α-Helix unfolding in simple shear flow†
Innocent B.
Bekard
a,
Kevin J.
Barnham
b,
Lee R.
White
c and
Dave E.
Dunstan
*a
aDepartment of Chemical and Biomolecular Engineering, The University of Melbourne, Melbourne, Victoria 3010, Australia
bDepartment of Pathology, Bio21 Institute, The University of Melbourne, Melbourne, Victoria 3010, Australia
cSchool of Mathematics and Statistics, University of South Australia, Mawson Lakes, 5095, Australia. E-mail: davided@unimelb.edu.au; Tel: +61 (03)8344 8261
First published on 13th October 2010
Abstract
The unfolding dynamics of the α-helical poly-L-lysine (α-PLL) in Couette flow is reported. Real-time circular dichroism measurements for a range of molecular weights and shear rates have been made. The PLL molecules show a time- and shear rate-dependent unfolding in simple shear flow with a critical strain (![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) tc) value of ∼105. This strain value is found to be independent of the chain-length of the α-helices. The extent of unfolding is less pronounced with increasing molecular weight (M) for a given strain, showing a linear dependence of the remaining helix, α, on M: α ≈ M. Furthermore, the helix content, α, is found to show a power law dependence with strain: α ≈ (t)−1/2. A shear-induced rapid unfolding of short chain α-PLL molecules in the flow field occurs. The shear-stability of the larger molecular weights is due to the cohesive forces stabilizing the helix, combined with the associated hydrodynamic screening of helical segments from the full effect of the drag in the flow field. The data are compared with recent molecular dynamics simulations of the dynamics of dilute polymer solutions in shear flow and scaling arguments are used to interpret the trends in the data.
tc) value of ∼105. This strain value is found to be independent of the chain-length of the α-helices. The extent of unfolding is less pronounced with increasing molecular weight (M) for a given strain, showing a linear dependence of the remaining helix, α, on M: α ≈ M. Furthermore, the helix content, α, is found to show a power law dependence with strain: α ≈ (t)−1/2. A shear-induced rapid unfolding of short chain α-PLL molecules in the flow field occurs. The shear-stability of the larger molecular weights is due to the cohesive forces stabilizing the helix, combined with the associated hydrodynamic screening of helical segments from the full effect of the drag in the flow field. The data are compared with recent molecular dynamics simulations of the dynamics of dilute polymer solutions in shear flow and scaling arguments are used to interpret the trends in the data.
Introduction
Astbury was the first to show molecular level conformational changes in proteins due to applied stress.1 Elastic stretching of keratin fibres in combination with X-ray scattering led Astbury and Woods to determine that a helix to β-sheet transition occurred under large strain.1 This insight led Pauling and Corey to later determine the helical structure accurately using X-ray scattering.2,3 Since the seminal study by Astbury and co-workers, a large body of work has been directed to understanding the effect of stresses on synthetic and biological molecules. The work of Smith et al. and LeDuc et al. has been instrumental in developing our understanding of the effect of hydrodynamic forces on DNA molecules in solution.4,5 A number of recent publications have shown that hydrodynamic forces do indeed induce conformational changes in proteins and cause unfolding events to occur.6–10 Given that protein conformation is critical in their function, these findings have significant implications for much of biology. Furthermore, the effects of agitation and shear flow have also been known to accelerate amyloid fibril formation.7,11–15 In physiology, haemodynamic shear stress is implicated in the formation of defective protein aggregates, a pathological signature of protein conformational disorders (e.g. Alzheimer's disease)16,17 and vascular disorders (e.g. atherosclerosis).18Several recent studies have shown the presence of α-helical intermediates in the pathway to fibril formation by several proteins.19–22 However, the role of the α-helical intermediate in amyloid formation is not understood. Given that α-helices are a very important and common secondary structural feature of a significant number of proteins,23 understanding their role in protein unfolding and fibril formation would seem a worthwhile endeavour. To this end, we have investigated the model system, poly-L-lysine, which forms predominantly α-helical structures at high pH.24,25 A range of molecular weights have been investigated by exposure to defined Couette flow while measuring the circular dichroism spectra in real time. Critically, we show that the α-helices unfold in flow where the strain is the key parameter in determining the unfolding. The findings have significant implications for proteins in flow. The specific nature of the protein tertiary structure will be instrumental in stabilising the helices against flow induced unfolding.
Experimental results demonstrate that partial unfolding of natively folded proteins, such as the helical segments of insulin, is a prerequisite for aggregation and amyloid fibril formation.19,26,27 Theoretically, stretching of protein molecules in extensional flow fields may well induce partially extended conformations,28 with solvent-exposed hydrophobic segments, leading to intra-/intermolecular complexation (aggregation) arising from hydrophobic interactions. The frequency of stretching events in simple shear flow amplifies as a function of shear rate (), and the duration (t) of its application.29 Nonetheless, the literature presents conflicting reports on the concept of protein unfolding in simple shear flow. For example, early reports which showed that fluid forces significantly impair the catalytic activity of several enzymes, even for shear rates as low as 9.15 s−1,30 could not be reproduced in later studies.31 For this reason, enzyme deactivation was attributed to secondary shear-associated effects such as gas–liquid interactions, oxidation, and metal contamination but not shear per se. However, a major limitation in these preliminary studies was the absence of sensitive optical techniques to directly probe structural changes in the globular proteins exposed to shear flow. Hence, indirect experimental evidence was used to examine shear denaturation.
Nonetheless, recent studies using real-time fluorescence spectroscopy, a sensitive structural technique, to directly monitor the shear stability of horse cytochrome c,32 and human von Willebrand factor (vWF),8 under hydrodynamic shear stress in capillary flow, gave opposing results. Whereas cytochrome c (Mw ≈ 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 384 Da) showed no structural changes for shear rates as high as 105s−1, vWF (Mw ≈ 2 × 107 Da) demonstrated structural instability at a threshold shear rate of 103s−1. Cathey and Fuller note that incomplete polymer chain extensions may occur in transient extensional flows because of the limited residence time a polymer spends in the flow field.33 More importantly, these results demonstrate that different protein molecules respond differently to shear, a feature attributable to variations in primary structure (i.e. surface properties), molecular weight, and solvent viscosity.29,32,34 Indeed for a given shear rate, the stress on protein molecules is purported to increase as a function of molecular weight and solvent viscosity.32 While fluorescence spectroscopy can be used as a diagnostic for the conformational state of protein systems, it is limited by its inability to determine and quantify the secondary structural components of such systems. Therefore, results obtained are only qualitative in nature.
384 Da) showed no structural changes for shear rates as high as 105s−1, vWF (Mw ≈ 2 × 107 Da) demonstrated structural instability at a threshold shear rate of 103s−1. Cathey and Fuller note that incomplete polymer chain extensions may occur in transient extensional flows because of the limited residence time a polymer spends in the flow field.33 More importantly, these results demonstrate that different protein molecules respond differently to shear, a feature attributable to variations in primary structure (i.e. surface properties), molecular weight, and solvent viscosity.29,32,34 Indeed for a given shear rate, the stress on protein molecules is purported to increase as a function of molecular weight and solvent viscosity.32 While fluorescence spectroscopy can be used as a diagnostic for the conformational state of protein systems, it is limited by its inability to determine and quantify the secondary structural components of such systems. Therefore, results obtained are only qualitative in nature.
By virtue of the structural complexity, or conformational heterogeneity, of protein systems, a number of experimental studies have used the homopolypeptide poly-L-lysine as a model system to investigate the conformational stability of proteins in shear flow. Immaneni and McHugh reported a flow-induced gelation and reversible α-helix to β-sheet transition of dilute PLL samples (437 kDa) initially in the α-helix conformation.35 The observed changes were attributed to shear-enhanced hydrophobic interactions between PLL molecules, under otherwise stable solution conditions. In an ensuing paper, Lee and McHugh reported a reversible shear-induced helix-to-coil transition in dilute PLL samples originally in a helix–coil equilibrium.36 They noted the occurrence of this transition above a critical shear rate of 300 s−1 in simple shear flow.
The commercial availability of various molecular weights of PLL, needless to say, of the same chemical composition, affords a direct comparison of the chain-length dependence of the polypeptide in response to perturbation. For example, thermal studies on selected molecular weights of PLL show that the helix-to-sheet transition temperature decreases with increasing chain-length.37
We have used real-time, far-UV circular dichroism (CD) measurements to examine the structural integrity of different chain-lengths of α-helical poly-L-lysine in Couette flow. CD spectra in the far-UV wavelength range (typically from ∼190 to 250 nm) can be used to provide quantitative estimates of the secondary structural content of protein solutions and have been successfully applied as a structural analysis technique in protein studies.38 Additionally, CD allows the use of low sample concentrations ≤0.1 mg ml−1,39 hence dilute protein solutions can be studied, limiting intermolecular interactions and consequent alterations in solvent viscosity. We have previously reported the shear-induced unfolding of bovine insulin in Couette flow using direct CD measurements.7
Methods and materials
Materials
Four molecular weights of poly-L-lysine hydrobromide were studied. The samples were purchased from Sigma-Aldrich (St. Louise, MO) with a supplier reported molecular weight average (by viscosity) of 15, 68.3, 205.7 and 381.2 kDa. The polypeptides were prepared by dissolution in a triple-distilled water/NaOH mixture, pre-cooled and filtered (0.22 µm), to give a final working concentration of ∼0.2 mg ml−1, pH 11.7. The solution concentration of the PLL samples was calculated using the UV absorption at 214 nm and methods described by Kuipers and Gruppen.40 The samples were kept at 4 °C but allowed to equilibrate to room temperature (∼20 °C) before use.Couette flow cell
Shear experiments were performed in a custom built quartz-flow-cell of Couette geometry with a gap size of 0.021 cm as previously described.7,12PLL samples were exposed to shear rates of 74, 117, 219, 302, 518 and 715 s−1 for 1 h. At these shear rates, laminar flow patterns are achieved in the quartz-flow-cell. The flow cell was mounted and aligned in a Jasco J-815 spectrometer (Tokyo, Japan) such that CD data were recorded in real-time during shearing. As mentioned above, we have successfully applied this technique in studying the structural dynamics of bovine insulin in Couette flow.CD measurements
Far-UV CD measurements were performed over the wavelength range of 190 to 240 nm in a temperature controlled room (∼20 °C) to limit thermal contributions to PLL deformation. CD data were collected at 1 min intervals at each shear rate using a data interval of 0.1 nm, a bandwidth of 1.0 nm, response time of 4 s, and a scanning speed of 100 nm min−1. Solvent spectrum was used as baseline for data collection.Calculations of helix content
The helix content of the four PLL chains were calculated based on methods developed by Chen et al.41 By applying the chain-length-dependent model, the molar ellipticity value (XHn) for a 100% helix content for the individual chains was predicted using the following equation:| XHn = XH∞(1 − k/n) | (1) |
500 deg cm2 dmol−1. The fractional helix (fh) or helix content was then estimated using the following relationship: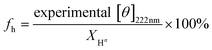 | (2) |
Results
The solution conformation of the four PLL samples was studied in situ for the selected shear rates over a period of 1 h. Under quiescent conditions, the CD spectral features of the four PLL samples, at 20 °C, were consistent with a classic α-helix structure with two minima at 208 and 222 nm. However, the magnitude of the negative intensity in the α-helix region was progressively higher with increasing chain-length, indicative of a growing helix content (Fig. 1, inset). Consistent with this observation, quantitative calculations showed an increase in the helix content of PLL as a function of chain-length (Fig. 1). For example, whereas the highest molecular weight (381.2 kDa) showed a 93.6% helix content, shorter chains (<205.7 kDa) contained significant amounts of random coils and turns.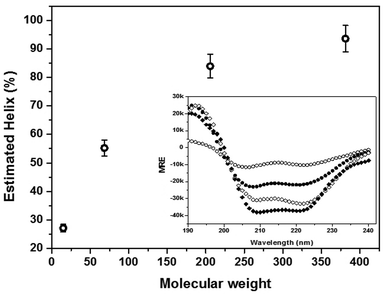 | ||
| Fig. 1 Chain-length dependence of the helix content of PLL. The figure shows that the helix content increases as a function of molecular weight. Inset: mean residue ellipticity (MRE) values of the wavelength scans for the four molecular weights: (○) 15 kDa, (●) 68.3 kDa, (◇) 205.7 kDa, and (◆) 381.2 kDa. The error bars represent ±standard deviation. | ||
While the PLL samples were stable under quiescent conditions over the 1 h period, structural changes in the α-helix region were observed upon exposure to shear. Representative CD spectra from the 205.7 kDa sample (Fig. 2) show that at the onset of shear, the intensity of the negative ellipticities at 209 and 222 nm diminished as a function of time, indicating a decline in the helix content. The observed change was more pronounced with increasing shear rate. For example, at a relatively low shear rate (117 s−1), there was no significant change in the spectral features of the wavelength scans. At the highest shear rate (715 s−1), however, a sharp decline in the two helix minima at 209 and 222 nm was observed. In addition, an isodichroic point in the region of 203 nm was observed in the high shear regime, which suggests that the shear-induced unfolding of the α-helical PLL structure proceeds via a two-state transition. This observation was substantiated by further analysis of the conformational transitions using phase diagrams (Fig. S1†). The phase plots were generated using the negative ellipticities at θ204nm and θ222nm. Clearly, the two linear segments in the plot for the high shear regime confirm the occurrence of a two state conformational transition and the existence of at least one intermediate state during the unfolding of the α-helix–PLL structure. It is noteworthy that the longer chain molecules (381.2 kDa) showed no such conformational transitions as the extent of unfolding was minimal even at 715 s−1.
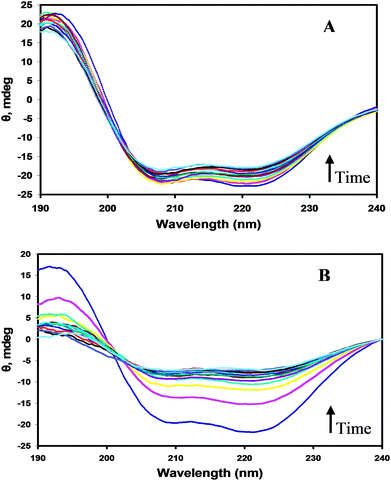 | ||
| Fig. 2 CD spectra of the 205.7 kDa PLL sample in simple shear flow. The spectra show wavelength scans from zero (no shear) to 60 min, at 5 min intervals, in the direction of the arrows for (A) 117 s−1 and (B) 715 s−1. | ||
The relationship between shear rate and the decline in negative ellipticity at 222 nm is shown in Fig. 3 using representative data from the 205.7 kDa sample. The kinetic graphs were obtained by plotting the ratio of the time-dependent change in the 222 nm minimum as a function of shear rate. The molar ellipticity value at 222 nm is widely used as a diagnostic for the α-helix structure.42 Clearly, as the magnitude of the shear rate increased, with the consequent increase in the hydrodynamic forces on the molecules, a sharp decline in the helix content was observed. The observed change was irreversible, which is possibly because the unfolding PLL molecules get trapped in a metastable conformation that is prone to aggregation.
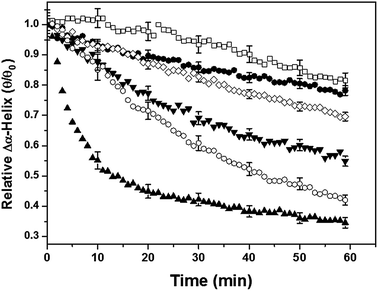 | ||
| Fig. 3 Change in the relative helix content of PLL (205.7 kDa) as a function of time. The shear rates applied were (□) 74 s−1, (●) 117 s−1, (◇) 219 s−1, (▼) 302 s−1, (○) 518 s−1 and (▲) 715 s−1. The figure shows that the unfolding of the α-helix–PLL structure depends on both the magnitude of shear and the duration of its application. The error bars represent ±standard deviation. | ||
To investigate the influence of chain-length on the shear-stability of the PLL molecules, we compared the extent of unfolding of the α-helix–PLL structure, after 1 h of shear exposure, for the four molecular weights as a function of shear rate (Fig. 4). The plot was generated by calculating the percentage change in the helix content of individual PLL chains, taking the initial helix content per chain under quiescent conditions to be 100%. It is important to note that the estimated helical content of the unsheared PLL is a function of molecular weight as shown in Fig. 1. Here, the extent of unfolding of the α-helix–PLL structure in simple shear flow was found to also depend on the chain-length of the PLL molecules. For example, at 715 s−1, only ∼24% of the initial helix content of the 381.2 kDa sample unfolded compared to ∼80% in the shortest chain (15 kDa). The data suggest that at a given shear rate, the shorter chain PLL molecules unfold more rapidly in the flow field relative to their longer chain counterparts. In addition, the extent of unfolding was more pronounced with increasing shear rate for all molecular weights, approaching equilibrium beyond 500 s−1.
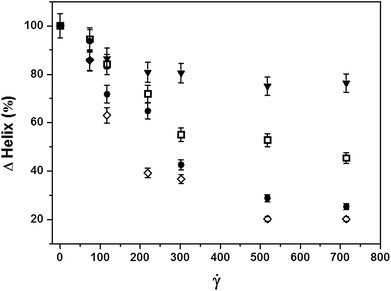 | ||
| Fig. 4 Molecular weight dependence of the extent of helix unfolding after 1 h in shear flow. Deformation of the α-helix–PLL structure was less pronounced with increasing molecular weight: (◇) 15 kDa, (●) 68.3kDa, (□) 205.7 kDa, and (▼) 381.2 kDa. | ||
The data were further analyzed by considering the molecular weight dependence of the helix content of the PLL chains at a given shear rate (Fig. 5). We considered the highest shear regime where the unfolding of the α-helix–PLL structure was more pronounced. The data show a strong linear correlation of the percentage change in helix as a function of molecular weight. Furthermore, the data clearly reveal the hysteresis in the long chain helices even in the highest shear regime applied.
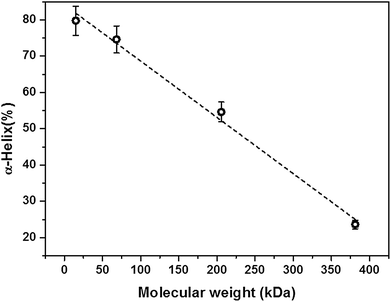 | ||
| Fig. 5 Molecular weight dependence of the helix content after 1 h of shearing at 715 s−1. The broken line is a linear fit to the data with an R2 value of 0.99. The error bars represent ±standard deviation. | ||
Interestingly, using representative data from the 68.3 kDa sample, a plot of the change in helix content against shear strain (log [t]) also showed a strong correlation, typical for the four molecular weights studied (Fig. 6). From this plot, the unfolding transition of the α-helix–PLL structure occurred at a strain value in the region of 105, and was found to be independent of the chain-length. By introducing the solvent viscosity, η (10−3 Pa s), a similar trend is observed if the change in helix content was plotted against the cumulative stress (log [ηt]) values (i.e. stress history). Here, the critical cumulative stress (τc) value for the deformation of the PLL helix structure is of the magnitude 101 Pa s. In addition, by taking the time point where the two linear segments intersect in the phase diagram (Fig. S1†), a similar strain value is observed. This clearly shows that the conformational transition in the sheared PLL chains indeed begins at this strain value.
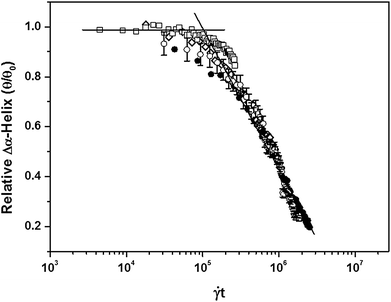 | ||
| Fig. 6 Change in the helix content of PLL as a function of shear strain. The figure shows that the α-helix–PLL structure (68.3 kDa) is stable below a strain value of 105. For clarity, the shear rates plotted are: (□) 74 s−1, (◇) 302 s−1, (○) 518 s−1, and (●) 715 s−1. For clarity, error bars representing ±standard deviation are shown only for the sample sheared at 518 s−1. | ||
Discussion
Studies of the shear-stability of dilute protein systems, and polymers at large, are of immediate relevance to both industry and medicine. The complexity of this subject has resulted in a greater number of theoretical studies, rather than experimental investigations, to appreciate the conformational dynamics of polymers in shear flow.28,29,43,44 The handful of experimental studies on protein systems in this field give conflicting reports.7,30–32 These differences can be ascribed to a number of factors including the type of flow applied, the duration of shear exposure (residence time), and solvent viscosity. More importantly, differences in the primary structure of protein systems have been found to influence their shear-stability,45 which further complicates data interpretation and extrapolation to other systems. The current study presents an ideal case where the unfolding dynamics of different molecular weights of the same system are compared, with the aim of understanding the shear-stability of an α-helical structure in shear flow. Needless to say, the α-helical structure is common to many peptides and proteins, and the structural destabilization of a native protein conformation is a prerequisite for aggregation and amyloid fibril formation.46Chain-length dependence of [θ]222nm (helix content)
At high pH, the lysyl residues making up poly-L-lysine are neutralized in solution. The α-helical conformation assumed by the homopolypeptide, below 30 °C, is attributable to intramolecular hydrogen bonds between the hydrophobic lysine side chains, and in the absence of electrostatic interactions, is primarily stabilized by intramolecular hydrophobic interactions.47 Theoretical studies suggest that, in addition to a stabilizing solvation effect, attracting forces within an α-helix provide extra stability to the overall helix structure.48 Thus, an increase in chain-length, with the consequent increase in α-helical turns, confers additional stability on a PLL chain. However, it is argued that a dipole moment induced clustering of helices in high molecular weight PLL may exclude hydrating water molecules from the inter-helical space, leading to a possible helix destabilization.37 This feature was purported to explain the thermal susceptibility of heavy PLL chains in the α-helix state. However, extension of polyalanine chain-length was found to favor α-helix conformations even in a hydrophobic environment.48The CD spectra of the four PLL molecular weights studied show α-helix secondary structural features at elevated pH. This was expected since the supplier reported degree of polymerization for the shortest chain (15 kDa by viscosity), given as 71, is sufficient for the formation of secondary structures such as α-helix and β-sheet. It is generally accepted that the molar ellipticity value at 222 nm ([θ]222nm) is directly proportional to the number of residues in a helix. However, this value is found to be influenced by the chain-length and dynamical motion of a polypeptide chain.42 That is, longer chain helices show a higher negative ellipticity at 222 nm relative to their shorter chain counterparts. This explains the observed increase in [θ]222nm as a function of molecular weight. Hence, by using the length dependent model (eqn (2)), which takes into account the end-effects of longer helices, it was not surprising that the calculated helix content increased with increasing chain-length. A simple explanation for this observation is that, as the PLL length grows, an increasing number of the lysine residues form part of the helical segments. Indeed the helicity and stability of synthetic peptides were found to increase with increasing chain-length.49 The conformational attributes of the four PLL chains studied, under quiescent conditions, are summarized in Table 1.
| M/kDa | DP | H (%) | Ha | Ca | Helix turnsb | Hc | Cc | L d/nm |
|---|---|---|---|---|---|---|---|---|
| a Number of lysine residues in a particular conformation. b Number of helix turns per chain assuming that one turn involves 3.6 Lys residues. c Total length (nm) of the helix and coil segments of the PLL chain assuming a helix pitch of 0.54 nm and the length of a Lys residue in the PLL chain to be 0.917 nm.50 d Combined length of the PLL chain. DP is the supplier reported degree of polymerization by viscosity. H and C represent the helix and coil conformations of native PLL chains. | ||||||||
| 15 | 71 | 27.1 | 19.2 | 51.8 | 5.3 | 2.9 | 47.5 | 50.3 |
| 68.3 | 327 | 55.2 | 180.5 | 146.5 | 50.1 | 27.1 | 134.3 | 161.4 |
| 205.7 | 984 | 83.9 | 825.6 | 158.4 | 229.3 | 123.8 | 145.3 | 269.1 |
| 381.2 | 1824 | 93.6 | 1707.3 | 116.7 | 474.2 | 256.1 | 107.0 | 363.1 |
Unfolding of poly-L-lysine molecules in simple shear flow
Like other polymers, poly-L-lysine chains exposed to simple shear flow are expected to undergo a combination of rotational and extensional dynamics, dictated by the flow field, even at moderate shear rates.28,51 In the extensional flow field, the molecules experience a hydrodynamic drag which counters the cohesive forces stabilizing the native α-helix conformation. Theoretical studies suggest that the frequency of stretching events, hence average polymer extension, in simple shear flow amplifies with increasing shear rate.5 This is not surprising since an increase in shear rate imparts a stronger hydrodynamic drag to the flow field. In contrast, the fluid forces acting on PLL molecules recede as the molecules orient in the rotational component of the flow field. As individual molecules randomly access the two flow fields, temporal fluctuations which involve periodic elongation, relaxation and tumbling occur.5,52 For this reason, sharp conformational transitions, such as helix-to-coil, are not expected in simple shear flow.28 This is consistent with molecular dynamics simulations which reveal that, unlike pure extensional flow, the coil–stretch transition in simple shear flow involves intermediate states.28,53The four PLL molecular weights studied showed a time- and shear rate dependent decline in the molar ellipticity at [θ]222nm, which clearly shows a loss in helicity. Unfolding of the α-helix–PLL structure is expected to occur when molecules are aligned in the extensional flow field, and the magnitude of the hydrodynamic drag overwhelms the restoring (cohesive) forces stabilizing the PLL chains. At lower shear rates, only moderate losses in helicity occurred. In high shear regimes, a two-state conformational transition was observed in the PLL chains except for the heaviest chain (381.2 kDa) where the change in the helix content ([θ]222nm) was minimal. This is consistent with the notion that the hydrodynamic drag is enhanced as the shear rate increases. The existence of intermediate states during PLL unfolding is also consistent with theoretical predictions. On comparing the extent of structural deformation in the four molecular weights, the flow-induced unfolding of the α-helix–PLL structure was found to be less pronounced with increasing chain-length. In fact, in the highest shear regime, a strong linear correlation was observed between the percentage change in helix and chain-length after 1 h of shearing. This implies that the shorter chain helices unraveled more rapidly, with increasing shear rate, relative to the heavy chain helices.
Although a number of theoretical studies calculate the helix–coil transitions in polymers, especially homopolypeptides, under stress, little work has been expended on the molecular-weight-dependence of this phenomenon.54–56 To explain our results, we invoke simple theory regarding polymer dynamics in flow. Firstly, we consider the PLL as a rod-like structure. Here, the hydrodynamic forces on the rod will increase with both molecular weight and shear rate. Following the arguments of Bruinsma57 for the stress on a rod of “hydrodynamic beads,” we have derived the stress on the helix as:
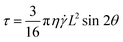 | (3) |
is the shear rate, L is the length, and θ is the angle subtended to the flow direction. As the length L is directly proportional to the molecular weight, the strain will be proportional to M2 and the remaining helix, α, will be inversely proportional to the strain, α ≈ M−2. Therefore, classical theory would predict that the deformation (unfolding) of the PLL chains increases with increasing chain-length. This is not observed in our results which show an α ∼ M curve (Fig. 4). The key experimental results are summarized as follows: (i) α ∝ (t)−1/2, (ii) α ∝ M, and (iii) tc ≈ 105, where tc is the critical strain.
More recently, theoretical studies of the dynamic instabilities of collapsed polymers in simple shear flow provide some simple scaling arguments worth noting.29 By taking into account the opposing effects of the fluid drag and the cohesive forces stabilizing a polymer chain, the authors predict the monomer size dependence of the critical strain rate required to induce unfolding by the following scaling relationship: c ∝ R, where R is the radius of the collapsed globule. Here, the critical strain rate, required for deformation/unfolding, varies linearly with the hydrodynamic radius of a given polymer chain. Hence, it is expected that shorter PLL chains would show shear-instabilities at relatively low strain rates compared to heavy chains. This is explained by the phenomenon of hydrodynamic screening, where the hydrodynamic interactions between lysyl side chains lead to a reduction of the fluid shear rate, hence fluid forces, acting on the surface of individual PLL globules. In addition, only the outer segments of the globules are directly exposed to the fluid drag in the flow field.28 Therefore, as R grows, the critical shear rate required to unfold the PLL globule increases. However, the hydrodynamic interactions between lysyl side chains are reduced once the globule begins to unravel, leading to rapid unfolding, especially in a purely elongational flow field.28,58
Simply put, at a given shear rate, enough to initiate unfolding, it is expected that short-chain PLL helices will unfold more rapidly in the flow field. This conclusion is further buttressed by computational predictions of the chain-length effect of dilute polymer solutions in elongational flow.59 The authors observed that relative to shorter chains, longer chains remain in a coil-like state for larger strains. A schematic depicting this conclusion is shown in Fig. 7. The figure illustrates that whereas the short chain helices stretch easily in the extensional flow component of the flow field, the heavy chains show greater shear-stability.
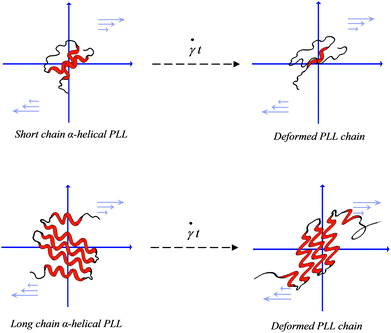 | ||
| Fig. 7 Schematic of the shear-induced unfolding of the α-helix–PLL structure in simple shear flow. Unfolding of the short chain α-PLL is shown on the left and that of the heavy chain on the right. The figure illustrates a rapid unfolding of short chain helices upon shear exposure whereas the heavy chain helices experience structural distortions with minimal unfolding. | ||
A priori, it is reasonable to ascribe the hysteresis observed in the heavy chain PLL to the greater stability of the helix-structure originating from the large network of hydrophobic interactions in concert with attracting forces within the helices.49,60 Hence, even at 715 s−1, the restoring force in the 381.2 kDa sample counterbalance the fluid drag in the extensional flow cycle and only ∼24% of the initial helix content is lost over the 1 h period. In fact, de Gennes notes that in flow fields such as simple shear flow, where the coil–stretch transition is discontinuous, hysteresis should often be observed.28 In addition, the compact folding of the heavy PLL chain, perhaps giving rise to a tertiary configuration, provides hydrodynamic screening for the hydrophobic helical segments concealed in the PLL matrix.28,29,61,62 Hence, only the outer segments are exposed directly to the flow field. This in effect reduces the hydrodynamic drag acting on the entire chain, leading to a minimal chain stretching in the extensional flow field. Although dilute PLL samples were used in this study, the possibility of tangling of partially extended conformations during the periodic end-over-end tumbling in the flow field cannot be ruled out. The occurrence of this event may give rise to conformational distortions resulting in kinked states,58,63 especially at high shear rates, which may slow the unraveling of the PLL helix. This in turn may enhance intermolecular hydrodynamic interactions, providing additional hydrodynamic shielding of helical segments from the fluid drag. In fact theoretical28 and molecular dynamics simulations64 predict the opposing effect of hydrodynamic interactions to protein unfolding in both uniform and elongational flow fields.
It is interesting to note that the four PLL molecular weights gave a similar strain value of 105, below which the helix structures were intact, although the extent of unfolding was less pronounced with increasing molecular weight. The data reveal that although the intrinsic properties of the PLL helix are independent of chain-length, its shear-stability arises from the hydrodynamic screening effect of the heavy chains. More importantly, the data suggest that the strain rate is not as critical as the duration of its application. That is, provided the PLL chains, irrespective of the molecular weight, are exposed to a fixed strain rate for a sufficient amount of time, the helical segments will eventually unfold at a critical strain value ≥105. This makes the idea of a critical shear rate only arbitrary. In fact, a similar shear strain value had previously been reported for globular proteins of varying molecular weights.30,65
It is noteworthy that proteins present a more complex network of intra-/intermolecular interactions compared to a homopolypeptide like poly-L-lysine. However, we have previously observed a similar shear effect on the helical structures of bovine insulin7 and bovine serum albumin66 even at a moderate shear rate of 300 s−1. In the insulin sample, fibrillar species were observed in high shear regimes even at room temperature. Indeed, several studies show that shear flow enhances amyloid fibril formation.12–14,67 Hence, if this observation pertains to the majority of protein systems, the shear effect could enhance the formation of β-sheet structures under favourable solution conditions such as elevated concentrations and temperature. This has serious implications in amyloid related diseases and vascular disorders, as well as quality control during the commercial isolation and purification of protein products.
Conclusion
We have shown that the α-helical–PLL structure unfolds in simple shear flow. The extent of unfolding is dependent on the monomer size of the PLL chains, as well as shear rate and the duration of its application. The shear-stability of the α-helical–PLL structure increased with increasing chain-length. However, the shear strain value of ∼105, required to unfold the α-helical–PLL structure, was independent of the PLL molecular weights. We observe the following dependence of the remaining helix (α): (i) α ∝ (t)−1/2 for all molecular weights measured and for strain values above the critical value of tc ≈ 105, and finally (ii) α ∝ M where the remaining helical content is proportional to the molecular weight.
Acknowledgements
We thank Dr Deborah Tew for assistance with the CD instrument. We also thank Sian Yang for assistance with the schematic diagram. Funding for this project was from the Australian Research Council. Helpful discussions with Professor Erik van der Linden are acknowledged.References
- W. T. Astbury and H. J. Woods, Philos. Trans. R. Soc. London, Ser. A, 1934, 232, 333–394 CrossRef.
- L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CAS.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 235–240 CAS.
- P. LeDuc, C. Haber, G. Bao and D. Wirtz, Nature, 1999, 399, 564–566 CrossRef CAS.
- D. E. Smith, H. P. Babcock and S. Chu, Science, 1999, 283, 1724–1727 CrossRef CAS.
- C. B. Elias and J. B. Joshi, Adv. Biochem. Eng./Biotechnol., 1998, 59, 47–71 Search PubMed.
- I. B. Bekard and D. E. Dunstan, J. Phys. Chem. B, 2009, 113, 8453–8457 CrossRef CAS.
- S. W. Schneider, S. Nuschele, A. Wixforth, C. Gorzelanny, A. Alexander-Katz, R. R. Netz and M. F. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7899–7903 CrossRef CAS.
- C. A. Siedlecki, B. J. Lestini, K. Kottke-Marchant, S. J. Eppell, D. L. Wilson and R. E. Marchant, Blood, 1996, 88, 2939–2950 CAS.
- I. Singh, E. Themistou, L. Porcar and S. Neelamegham, Biophys. J., 2009, 96, 2313–2320 CrossRef CAS.
- L. Nielsen, R. Khurana, A. Coats, S. Frokjaer, J. Brange, S. Vyas, V. N. Uversky and A. L. Fink, Biochemistry, 2001, 40, 6036–6046 CrossRef CAS.
- E. K. Hill, B. Krebs, D. G. Goodall, G. J. Howlett and D. E. Dunstan, Biomacromolecules, 2006, 7, 10–13 CrossRef CAS.
- C. Akkermans, P. Venema, S. S. Rogers, A. J. van der Goot, R. M. Boom and E. van der Linden, Food Biophys., 2006, 1, 144–150 CrossRef.
- C. Akkermans, A. J. van der Goot, P. Venema, E. van der Linden and R. M. Boom, Food Hydrocolloids, 2008, 22, 1315–1325 CrossRef CAS.
- T. R. Serio, A. G. Cashikar, A. S. Kowal, G. J. Sawicki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf and S. L. Lindquist, Science, 2000, 289, 1317 CrossRef CAS.
- J. C. de la Torre, Lancet Neurol., 2004, 3, 184–190 CrossRef.
- C. Jack, Neurol. Res., 2004, 26, 517–524 Search PubMed.
- Y. Jiang, K. Kohara and K. Hiwada, Stroke, 2000, 31, 2319–2324 CAS.
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 10737–10744 CrossRef CAS.
- M. D. Kirkitadze, M. M. Condron and D. B. Teplow, J. Mol. Biol., 2001, 312, 1103–1119 CrossRef CAS.
- A. Abedini and D. P. Raleigh, Phys. Biol., 2009, 6, 015005–015010 Search PubMed.
- V. N. Uversky and A. L. Fink, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1698, 131–153 CrossRef CAS.
- J. T. Yang and P. Doty, J. Am. Chem. Soc., 1957, 79, 761–775 CrossRef.
- N. J. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108–4116 CrossRef CAS.
- O. Kambara, A. Tamura, A. Naito and K. Tominaga, Phys. Chem. Chem. Phys., 2008, 10, 5042–5044 RSC.
- A. Ahmad, I. S. Millett, S. Doniach, V. N. Uversky and A. L. Fink, Biochemistry, 2003, 42, 11404–11416 CrossRef CAS.
- I. B. Bekard and D. E. Dunstan, Biophys. J., 2009, 97, 2521–2531 CrossRef CAS.
- P. G. De Gennes, J. Chem. Phys., 1974, 60, 5030–5042 CrossRef CAS.
- A. Alexander-Katz and R. R. Netz, Macromolecules, 2008, 41, 3363–3374 CrossRef CAS.
- S. E. Charm and B. L. Wong, Biotechnol. Bioeng., 1970, 12, 1103–1109 CrossRef CAS.
- C. R. Thomas and P. Dunnill, Biotechnol. Bioeng., 1979, 21, 2279–2302 CrossRef CAS.
- J. Jaspe and S. J. Hagen, Biophys. J., 2006, 91, 3415–3424 CrossRef CAS.
- C. A. Cathey and G. G. Fuller, J. Non-Newtonian Fluid Mech., 1990, 34, 63–88 CrossRef CAS.
- A. Oliva, A. Santoveña, J. Fariña and M. Llabrés, J. Pharm. Biomed. Anal., 2003, 33, 145–155 CrossRef CAS.
- A. Immaneni and A. J. McHugh, Biopolymers, 1998, 45, 239–246 CrossRef CAS.
- A. T. Lee and A. J. McHugh, Biopolymers, 1999, 50, 589–594 CrossRef CAS.
- W. Dzwolak, T. Muraki, M. Kato and Y. Taniguchi, Biopolymers, 2004, 73, 463–469 CrossRef CAS.
- S. M. Kelly and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 1997, 1338, 161–185 CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 119–139 CrossRef.
- B. J. H. Kuipers and H. Gruppen, J. Agric. Food Chem., 2007, 55, 5445–5451 CrossRef CAS.
- Y. H. Chen, J. T. Yang and K. H. Chau, Biochemistry, 1974, 13, 3350–3359 CrossRef CAS.
- J. D. Hirst and C. L. Brooks, J. Mol. Biol., 1994, 243, 173–178 CrossRef CAS.
- P. Szymczak and C. Marek, J. Chem. Phys., 2006, 125, 164903 CrossRef CAS.
- J. G. H. Cifre and J. G. de la Torre, J. Rheol. (Melville, NY, U. S.), 1999, 43, 339 Search PubMed.
- C. C. H. Yuh-Fun Maa, Biotechnol. Bioeng., 1997, 54, 503–512 CrossRef CAS.
- E. McNally and C. Lockwod, in Protein Formulation and Delivery, ed. E. McNally, Marcel Dekker, New York, 2000, pp. 111–138 Search PubMed.
- H. A. Arfmann, R. Labitzke and K. G. Wagner, Biopolymers, 1975, 14, 1381–1393 CrossRef CAS.
- C. Park and W. A. Goddard, J. Phys. Chem. A, 2000, 104, 7784–7789 CAS.
- J. Y. Su, R. S. Hodges and C. M. Kay, Biochemistry, 1994, 33, 15501–15510 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman and Co, New York, 5th edn, 2002 Search PubMed.
- J. L. Lumley, Annu. Rev. Fluid Mech., 1969, 1, 367–384 CrossRef CAS.
- A. Alexander-Katz, M. F. Schneider, S. W. Schneider, A. Wixforth and R. R. Netz, Phys. Rev. Lett., 2006, 97, 138101–138104 CrossRef CAS.
- A. S. Lemak, J. R. Lepock and J. Z. Y. Chen, Proteins: Struct., Funct., Genet., 2003, 51, 224–235 CrossRef CAS.
- M. N. Tamashiro and P. Pincus, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2001, 63, 21909 CrossRef CAS.
- S. Courty, J. L. Gornall and E. M. Terentjev, Biophys. J., 2006, 90, 1019–1027 CrossRef CAS.
- A. Buhot and A. Halperin, Macromolecules, 2002, 35, 3238–3252 CrossRef CAS.
- R. Bruinsma, W. Gelbart and A. Ben-Shaul, J. Chem. Phys., 1992, 96, 7710–7727 CrossRef CAS.
- R. G. Larson, Rheol. Acta, 1990, 29, 371–384 CAS.
- P. Sunthar and J. R. Prakash, ANZIAM J., 2004, 46, C320–C335 Search PubMed.
- K. Fukushima, T. Sakamoto, J. Tsuji, K. Kondo and R. Shimozawa, Biochim. Biophys. Acta, Proteins Proteomics, 1994, 1191, 133–140 CAS.
- U. S. Agarwal and R. A. Mashelkar, J. Chem. Phys., 1994, 100, 6055–6061 CrossRef CAS.
- T. G. M. van de Ven, Colloidal Hydrodynamics, Academic Press, London, 1989 Search PubMed.
- J. M. Rallison and E. J. Hinch, J. Non-Newtonian Fluid Mech., 1988, 29, 37–55 CrossRef CAS.
- P. Szymczak and M. Cieplak, J. Phys.: Condens. Matter, 2007, 19, 285224–285235 CrossRef.
- M. Tirrell and S. Middleman, Biotechnol. Bioeng., 1975, 17, 299–303 CrossRef CAS.
- P. Asimakis; I. Bekard; C. L. Teoh; T. Ryan; G. J. Howlett; J. Bertolini; D. E. Dunstan, unpublished work.
- P. Hamilton-Brown, I. Bekard, W. A. Ducker and D. E. Dunstan, J. Phys. Chem. B, 2008, 112, 16249–16252 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Phase diagram showing a two-state conformational transition during α-PLL unfolding in strong shear regimes. See DOI: 10.1039/c0sm00692k |
| This journal is © The Royal Society of Chemistry 2011 |