaging will be also hampered by the need for an O2-impermeable capillary material, such as glass, to contain the ink.
We have reported recently a series of UV-activated O2indicator ink films26–32 consisting of: a redox dye, such as methylene blue (MB), a sacrificail electron donor, i.e. SED, and nanoparticles of a UV-sensitive semiconducting photocatalyst (SC), all encapsulated within a polymer. Upon UV exposure, electrons are promoted from the valence to the conduction band within the SC, to create electron-hole pairs. The SED is then itself oxidised, to SEDox, upon reacting with the photogenerated holes, leaving the photogenerated electrons free to reduce the redox dye (Dox, MB) to its reduced, colourless form (Dred, LMB), as illustrated by step (1) in the schematic outline mechanism in Fig. 1. LMB is oxygen-sensitive so that in the absence of oxygen it remains in its colourless, reduced form, but upon exposure to oxygen, the colourless LMB is re-oxidised to its highly absorbing blue, MB form, thereby acting as an O2indicator, as illustrated by the ‘dark’ O2-sensitive step (2) of Fig. 1.
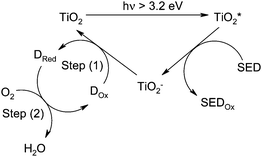 | ||
| Fig. 1 General mechanism of a UVA-activated O2 indicating ink, comprising a semiconductor photocatalyst, such as TiO2, a redox dye (Dox, such as methylene blue) and a sacrificial electron donor (SED, such as DL-threitol). | ||
A water-based version of this ink, when cast down as a film27,28 has been shown to be very responsive to O2, with LMB recovering to MB in ∼6 min in ambient air. Others33,34 have recently investigated the replacement of MB in this ink with polyviologens in order to decrease the sensitivity of a water-based O2indicator film towards low levels of oxygen, since polyviologens have more anodic reduction potentials than methylene blue, and so are less sensitive to oxygen and therefore respond to [O2] levels of 0.5–5.0% at a much reduced rate.
It is an intriguing challenge to move from the use of such inks for printing on food packages to incorporating the technology in the food packaging material itself. Thus, this paper describes the coating of titania nanoparticles with a solution comprising MB and DL-threitol, a SED, to create an O2-sentive pigment which is then incorporated into a thermoplastic polymer, via an extrusion process, to create a UV-activated, O2-sensitive smart plastic film for possible use in the food packaging industry.
2. Experimental
Aeroxide® TiO2 P25 was purchased from Evonik Degussa UK, and comprised 30 nm particles of titania (80% anatase; 20% rutile) with a specific surface area of 50 m2g−1. The low-density polyethylene (LDPE) powder with a melt flow index (MFI) of 20 was supplied by PW Hall UK and LDPE MFI 4 (LUPOLEN 3020 K) pellets were supplied by Ultrapolymers UK. All other chemicals were obtained from Aldrich Chemicals (Gillingham, Dorset, UK), and were used without further purification. All gases were obtained from BOC (London, UK).2.1 Preparation of the pigments
The pigments were prepared by dispersing 160 mg MB, 200 mg DL-threitol and 4 g P25 TiO2 in 30 mL ethanol and mixing thoroughly until a uniform dispersion was obtained. The solvent was then removed by evaporation under a reduced pressure using a rotary evaporator (Buchi, Rotavapor R-3), to produce nano-particulate pigment particles, comprising a mixture of MB and DL-threitol coated onto the insoluble P25 TiO2 particles, as represented in Fig. 2. The blue, UV-activated, O2-sensitive pigment was dried in an oven at 60°C for one hour and ground up to a fine powder using a mortar and pestle.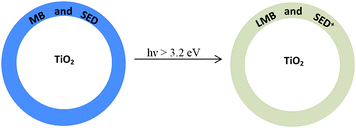 | ||
| Fig. 2 Cross-sectional schematic illustration of a nano-particulate TiO2 pigment particle composition, before and after UVA-activation. | ||
2.2 Extrusion of plastic films
The O2 smart plastic films were manufactured by first creating a pelletised masterbatch of pigment particles in LDPE and then extruding this masterbatch to form a thin O2-sensitive smart plastic film. In the generation of the latter, an LDPE strand containing 10% of the pigment (20 g) dispersed in LDPE MFI 20 powder (180 g) was first extruded using a single-screw Betol BK32 extruder (24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 L:D barrel) and pelletised using a Rondol pelletising unit. The temperatures across the zones of the extruder barrel were 155, 155, 155, 150 and 150°C from feed end to strand die, with the main barrel speed fixed at 20 RPM (2.09 rad s−1) with an output of 34 g min−1. The pelletiser cutter rotation speed was set at 240 RPM (25.1 rad s−1) to create pellets with an average length of 3.2 mm and an average diameter of 2.7 mm. The collected pellets (∼190 g) were then added to the LDPE MFI 4 pellets as a 50:50 (w/w) mixture and extruded as a strand and pelletised. In order to improve the pigment dispersion within the LDPE these pellets were then extruded a third time without any further dilution of the pigment, resulting in a masterbatch of extruded blue pellets containing 5% pigment. Hence the pphr (parts per hundred resin) of the masterbatch containing LDPE, TiO2, DL-threitol and MB is: 100/4.83/0.25/0.19 pphr, where the resin is LDPE.
:1 L:D barrel) and pelletised using a Rondol pelletising unit. The temperatures across the zones of the extruder barrel were 155, 155, 155, 150 and 150°C from feed end to strand die, with the main barrel speed fixed at 20 RPM (2.09 rad s−1) with an output of 34 g min−1. The pelletiser cutter rotation speed was set at 240 RPM (25.1 rad s−1) to create pellets with an average length of 3.2 mm and an average diameter of 2.7 mm. The collected pellets (∼190 g) were then added to the LDPE MFI 4 pellets as a 50:50 (w/w) mixture and extruded as a strand and pelletised. In order to improve the pigment dispersion within the LDPE these pellets were then extruded a third time without any further dilution of the pigment, resulting in a masterbatch of extruded blue pellets containing 5% pigment. Hence the pphr (parts per hundred resin) of the masterbatch containing LDPE, TiO2, DL-threitol and MB is: 100/4.83/0.25/0.19 pphr, where the resin is LDPE.
An O2 smart plastic film was obtained by extruding the masterbatch pellets on a PRISM TSE16TC twin-screw extruder (co-rotating, 25:1 L:D barrel). The temperatures across the zones of the extruder barrel were 50, 135, 145, 150, 155, 155 and 155°C from feed end to film die. The main barrel speed was fixed at 180 RPM (18.8 rad s−1), the feed hopper speed set to 80 RPM (8.38 rad s−1) with an output of 8 g min−1 and the film take off roller speed was fixed at 99 RPM (10.4 rad s−1) producing 2.25 m min−1 of ∼12 cm wide polymer film, with an average central film thickness of 70 μm.
The extruded O2 smart plastic film produced was initially blue upon leaving the extruder, but when left to cool in the dark changed to purple as MB dimers and trimers formed within the film. However, upon 90 s exposure to UVA light (4 mWcm−2) and subsequent recovery in air in the dark for 3 h at 70°C (used to speed up the colour-recovery process) and then overnight at RT, the films return to a consistent blue colour regardless of any further treatment. Thus, subsequent photobleaching and recovery cycles of these UV/heat conditioned films showed the return of the same blue colour. As a consequence, all films described in this work were pre-conditioned as detailed above, prior to their subsequent characterisation and application.
2.3 Methods
All UV irradiations were carried out with a 2 × 8-W Vilber Lourmat black light blue (i.e.UVA) lamp (4 mW cm−2, except when the irradiance was deliberately varied) under otherwise ambient conditions of ∼21°C and ∼65% RH. Unless stated differently, CIELAB colour values (using standard illuminant D65) and diffuse reflectance spectra for the O2 smart plastic films were obtained using a hand-held Konica Minolta CM-2500d spectrophotometer.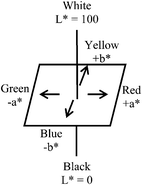 | ||
| Fig. 3 CIELAB colour space. | ||
Since the MB contained in the O2 smart plastic film is initially blue in colour, and upon photobleaching is converted to colourless LMB, the key parameter change is in −b*, where −Δb* is defined:
| −Δb* = b*dark − b*sample,t | (1) |
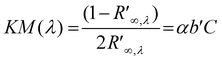 | (2) |
3. Results and discussion
3.1 Photo-activation step
As with the previously reported oxygen-indicating ink films,26–29,31,32 the extruded O2 smart plastic film requires activation by UVA light before oxygen can be detected. The overall process is summarised in Fig. 1. Thus, a typical O2 smart plastic film is activated in less than 90 s, i.e. photobleached, by exposing it to ultraviolet light from a 2 × 8-W UVA lamp (irradiance 4 mWcm−2) and Fig. 4 shows a sample of the extruded O2 smart plastic film, (a) before and (b) after such a UVA-activation step, with Fig. 2 providing a schematic illustration of the cross-section profiles of a typical TiO2/(MB/DL-threitol) nano-particulate pigment particle in the smart plastic film before and after its illumination. | ||
| Fig. 4 Photographs of a section of extruded O2-indicating plastic film (a) before photoactivation and (b) after 90s exposure to UVA with 4 mWcm−2 irradiance. | ||
The change in the diffuse reflectance of the O2 smart plastic film, as a function of irradiation time, was monitored and its KM spectrum recorded before and during the photo-activation process and the results are illustrated in Fig. 5(a). From this data it is interesting to note that the spectrum of the unbleached film shows the MB comprised of a combination of its monomer, dimer and trimer forms,40 most notably the trimer (λmax = 580 nm), which mostly masks the presence of the dimer (λmax = 610 nm). However, during the rapid, initial photobleaching stages the trimer peak appears to be photo-bleached first, to reveal the slightly more recalcitrant dimer and monomer (λmax = 660 nm) peaks. The monomer is consistently and clearly seen during the photobleaching process as a shoulder at 660 nm and, as a result, KM (660 nm) was monitored as a function of time during photobleaching by UVA-light. A plot of KM (660 nm) vs. irradiation time is illustrated in Fig. 5(b), alongside a plot of the variation in −Δb* for the same sample. The insert plot demonstrates the linear relation between −Δb* and KM (660 nm), and since KM (λ) provides a measure of [MB], as indicated by eqn (2), the insert plot suggests that in our system −Δb* also provides a direct measure of [MB]. As a result, −Δb* is used subsequently throughout this work as a measure of [MB].
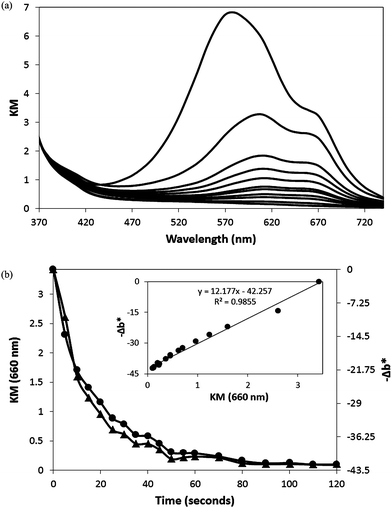 | ||
| Fig. 5 (a) Plot of KM spectra of the oxygen-indicating plastic film, before and during UVA light activation (90 s at irradiance of 4 mW cm2). The irradiation times are (from top to bottom): 0, 5, 10, 15, 20, 25, 30, 40, 50, 60 and 90 s respectively. (b) KM (660 nm) (▲) and −Δb* (●) as a function of irradiation time during photoactivation. The insert plot shows the linear correlation between KM (660 nm) and −Δb*. | ||
The initial rate of photobleaching for a typical O2 smart plastic film was studied as a function of the incident light irradiance and Fig. 6(a) illustrates a plot of a series of recorded −Δb* vs. irradiation time profiles for different UVA light irradiances. For most photocatalytic systems, at low irradiances, the initial rate is found to be proportional to the irradiance, I, (indicating that electron–hole recombination is not significant) and proportional to I1/2 at high irradiance values (indicating that recombination is the predominant fate of photogenerated electron–hole pairs).41 Often,42 at intermediate irradiances, the initial rate is found to be proportional to IΘ, where 0.5 < Θ < 1, and the results of this work is an example of such a case, with Θ = 0.84, as indicated by the plot of the data in Fig. 6(b). Under a modest irradiance (I = 4 mWcm−2) the quantum efficiency43 of the photobleaching process was calculated to be 12%, based on an initial rate of photobleaching at 660 nm of 0.162 KM units/s and a film thickness of ca. 70.0 μm. This is the maximum quantum efficiency, assuming no loss of MB during the mixing and extrusion process and its high value reveals the photocatalytic photoactivation (bleaching) process to be very efficient.
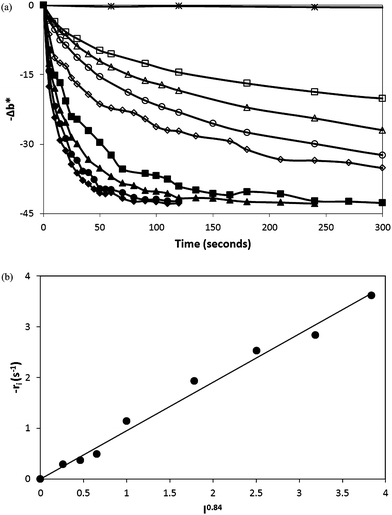 | ||
| Fig. 6 (a) −Δb* vs. time profiles for a typical O2 smart plastic film, photobleaching as a function of different UVA irradiances. The irradiances used were: 0 (*), 0.2 (□), 0.4 (△), 0.6 (○), 1.0 (◇), 2.0 (■), 3.0 (▲), 4.0 (●) and 5.0 (◆) mWcm−2. (b) The initial rate of dye photobleaching (d − Δb*/dt = ri) as a function of the incident irradiance to the power 0.84. | ||
3.2 Dark recovery step
After UVA-activation, the O2 smart plastic film will only return to its original blue colour if an oxidising gas, such as O2 is present. In this case the colour recovery is due to step (2) in Fig. 1, as a consequence of the O2-sensitivity of LMB. This step is irreversible and so any blue colour, due to step (2), will persist even after the source of oxygen is removed, unless or until the film is re-activated with UVA light. The reaction equation for the dark recovery is as follows:| 2LMB + O2 → 2MB+ + 2OH− | (3) |
In contrast to the relatively fast photoactivation step (<90 s at UVA irradiance of 4 mWcm−2), the dark recovery step, back to the O2 smart plastic film's initial blue colour, takes ∼2.5 days in air under ambient conditions (∼21°C, ∼65% RH). This recovery time is significantly slower than that observed for a water-based O2-indicating ink, which recovers in ∼6 min!27 From other work on a similar system in a hydrophobic medium,31 it is suggested that this lengthy recovery time is at least in part due to the formation of the charged species, MB+ and OH−, as seen in eqn (3), being hindered by the more hydrophobic film medium (in this case LDPE) compared to the more hydrophilic medium of a water-based ink film.
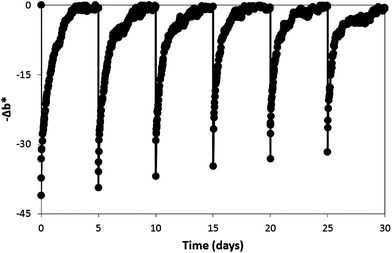 | ||
| Fig. 7 A series of six photoactivations (each: 120 s duration, 4.0 mW cm−2UVA) and subsequent dark recoveries of a single section of extruded O2 smart plastic film, with data collected every 15 min. | ||
The complete recovery of the colour of the film, each time it is UVA activated, suggests the dye is not photochemically or photocatalytically degraded as a consequence of this process; in particular, the photocatalytic oxidative degradation of the dye is prevented by the excess SED, DL-threitol, present which scavenges the photogenerated holes. The slight upward shift in −Δb* values following each irradiation is probably due to the partial consumption of the SED and can be compensated for by using a slightly longer UVA activation time.
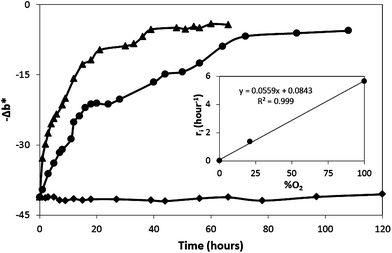 | ||
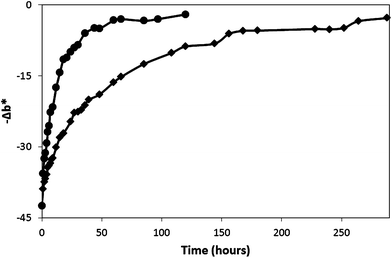
| Fig. 8 Dark recovery profiles of O2 smart plastic film in jars flushed with dry N2 (◆), dry air (●) and dry O2 (▲) gases at 21°C. The insert plot shows the initial rates of recovery as a function of %O2. | ||
 | ||
| Fig. 9 Dark recovery profiles of O2 smart plastic film, stored in a fridge at 5°C (◆) and in a cupboard at 21°C (●). | ||
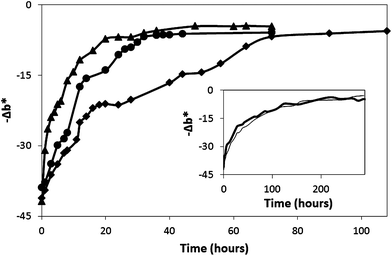 | ||
| Fig. 10 Dark recovery profiles of an O2 smart plastic film in jars flushed with dry air (◆), ambient air (●) and air at 100% RH (▲), all at 21°C. The insert plot shows the dark recovery profiles of an O2 smart plastic film in a fridge at ambient humidity (—) and in a high-humidity fridge (—), both at 5°C. | ||
The recovery of a photobleached O2 smart plastic film at high humidity and low temperature was also monitored by storing a photobleached film in a 5°C fridge containing a water-filled basin to create a RH of 100% at 5°C. The recorded recovery profile was compared to that of a film in a 65% RH atmosphere, also at 5°C, and both recovery profiles are shown in the insert plot of Fig. 10. These results reveal that the change from 65% to 100% RH at 5°C does not significantly affect the performance of the O2 smart plastic film at this temperature. This is possibly not too surprising given the overall low sensitivity of the O2 smart plastic film toward humidity and the low value of the actual (not relative) humidity at 5°C compared to that at room temperature (i.e. 3 times lower).
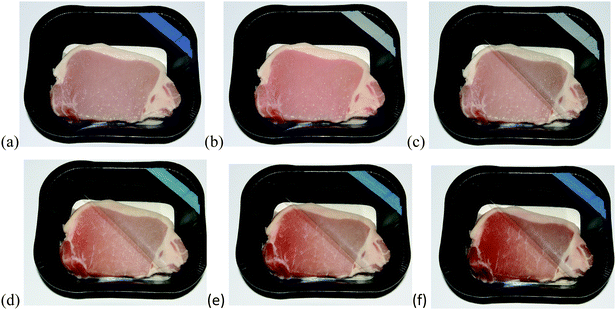 | ||
| Fig. 11 Photographs of pork, packaged along with an O2 smart plastic film, heat sealed to the inside of the PET packaging lid. The package was sealed in the absence of oxygen (a) and the indicator activated (b). The packaging lid was then broken and partly lifted back, and photographed immediately (c), and after 1 day (d), 2 days (e) and 4 days (f) in a fridge at 5°C. | ||
It should be possible to speed up the response of the indicator film by depositing Pt nanoparticles onto the surface of the titania. It should be possible to decrease the response of the film by using a different dye with a more positive redox potential, as found by others.33,34
4. Conclusion
A novel, water-proof, irreversible, reusable O2 smart plastic film containing nano-particulate pigment particles, consisting of MB and DL-threitol coated onto P25 TiO2, has been extruded in LDPE, and subsequently characterised. The blue-coloured indicator is photobleached in <90 s at 4 mWcm−2 and recovers in ∼2.5 days in air under ambient conditions (∼21°C, ∼65% RH). The rate of recovery is linearly dependent upon the %O2 and reduced at a lowered temperature of 5°C, with a calculated Ea = 28 kJmol−1. The rate of colour recovery is moderately dependent upon humidity at 21°C, but is not significantly dependent upon humidity at 5°C. The O2 smart plastic film has potential as a ‘consume-within’ indicator in the food packaging industry.References
- M. L. Rooney, Active Food Packaging, Blackie, London, 1995 Search PubMed.
- A. L. Brody, B. R. Strupinsky and L. R. Kline, Active Packaging for Food Applications, Technomic Publishing Co., Lancaster, 2001 Search PubMed.
- A. Mills, Chem. Soc. Rev., 2005, 34, 1003–1011 RSC.
- J. Kerry and P. Butler, ed., Smart Packaging Technologies for Fast Moving Consumer Goods, John Wiley & Sons Ltd., Chichester, 2008 Search PubMed.
- D. L. Putnam and T. Hubbard, US Pat., 6 794 191, 2004.
- F. C. O'Mahony, T. C. O'Riordan, N. Papkovskaia, J. P. Kerry and D. B. Papkovsky, Food Control, 2006, 17, 286–292 CrossRef CAS.
- C. Baleizão, S. Nagl, M. Schäferling, M. N. Berberan-Santos and O. S. Wolfbeis, Anal. Chem., 2008, 80, 6449–6457 CrossRef.
- http://www.oxysense.com/(accessed 02/08/2011).
- http://www.mocon.com/ (accessed 02/08/2011).
- http://www.freshpoint-tti.com/(accessed 02/08/2011).
- M. A. Pascall, U. Fernandez, R. Gavara and A. Allafi, J. Food Eng., 2008, 86, 501–507 CrossRef.
- R. C. Evans, P. Douglas, J. A. G. Williams and D. L. Rochester, J. Fluoresc., 2006, 16, 201–206 CrossRef CAS.
- X.-D. Wang, X. Chen, Z.-X. Xie and X.-R. Wang, Angew. Chem., Int. Ed., 2008, 47, 7450–7453 CrossRef CAS.
- X.-D. Wang, H.-X. Chen, Y. Zhao, X. Chen and X.-R. Wang, TrAC, Trends Anal. Chem., 2010, 29, 319–338 CrossRef CAS.
- Y. Yoshikawa, T. Nawata, M. Goto and Y. Fujii, US Pat., 4 169 811, 1979.
- Y. Yoshikawa, T. Nawata, M. Goto and Y. Kondo, US Pat., 4 349 509, 1982.
- H. Nakamura, N. Nakazawa and Y. Kawamura, JP Pat., 62 183834, 1987.
- K. C. Krumhar and M. Karel, US Pat., 5 096 813, 1992.
- T. Mattila-Sandholm, R. Ahvenainen, E. Hurme and I. T. Järvi-Kääriäinen, EP Pat., 0 666 977, 1995.
- C. D. Garner and E. S. Davies, UK Pat., 2 298 273, 1996.
- H. Hatakeyama, S. Tobari, S. Iwauchi and M. Ohsawa, US Pat., 6 676 901 2000.
- M. Sumitani, H. Inoue and K. Sugito, US Pat., 6 703 245, 2004.
- http://www.mgc.co.jp/eng/products/abc/ageless/eye.html (accessed 02/08/2011).
- S. Wilhelm and O. Wolfbeis, Microchim. Acta, 2010, 171, 211–216 CrossRef CAS.
- S. Wilhelm and O. S. Wolfbeis, Sens. Actuators, B, 2011, 153, 199–204 CrossRef.
- A. Mills and S.-K. Lee, Int. Pat., 021 252, 2003.
- S.-K. Lee, A. Mills and A. Lepre, Chem. Commun., 2004, 1912–1913 RSC.
- S.-K. Lee, M. Sheridan and A. Mills, Chem. Mater., 2005, 17, 2744–2751 CrossRef CAS.
- A. Mills and D. Hazafy, Analyst, 2008, 133, 213–218 RSC.
- A. Mills and D. Hazafy, Sens. Actuators, B, 2009, 136, 344–349 CrossRef.
- A. Mills, D. Hazafy and K. Lawrie, Catal. Today, 2011, 161, 59–63 CrossRef CAS.
- A. Mills and K. Lawrie, Sens. Actuators, B, 2011, 157, 600–605 CrossRef CAS.
- L. Roberts, R. Lines, S. Reddy and J. Hay, Sens. Actuators, B, 2011, 152, 63–67 CrossRef.
- L. Roberts, R. Lines, S. Reddy and J. Hay, Sens. Actuators, B, 2011, 159, 342–344 CrossRef CAS.
- CIE, Colorimetry, Third edn, Commission internationale de l'Eclairage, CIE Central Bureau, 2004 Search PubMed.
- HunterLab, in Applications Note, Insight on Color, 2008, vol. 8, pp. 1–4 Search PubMed.
- P. Kubelka, J. Opt. Soc. Am., 1948, 38 CAS , 448–457 & 1067.
- M. P. Fuller and P. R. Griffiths, Anal. Chem., 1978, 50, 1906–1910 CrossRef CAS.
- A. J. Julson and D. F. Ollis, Appl. Catal., B, 2006, 65, 315–325 CrossRef CAS.
- E. Braswell, J. Phys. Chem., 1968, 72, 2477–2483 CrossRef CAS.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- J. M. Herrmann, Top. Catal., 2005, 34, 49–65 CrossRef CAS.
- N. Serpone, R. Terzian, D. Lawless, P. Kennepohl and G. Sauvé, J. Photochem. Photobiol., A, 1993, 73, 11–16 CrossRef CAS.
- M. Sheridan, PhD Thesis, University of Strathclyde, 2008.
- A. Vishnyakov and A. V. Neimark, J. Phys. Chem. B, 2008, 112, 14905–14910 CrossRef CAS.
- S. N. Dirim, H.Ö. Özden, A. BayIndIrlI and A. Esin, J. Food Eng., 2004, 63, 9–13 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |