Anion tuning of the rheology, morphology and gelation of a low molecular weight salt hydrogelator†
Gareth O.
Lloyd
*a and
Jonathan W.
Steed
*b
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: gol20@cam.ac.uk; Fax: +44 (0)1223 336 017; Tel: +44 (0)1223 763 989
bDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: jon.steed@dur.ac.uk; Fax: +44 (0)191 384 4737; Tel: +44 (0)191 334 2085
First published on 30th September 2010
Abstract
1-(3-Methyl-1H-pyrazol-5-yl)-3-(3-nitrophenyl)urea (1) forms hydrogels in a range of acids at pH 1–2. The morphology and rheology of these gels are dependent on the identity of the anion and represent a way of tuning the gels' physical properties. The magnitude of the elastic storage modulus of the gels follows the trend according to the acid used to protonate the gelator H2SO4 > MePO3H2 > H3PO4 ≈ HBF4 ≈ HPF6 > EtPO3H2. Chloride salts do not form gels and the nitrate gels are unstable with respect to crystallisation. Salting out or addition of methanol results in the characterisation of five salts of 1 which reveals extensive solvation in the solid state and the absence of urea⋯urea hydrogen bonding in contrast to the well known bis(urea) gelators.
Introduction
Gels are solid, jelly like materials formed from colloidal mixtures comprising a liquid dispersed within a solid continuous gelator phase. What makes gels so interesting is that by weight and volume they are mostly liquid, yet they behave like a solid. Of the many substances that can be used as gelators, low molecular weight gelators (LMWGs) represent soft materials that can be prepared by the bottom-up assembly of designed small molecules. These small molecules assemble into nanoscale fibrils which bunch to form micro-fibres which, in turn entangle and cross-link via interconnected nodes to form macro-sized gels that comprise a 3D sample-spanning network with the liquid immobilised in continuous microchannels, usually as a result of surface tension effects.1–6 Hydrogels are gels that are formed with water as the major constituent of the solvent. The biological and technological relevance of these gels has lead to a myriad of applications, such as drug delivery, catalysis, scaffolds for tissue engineering, templates for materials synthesis, lithography and as separation materials.7–11 LMWGs are supramolecular in nature, and as such, are easily tunable by both synthetic modification and external stimuli. The external stimuli may be mechanical, thermal, electrochemical, electromagnetic or chemical. Chemical stimuli, as additives, have been reported, and include examples of variation in pH, addition of/response to biologically relevant compounds, the incorporation of metals or anions.6,12 Anion tuning in particular has become a highly topical subject within the field of tunable LMWGs because anions act as effective hydrogen bond acceptors often in competition with fibre formation.13,14 Even so, the effects that anions can have on LMWG assembly are still not fully understood.14–19 This uncertainty is partially due to the fact that in most cases the LMWG only gels in the presence of a few anions or gelation has only been studied with a limited range of salts. Moreover, much recent activity has focused on neutral LMWGs that bind anions in the presence of a corresponding charge-balancing cation, further complicating the picture.6,13,14 In the present work we target a range of salt LMWGs in which there is the possibility of tuning the gel properties of one particular LMWG cation by varying the identity of the anion. We have chosen to study pyrazole-appended ureas in which the urea group can contribute to gel fibre assembly20–24 while the basic pyrazole group acts as a protonation site. There are very few examples of pyrazole containing LMWGs in the literature,25 despite their resemblance to the better studied pyridyl ureas.6,26–31 The protonated pyrazole unit is also relatively sterically unencumbered, enhancing potential hydrogen bond donor characteristics and hence a protonated pyrazole urea may represent an alternative to a bis(urea) gelator.Results and discussion
Synthesis
1-(3-Methyl-1H-pyrazol-5-yl)-3-(3-nitrophenyl)urea (1) was synthesised from 3-amino-5-methylpyrazole and m-nitrophenyl isocyanate, viatert-butyloxycarbonyl (BOC) protection of the pyrazolyl group (Scheme 1). The BOC protection is necessary as the direct reaction between an aminopyrazole and an isocyanate produces a carboxamide.32–34 Proof of the BOC protection occurring on the pyrazolyl ring comes in the form of analytical data and the crystal structure of the BOC protected pyrazole in which the BOC group is bound to the 3 nitrogen position of the pyrazolyl ring, in agreement with the literature (ESI†).35,36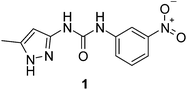 | ||
| Scheme 1 1-(3-Methyl-1H-pyrazol-5-yl)-3-(3-nitrophenyl)urea (1). | ||
Testing for gel formation was carried out by dissolving 1 in 1–2 mL of water at controlled pH in the presence of a variety of acids with a view to forming salt hydrogelators of the type (1H++)(X−) where X− is the conjugate anion of the acid in question. Solutions were cooled to room temperature both with and without sonication37 and were tested for gelation by performing the “inversion test”. Table 1 summarizes the acids used to adjust the pH and the resultant characteristics of the materials formed.
| Acid used | H2SO4 | HBF4 | HPF6 | HNO3 | HCl | H3PO4 | MePO3H2 | EtPO3H2 |
|---|---|---|---|---|---|---|---|---|
| a % critical gel concentration (CGC) in weight percentage (g mL−1). b Order of 1 to 8 being from kosmotropic (strongly hydrated anions) to chaotropic (weakly hydrated anions). c Sequence from 1 strongest to 6 weakest, according to G′ value and “yield stress”. | ||||||||
| Appearance | Clear to opaque yellow gel | Opaque cream gel | Opaque cream gel | Clear yellow gel that breaks down to yellow crystalline powder | Cream crystalline powder | Clear to opaque yellow gel | Opaque cream gel | Clear, yellow weak gel |
| CGC a | 0.12 | 0.30 | 0.20 | n/a | n/a | 0.11 | 0.13 | 0.18 |
| Gelation pH range | 1.0–2.0 | 1.0–2.0 | 1.0–1.7 | 1.0–2.5 | n/a | 1.0–2.1 | 1.0–2.0 | 1.0–2.0 |
| Morphology | Sponge-like | Particulate | Particulate | Fibre to crystalline | Crystalline | Sponge-like | Sponge-like | Sponge-like |
| Crystal structure | (1H++)2(SO42−)·7H2O | (1H++)(BF4−)·H2O and (1H++)(BF4−)·1 | n/a | (1H++)(NO3−)·MeOH | 2[(1H++)(Cl−)]·MeOH·H2O | n/a | n/a | n/a |
| Hofmeister series orderb | 1 | 8 | 7 | 6 | 5 | 3 | 2 | 4 |
| Rheological strength orderc | 1 | 5 | 3 | n/a | n/a | 4 | 2 | 6 |
The results indicate that a narrow pH range of ca. 1–2 is required for gel formation to occur, thus making compound 1 a pH tunable LMWG.20,28,38–57 At the low pH used to induce gel formation in 1, it is expected that the pyrazolyl group will be protonated to give a cationic species (pyrazoles are strongly basic with pKaca. 2.5).58 This protonation allows systematic study of the effect of the variation in anion on the gel properties, by changing the acid used to acidify the water.
Visual inspection of the resultant materials shows that there are changes in the type of materials produced, according to the identity of the anion. Of the 13 acids tested, gels were formed by the acids H2SO4, H3PO4, HBF4, HPF6, MePO3H2 and EtPO3H2 (Fig. 1, S1 and S2†). HNO3 gave an unstable gel, while HCl, CH3COOH, CF3COOH, MeSO3H, CF3SO3H and p-toluene sulfonic acid all gave precipitates (powder patterns of HNO3 and HCl experiments, Fig. S21 and S22†). The HNO3 acidified solution formed a weak gel upon slow cooling, however, the gel breaks down within a few minutes.59,60 The gel and precipitate formation is reversible, in that the solution can be heated and cooled repeatedly giving the same results each time. As has been seen in other classes of LMWGs, the gelation ability of 1 was found to be enhanced when hot solutions were sonicated to give more uniform gels, especially, in the cases when HBF4 and HPF6 were used.37
 | ||
| Fig. 1 Photograph of gels of 1H++ at 1% by weight in water acidified with, from left to right, EtPO3H2; MePO3H2; H3PO4; H2SO4; HPF6 and HBF4. | ||
Tuning of the gel state by addition of anions has become a focused subject within the field of LMWGs. Since 1 does not gel in the presence of HCl, chloride salts (NaCl and tetrabutylammonium chloride, TBA+Cl−) were added to modify the strength of gels of 1. When one equivalent of anion (in relation to 1) was added to gels, breakdown of the gel to a precipitate was observed irrespective of the counterion forming the gel. This precipitation caused by the addition of Cl− provides the ability to tune a gel of 1 and represents a kind of salting out effect.41,51,57,61
Microscopy
Visual inspection of the gels reveals a notable difference in the appearance and colour. The morphology of the gels was determined using scanning electron microscopy (SEM) and tunneling electron microscopy (TEM).The morphology of the H2SO4 gels (which are also representative of the H3PO4 (Fig. S10†), MePO3H2 and EtPO3H2 gels) was studied by SEM and TEM imaging (Fig. 2). Samples of gels of 1 acidified with H2SO4 to a pH of 1.0 for SEM imaging were dried under vacuum and then coated with a thin layer of platinum metal. As can be seen from Fig. 2a the imaging reveals a porous sponge-like morphology. The Pt coating has created a smooth surface on the gel struts with the pores of the gels appearing darker. The gel morphology from the TEM images, as shown in Fig. 2b, appears to be the same as that seen in the SEM images. The gel material breaks down under the electron beam, but the porous sponge-like morphology of the gel structure remains visible. The visible edge of a large circular pore, which the gel transverses, is the edge of the carbon mesh grid used to hold the gel sample. Electron energy-loss spectroscopy (EELS) was run on the TEM samples to confirm the presence of SO42− or HSO4− within the gel struts (Fig. S6†). The evidence of sulfur within the sample confirms the presence of the sulfate anion and hence a salt gel.
 | ||
| Fig. 2 (a) SEM image of dried gels of 1 in water acidified with H2SO4 to a pH of 1.0 that has been covered with a thin layer of Pt. (b) TEM image of a gel of 1 in water acidified with H2SO4 to a pH of 1.0. | ||
TEM imaging of the HBF4 and HPF6 acidified gels reveals that the microstructure is different to that of the H2SO4 acidified gels (Fig. 3). Both gels showed a particle-based gel structure.62–64 The images of the HBF4 acidified gels showed a porous structure with struts consisting of small particles ranging in size from 10 to 50 nm with the gel spanning the pores of the carbon mesh holding the samples. The images of the HPF6 gels showed a similar gel structure with struts consisting of nano-sized particles. The particles of the HBF4 and HPF6 acidified gels were crystalline in nature as shown by their ability to diffract the electron beam in diffraction mode on the TEM instrument (Fig. S8†). The d-spacings do not much up with any of the known crystal structures or powder patterns of 1 or dried samples of gels of 1 (Fig. S17 and S18†). SEM images of gel samples acidified with HBF4 and HPF6 that had been dried under high vacuum and covered in a thin layer of Pt showed that the HBF4 acidified gels form crystalline flakes upon drying (Fig. 4a). The SEM images of the HPF6 acidified gels showed porous structures which are similar to those seen for the H2SO4 acidified gels but with a much smaller scale in size (Fig. 4b).
 | ||
| Fig. 3 TEM images of gels of 1 acidified with (a and b) HBF4 and (c and d) HPF6. | ||
 | ||
| Fig. 4 SEM images of dried gels of 1 acidified with (a) HBF4 and (b) HPF6. | ||
The transient nature of the nitrate-acidified gel, that turns into a precipitate, meant that it could not be studied using rheology. However, TEM images of a gel that had just formed allowed for some characterisation of the gel (Fig. 5).60 Interestingly, the transformation from gel to crystal could be observed directly, as clusters of nanocrystals grew on what appeared to be the gel fibres.
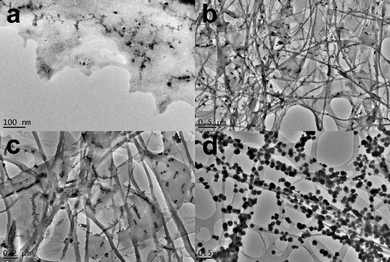 | ||
| Fig. 5 TEM images of the gels from acidification of a water solution of 1 with HNO3. (a) and (b) are from 1 with HNO3 gel sample. (c) and (d) are from a gel of 1 with HNO3 that had started to break down to a precipitate. | ||
Rheology
The rheological characteristics of a gel can be used to identify important changes that occur in its structure as a function of the identity of the anion.65–71 Gels of protonated 1 were characterised by stress sweep and frequency sweep rheometry. When a frequency sweep was performed with a small amplitude stress (Fig. 6, S7 and S9†), with a number of different samples varying in concentration, the solid-like nature at 20 °C was reflected in the storage modulus, G′, typically two to five times greater than the loss modulus, G″, thus demonstrating the elastic behaviour of the systems, albeit in robust gels an order of magnitude difference is expected.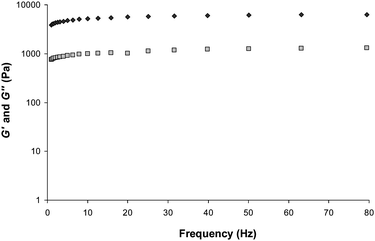 | ||
| Fig. 6 Frequency sweep rheometry of a gel formed by the sulfate salt of protonated 1 (0.1% by weight, pH 1.0); G′ (dark grey filled ◇) and G′′ (light grey filled □). | ||
This viscoelastic behaviour is associated with classical gels, and therefore supports the notion that the cooling of these samples from a solution to a solid-like material resulted in a true gel state.72 The effect of the concentration on the gel strength, in the form of G′ and “yield strength” (Fig. S3†) values, was also investigated using rheology to ascertain the morphology of the gel. The two theoretical models put forward for the mechanical properties of gels73 that are pertinent to LMWGs are the colloidal gel description73,74 and the cellular solid model.65,73,75,76
The dependence of the strength of the gel of 1, in water acidified to pH 1 with H2SO4, and the concentration of 1 proved to fit well to the relationships G′ ∝ [conc]2.2 (Fig. S4†) and “yield stress” ∝ [conc]1.5 (Fig. S5†). This fit is in agreement with the cellular solid model76 which is predicted to have a relationship of G′ ∝ [conc]2 and “yield stress” ∝ [conc]n where n can vary between 1 and 2, with the colloidal gel description giving n > 2.65,76 The cellular solid model describes an open-cell cellular material which consists of load bearing struts interconnected via crosslinks or junction points which deform by bending, which agrees with the morphologies seen by microscopy.
A comparison of all the gels, at a fixed 1% by weight, by stress sweep rheometry reveals the tuning of the rheological characteristics of gels formed by 1 by changing the identity of the anion (Fig. 7). The plateau G′ value for each of the different salt gels shows the trend in strength to be protonation by H2SO4 > MePO3H2 > H3PO4 ≈ HBF4 ≈ HPF6 > EtPO3H2. The “yield stress” follows a similar trend H2SO4 > MePO3H2 > HPF6 > H3PO4 > HBF4 > EtPO3H2. This ranking parallels the well-known Hofmeister series77–79 which ranks anions from kosmotropic (strongly hydrated anions) to chaotropic (weakly hydrated anions) in the order: SO42− ≈ MePO32− ≈ HPO42− > EtPO32− > Cl− > NO3− > PF6− > BF4−. Kosmotropic relates to the ‘salting-out’ anions and chaotropic behaviour applies to ‘salting-in’ anions. When the kosmotropic anions, from the acids H2SO4, MePO3H2, H3PO4, EtPO3H2, were used, gel formation was seen, with the SO42− and MePO32− based gels being the strongest, whereas the chaotropic anions are weaker. Gelation by nitrate is borderline. The chaotropic anion gels (BF4−, PF6− and the nitrate material) have rather different morphologies to the kosmotropic anion based materials. The hydration of the anions, therefore, possibly plays a role in the morphology of the gel.
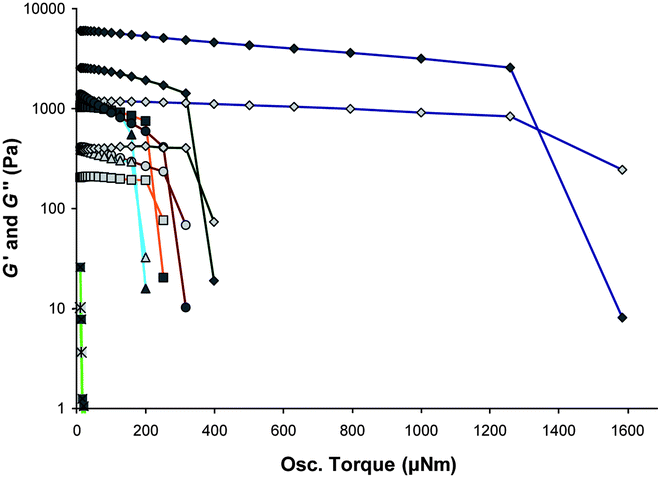 | ||
| Fig. 7 Anion tuning of the rheometric characterisations of gels of 1 shown by stress sweep rheometry of the hydrogels of 1 at 1% by weight acidified with H2SO4 (blue ◇), H3PO43 (orange □), MePO3H2 (green ◇), HBF4 (light blue △), HPF6 (brown ○) and EtPO3H2 (light green □). | ||
Crystallography
The transient nature of the HNO3 acidified gels and the precipitation upon addition of Cl− reveal a close connection between the stabilities of the gel and crystalline states.80–82 Characterisation of the crystalline materials is expected therefore to lead to a better understanding of the nature of the anion–gel interactions as well as fully characterising the salts of 1. The formulae of all of the crystalline products isolated are given in Table 1 and it is interesting that all proved to be highly solvated.The crystal structure of parent compound 1 was determined with the crystals grown from water at pH 7 (Fig. 8). The structure exhibits an intramolecular hydrogen bond between N4 and N2, within the asymmetric unit of 1, which results in an anti conformation for the urea group. A few 2-pyridylureas and 2-pyrrolylurea derivatives exhibit similar conformations.83–86 The rest of the urea group that is not involved in the intra-molecular hydrogen bond forms a dimer with the neighbouring molecule via an R22(8) hydrogen bond motif, in a similar way to related compounds.87,88 The dimers further aggregate to give a 1D hydrogen bonded chain via a N1–H1⋯O3 hydrogen bond between the pyrazole and nitro groups (Fig. S12†). These flat chains stack one on top of each other resulting in 2D stacks (inter-planar distance 3.3 Å). These stacks run perpendicular to each other (Fig. S13†). This stacking is possible due to the flatness of 1, RMS value of 0.0694 Å.
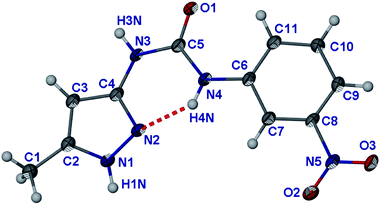 | ||
| Fig. 8 Molecular structure of 1. Atoms are shown as ellipsoids at 50% probability. | ||
The gels of 1 formed in water acidified at pH 1.0 with H2SO4 were induced to crystallise by addition of salt in the form of Na2SO4 or by mechanical stress. Stirring with a spatula, which creates nuclei for crystallisation to occur, resulted in crystallisation after a few weeks with the gel slowly converting to the crystal form.60 The crystals were dissolved by heating the solution, and upon cooling, the gel reformed. The structure of these crystals (1H++)2(SO42−)·7H2O provides a “snapshot” into the structural characteristics of the hydrogel.28
The asymmetric unit comprises two molecules of protonated 1 (1H++), an SO42− anion and seven water molecules (Fig. 9). The additional proton is located on the pyrazole group resulting in the breaking of the intra-molecular hydrogen bond seen in the crystal structure of 1 and hence imparting a syn conformation to the urea group. The 1H++ cations are flat (RMS deviation of 0.102 Å and 0.08 Å for the molecules with atoms C1 and C12, respectively). There are intramolecular hydrogen bonds between the carbonyl group of the urea and the pyrazole NH nearest the urea group as well as between the carbonyl group of the urea and the CH moiety on the nitrophenyl group. This type of CH⋯O intermolecular interaction is expected for ureas with electron withdrawing substituents on the aryl rings.89
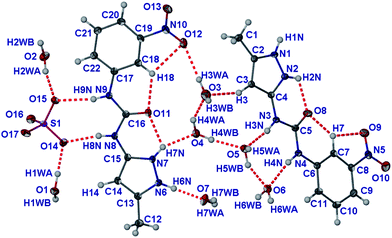 | ||
| Fig. 9 Asymmetric unit of (1H++)2(SO42−)·7H2O. Atoms are shown as ellipsoids at 50% probability. | ||
The large number of water molecules form an extensive hydrogen bonded network (ESI†). There are just three hydrogen bonds that do not involve water molecules; the hydrogen bonding of the urea group (N8 and N9) to the SO42− anion in a R22(8) motif, and hydrogen bonding of the protonated pyrazole (N1) to the SO42− anion. The overall packing of the 1H++ cations forms a one on top of each other π-stacking arrangement with interplanar distance varying from 3.33 Å to 3.49 Å to give columns that contain the SO42− and water molecules surrounded by four stacks of 1H++ (Fig. 10). This packing is similar to that seen by Dastidar, Das and coworkers in a bispyridyl urea hydrogelator with occluded ethylene glycol and water solvent. These authors suggested that the structure represents a “snapshot” of the gelator interacting with the solvent it gels.28
![Packing diagram of (1H++)2(SO42−)·7H2O showing the stacking of 1H++ one on top of each other resulting in a column in which the SO42− and water are located. Packing viewed down [100].](/image/article/2011/SM/c0sm00594k/c0sm00594k-f10.gif) | ||
| Fig. 10 Packing diagram of (1H++)2(SO42−)·7H2O showing the stacking of 1H++ one on top of each other resulting in a column in which the SO42− and water are located. Packing viewed down [100]. | ||
1D growth of gel fibres due to 1D supramolecular bonds is considered important consideration in designing LMWGs.80 Keeping this in mind, the needle-shaped crystals of (1H++)2(SO42−)·7H2O were face indexed to determine what the driving force is for the “quicker” growth of the face that determines the needle morphology. This quicker growing face was determined to be (100). The packing of the 1H++ molecules within the structure one on top of each other through π-stacking is in the [100] direction which correlates with the (100) quicker growing face and hence the π-stacking interaction of the planarised 1H++ drives 1D growth of the crystal and may well also do so in gel fibre formation in an aqueous environment where hydrophobic interactions are dominant.90 Unfortunately the hydroscopic nature of the compound meant that no XRPD pattern of the H2SO4 gel samples could be obtained for comparison with the single crystal data.
Crystallisation of the gels obtained from HBF4 and HPF6 were attempted by inducing salt formation by adding salts such as NaCl and Na2HPO4 to the gels and by growing single crystals from MeOH solutions of 1 acidified with the appropriate acids. No single crystals were obtained from HPF6 but the HBF4 sample yielded block-shaped crystals of formula (1H++)(BF4−)·H2O from a MeOH solution acidified with HBF4 that was allowed to evaporate for one week, as well as when a gel was stirred vigorously with a spatula (Fig. 11). The structure comprises a 1H++ cation in which the nitrophenyl group is twisted away from the plane defined by the pyrazole and urea groups, the torsion angle ∠C5–N4–C6–C7 = −30.1° compared to less than 12° in the other structures discussed herein, and as little as 1° in the structure of (1H++)2(SO42−)·7H2O.
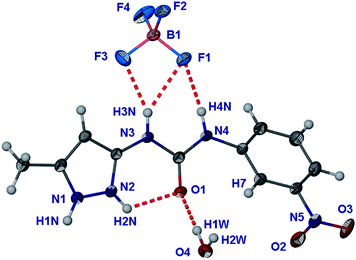 | ||
| Fig. 11 Molecular structure of (1H++)(BF4−)·H2O. Atoms are shown as ellipsoids at 50% probability. | ||
The RMS deviation from planarity of 0.308 Å for the 1H++ molecule within the (1H++)(BF4−)·H2O structure also highlights this twisting. The twisted conformation of the molecule 1 disrupts the intra-molecular hydrogen bonds involving the carbonyl group of the urea, in particularly the hydrogen bond with the nitrophenyl group, in comparison to the structures of 1 and (1H++)2(SO42−)·7H2O; N2⋯O1 = 2.626(2) Å; ∠N2–H2N⋯O1 = 119.0°, C7⋯O1 = 2.905(18) Å; ∠C7–H7⋯O1 = 111.5°. Moreover, the H⋯O distance is longer at 2.42 Å (compared to the 2.24 Å and 2.12 Å of the SO42− structure).
The pseudo-R22(8) hydrogen bonding to the BF4− anion through the urea group is shifted making F1 a bifurcated acceptor (Fig. 12). There is one water molecule that hydrogen bonds to both the 1H++ (as both a donor and an acceptor) and the BF4− anion. The intra-molecularly hydrogen bonded N2–H2N also hydrogen bonds to the BF4− anion. Due to the twisting of the nitrophenyl group in 1H++, the 1H++ cations do not quite pack one on top of each other as seen in the other structures containing 1H++ (Fig. 12a). The π–π stacking of the nitrophenyl groups down (010) is therefore rather different in character to the sulfate salt (Fig. 12b, the c axis length of 7.7509(4) Å is double the π-stacking distance).
 | ||
| Fig. 12 (a) Structure of 1H++BF4−·H2O as viewed down (100). (b) Structure of 1H++BF4−·H2O as viewed down (010). Hydrogen bonds not shown for clarity. | ||
A second structure of a co-crystal91–95 (1H++)(BF4−)·1 was obtained by adding Na2HPO4 (50 mg to a 3 mL 0.2% by weight gel) to a gel in water acidified with HBF4. The structure comprises both protonated and unprotonated 1 (Fig. 13). The acidic proton is disordered equally between N2 and N7 of neighbouring molecules of 1 with the hydrogen bonding arrangement between the two compounds changing with the position of the proton. The two molecules of 1 are both planar but the nitrophenyl groups are twisted slightly due to steric hindrance arising from the interactions between the pyrazole groups, RMS deviations of 0.119 Å (molecule 1 based on C1) and 0.216 Å (molecule 2 based on C12). Indeed, the nitrophenyl group of molecule 2 has a “transoid” orientation in relation to the pyrazole group orientation rather than the “cisoid” orientation seen in all the other structures of 1.
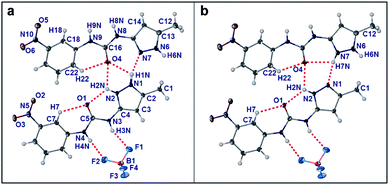 | ||
| Fig. 13 Molecular structure of (1H++)(BF4−)·1. Atoms are shown as ellipsoids at 50% probability. (a) Disordered proton situated on N1. (b) Alternate arrangement with disordered proton situated on N7. | ||
When the proton is on N2, in addition to an N2⋯N7 hydrogen bonding interaction, the pyrazole group forms a R22(8) hydrogen bond with the carbonyl oxygen atom of the neutral molecule of 1 helping to disrupt the intramolecular hydrogen bond seen in the crystal structure of native unprotonated 1. When the proton is on N7, N1 becomes an acceptor resulting in an intra-molecular hydrogen bond to the urea carbonyl group as seen in the other structures. The intra-molecular hydrogen bonds from the nitrophenyl moiety are slightly different due to the twisting of the nitrophenyl group described above. Hydrogen bonding between neutral 1 species and a protonated cation via their carbonyl and pyrazole groups results in the NH groups of the urea moieties and one NH group of the pyrazole group, N6–H6N, being available as hydrogen bond donors and hence the urea groups hydrogen bond to the BF4− with the N3–H3N and N4–H4N groups forming a R22(8) hydrogen bond pattern. The other urea group (N8 and N9) hydrogen bonds to F4 on two symmetry independent BF4− ions. The free NH group of the pyrazole group hydrogen bonds to O3 of neighbouring nitro groups which results in a hydrogen bonded thread of 1H++·1 units. These threads are stacked one on top of each other (the stacking distance being a quarter of the a–c unit cell diagonal distance) giving a 2D dimensional layer that alternates with layers of the BF4− ions (Fig. S14†). The formation of this co-crystal may be due to the change in pH induced by the addition of basic Na2HPO4. Simulation of the PXRD patterns of either of these BF4−1H++ single crystal structures resulted in no confirmed match with the d-spacings found from either the electron diffraction of the particles of the TEM gel samples (Fig. S8†) or the XRPD patterns measured of dried gel samples (Fig. S18†). The dried gel PXRD patterns of the HBF4 and HPF6 acidified gels show some similarities, but are not perfectly matched.
As no single crystals of the crystalline materials from the HNO3 and HCl acidified solutions could be obtained from acidified water, a polar organic solvent was added to slow down crystal formation. This resulted in the formation and characterisation of a methanol solvate of the chloride salt of 1H++ and a methanol/water solvate of the nitrate salt when 1 was crystallised from a MeOH solution acidified with an equal volume of the concentrated acids.
The (1H++)(NO3−)·MeOH structure has an asymmetric unit comprising two 1H++ cations, two NO3− anions and two molecules of MeOH, i.e.Z′ = 2 (Fig. 14).96 Once again, the 1H++ cations are planar with the pyrazole being protonated, RMS deviations of 0.105 Å (molecule based on C1) and 0.040 Å (molecule based on C12). The intra-molecular hydrogen bonds between the pyrazole and the carbonyl of the urea and the carbonyl group of the urea and a CH unit on the nitrophenyl group contribute to the planarisation of the 1H++ cation. One of the nitrate anions is asymmetrically chelated between two urea groups in an asymmetric R22(8) hydrogen bonded motif involving O9 as a bifurcated acceptor.97 The R22(8) motif of the urea group containing N3 and N4 is symmetrical, however.98 The pyrazole group containing N1 and N2 hydrogen bonds to the second nitrate anion, but due to the divergent character of the pyrazole NH groups only the N–H⋯O11 hydrogen bond is short. The MeOH molecules hydrogen bond (as donors) to this same nitrate anion and are hydrogen bonded (as acceptors) to the other pyrazole NH group. The planar 1H++ molecules, once again, stack one on top of each other, but in an alternating pattern (Fig. S15†). There are four stacks of 1H++ that surround a column of methanol and nitrate anions.
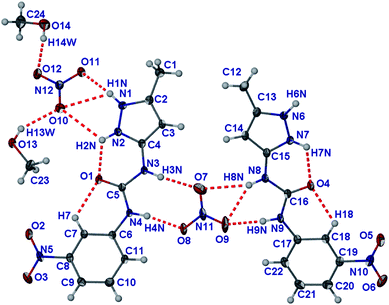 | ||
| Fig. 14 Molecular structure of (1H++)(NO3−)·MeOH. Atoms are shown as ellipsoids at 50% probability. | ||
The crystallization of 1 from aqueous HCl gives a fine precipitate. Crystallisation of 1 from methanolic HCl gives a different crystalline material to that from water that is less fine and has large crystallites allowing for the determination of the structure using single crystal methods. While the crystallographic data are of low precision, the qualitative details of the structure of this product, (1H++)2(Cl−)2·MeOH·H2O, are clear. The material is similar to the structure of (1H++)NO3−·MeOH (Fig. 15). The 1H++ cations are planar with the pyrazole protonated, RMS deviations of 0.053 Å (molecular based on C1) and 0.100 Å (molecule based on C12). The intra-molecular hydrogen bonds between the protonated pyrazole group and the carbonyl oxygen atom of the urea, and between the carbonyl group of the urea and a CH unit on the nitrophenyl group contribute to the planar conformation. As with the nitrate structure, the anion is chelated in between the two urea groups of 1H++. The R12(6) hydrogen bonding by both ureas is symmetrical. The N1–H1N group of the pyrazole hydrogen bonds to the second chloride ion. The other pyrazole group has hydrogen bonds to the MeOH and water molecules. The MeOH hydrogen bonds to the chelated Cl1. As seen with the other structures containing protonated 1, the 1H++ cations pack one on top of each other in an alternating fashion similar to the nitrate analogue to give the usual stacks of 1H++ with anion and solvent in between. As a result of the smaller size of chloride compared to nitrate, the anion column between groups of four cation stacks is shifted resulting in a kinked arrangement (Fig. S16†).
![Molecular structure of 2[(1H++)(Cl−)]·MeOH·H2O. Atoms are shown as ellipsoids at 20% probability.](/image/article/2011/SM/c0sm00594k/c0sm00594k-f15.gif) | ||
| Fig. 15 Molecular structure of 2[(1H++)(Cl−)]·MeOH·H2O. Atoms are shown as ellipsoids at 20% probability. | ||
Conclusions
Protonation of compound 1 resulted in the formation of hydrogels in the pH range of 1 to 2, thus making compound 1 a hydrogelator exhibiting pH tunable gelation.20,28,38–57 Rheological characterization, in conjunction with TEM and SEM imaging, of the acidified gels indicates a cellular solid gel material that consists of load bearing struts interconnected via cross-links or junction points which deform by bending. In addition, SEM and TEM imaging revealed a morphological difference between the gels with chaotropic anions BF4− and PF6−, which are particulate, and the gels synthesized with kosmotropic oxyanions: SO42−, HPO42−, EtPO32− and MePO32−. The anion tuning of the gel 1 characteristics was accomplished by varying the identity of the anion by changing the acid used to adjust the pH of the water solutions. Gel formation changed to crystallisation when Cl− or NO3− was introduced. This resulted in the ability to precipitate a gel by the addition of salt containing these anions. The smaller NO3− and Cl− are bound between pairs of 1H++ cations by the urea groups in the solid state, as shown by the crystal structures of (1H++)(NO3−)·MeOH and (1H++)2(Cl−)2·MeOH·H2O. Characterisation of the crystalline salts of 1 indicates that the assembly of the gels may be based on the 1D stacking of the planar 1H++ cations driven by hydrophobic effects and π–π stacking. No urea⋯urea hydrogen bonding is observed, in contrast to the well known bis(urea) class of gelators.99 The anion induced change from gel to precipitate appears to be due to the ability of 1H++ to coordinate through multiple hydrogen bonds to the anion. These hydrogen bond patterns, in combination with other less well understood factors including lattice energy, nucleation and the thermodynamics of the systems, lead to the formation of the crystalline state instead of gelation. The crystal structures (1H++)(BF4−)·H2O and (1H++)2(SO42−)·7H2O reveal a slight change in the conformation of the 1H++ cation, and the level of hydration, respectively. In addition to the anion tuning from gel to crystal states, the rheological characteristics of gels of 1 were markedly influenced by the identity of the counter anion. The gels of 1 are ranked in terms of elastic modulus in the order of acid used to protonate the gelator: H2SO4 > MePO3H2 > HPF6 > H3PO4 > HBF4 > EtPO3H2. Neither the Hofmeister series and/or level of hydration of the anions conclusively explains the order of strength of these. The change in morphology of the gels, from the oxygen containing anions to BF4− and PF6−, may explain the inconsistency, as the structures are seen to be different due to the hydrogen bond propensity of these anions, as shown by the crystal structure (1H++)(BF4−)·H2O. Additionally, the crystallisation abilities of the salts are super-imposed onto the gelation of this series of anion containing gels. The tuning of the gel characteristics of gels of 1 is summarized in Table 1. This study, overall, shows that LMWG salts can be tuned in terms of gel rheology characteristics over a large range of gel strength, from strong gel to total loss of gel formation, by simply varying the identity of the anion or the pH.Experimental
General
All reagents and chemicals were obtained from commercial sources and used without further purification. SEM samples were partially dried under room temperature and atmosphere, and then dried under high vacuum on a piece of silicon wafer. They were then coated with a thin layer of platinum of 3–5 nm using a 308UHR Ultra High Resolution Coating System (Biology, Durham). Samples were imaged using an S-5200 UHR FE-SEM Hitachi Scientific instrument (Biology, Durham). TEM samples were deposited onto a holey carbon grid and were immediately examined in a JEOL 2100F FEG TEM (Physics, Durham) operating at 200 kV. Rheology experiments were performed using a TA Instruments Advanced Rheometer 2000. A parallel plate geometry (40 mm diameter) was used. Single crystal diffraction data were collected on a Bruker SMART 6K (6000 CCD) and solved and refined using the SHELX suite of programs100 with the aid of X-Seed.101,102 Powder samples were dried and processed through a 100 μm microsieve and run on silicon zero beckground sample holders using a Siemens D5000 instrument.Synthesis
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH-) 5.35 (2H, br s, -NH2). 13C{1H} NMR: (100 MHz, CDCl3, δ/ppm) 14.4, 28.0, 84.9, 89.3, 92.0, 150.7, 153.2. Anal. Calc. C, 54.8; H, 7.67; N, 21.3%. Found: C, 54.3; H, 7.61; N, 20.9%. Mp 120 °C. ES+-MS (m/z): 198 [100%] C9H15N3O2 + H+, 220 [5%] C9H15N3O2 + Na+, 395 [33%] 2C9H15N3O2 + H+. FT-IR: (ν, cm−1) 3441 (m), 3358 (w), 3285 (w), 3212 (w), 3161 (w), 3120 (w), 2982 (m), 2933 (w), 1710 (s), 1619 (s), 1553 (m).
CH-) 7.56 (1H, t, J = 8.4, -CH) 7.70 (1H, ddd, J = 8.4, 1.0, 2.2, -CH) 7.83 (1H, ddd, J = 8.4, 1.0, 2.2, -CH) 8.53 (1H, t, J = 2.2, -CH) 9.72 (1H, s, -NH-) 10.50 (1H, s, -NH-). 13C{1H} NMR: (100 MHz, d6-DMSO, δ/ppm) 13.8, 27.6, 85.1, 94.0, 96.1, 112.0, 116.7, 124.2, 130.1, 140.6, 141.8, 148.1, 150.1, 151.8. Anal. Calc. C, 53.2; H, 5.30; N, 19.4%. Found: C, 53.0; H, 5.54; N, 17.8%. Mp 176 °C. ES−-MS (m/z): 360.2 [51%] C11H11N5O3 − H+, 405.9 (80%) C11H11N5O3 + Cl−, 720.7 [25%] 2(C11H11N5O3) − H+, 566.2 (12%) 2(C11H11N5O3) + Cl−, FT-IR: (ν, cm−1) 3342 (w), 3272 (m), 3141 (w), 3092 (w), 3034 (w), 2989 (w), 2940 (w), 1694 (m), 1661 (m), 1608 (m), 1586 (w), 1564 (m).
CH-) 7.46 (1H, t, J = 8.0, -CH) 7.71 (1H, ddd, J = 8.0, 0.9, 2.2, -CH) 7.81 (1H, ddd, J = 8.0, 0.9, 2.2, -CH) 8.13 (1H, br s, -NHpyz) 8.55 (1H, t, J = 2.2, -CH) 10.10 (1H, br s, -NH-) 10.45 (1H, br s -NH-). 13C{1H} NMR: (100 MHz, d6-DMSO, δ/ppm) 10.8, 94.0, 112.0, 116.1, 124.2, 130.0, 141.2, 148.11, 152.0. Anal. Calc. C, 50.6; H, 4.24; N, 26.8%. Found: C, 50.4; H, 4.25; N, 26.2%. Mp 180 °C. ES−-MS (m/z): 260.2 [53%] C11H11N5O3 − H+, 306.0 (100%) C11H11N5O3 + Cl−,520.8 [25%] 2(C11H11N5O3) − H+, 566.2 (12%) 2(C11H11N5O3) + Cl−. FT-IR: (ν, cm−1) 3346 (m), 3042 (m), 3010 (w), 2936 (w), 1707 (s), 1611 (m), 1583 (m), 1544 (s), 1523 (s).
CH-) 5.35 (2H, br s, -NH2). 13C{1H} NMR: (100 MHz, CDCl3, δ/ppm) 14.4, 28.0, 84.9, 89.3, 92.0, 150.7, 153.2. Anal. Calc. C, 54.8; H, 7.67; N, 21.3%. Found: C, 54.3; H, 7.61; N, 20.9%. Mp 120 °C. ES+-MS (m/z): 198 [100%] C9H15N3O2 + H+, 220 [5%] C9H15N3O2 + Na+, 395 [33%] 2C9H15N3O2 + H+. FT-IR: (ν, cm−1) 3441 (m), 3358 (w), 3285 (w), 3212 (w), 3161 (w), 3120 (w), 2982 (m), 2933 (w), 1710 (s), 1619 (s), 1553 (m).
CH-) 7.56 (1H, t, J = 8.4, -CH) 7.70 (1H, ddd, J = 8.4, 1.0, 2.2, -CH) 7.83 (1H, ddd, J = 8.4, 1.0, 2.2, -CH) 8.53 (1H, t, J = 2.2, -CH) 9.72 (1H, s, -NH-) 10.50 (1H, s, -NH-). 13C{1H} NMR: (100 MHz, d6-DMSO, δ/ppm) 13.8, 27.6, 85.1, 94.0, 96.1, 112.0, 116.7, 124.2, 130.1, 140.6, 141.8, 148.1, 150.1, 151.8. Anal. Calc. C, 53.2; H, 5.30; N, 19.4%. Found: C, 53.0; H, 5.54; N, 17.8%. Mp 176 °C. ES−-MS (m/z): 360.2 [51%] C11H11N5O3 − H+, 405.9 (80%) C11H11N5O3 + Cl−, 720.7 [25%] 2(C11H11N5O3) − H+, 566.2 (12%) 2(C11H11N5O3) + Cl−, FT-IR: (ν, cm−1) 3342 (w), 3272 (m), 3141 (w), 3092 (w), 3034 (w), 2989 (w), 2940 (w), 1694 (m), 1661 (m), 1608 (m), 1586 (w), 1564 (m).
CH-) 7.46 (1H, t, J = 8.0, -CH) 7.71 (1H, ddd, J = 8.0, 0.9, 2.2, -CH) 7.81 (1H, ddd, J = 8.0, 0.9, 2.2, -CH) 8.13 (1H, br s, -NHpyz) 8.55 (1H, t, J = 2.2, -CH) 10.10 (1H, br s, -NH-) 10.45 (1H, br s -NH-). 13C{1H} NMR: (100 MHz, d6-DMSO, δ/ppm) 10.8, 94.0, 112.0, 116.1, 124.2, 130.0, 141.2, 148.11, 152.0. Anal. Calc. C, 50.6; H, 4.24; N, 26.8%. Found: C, 50.4; H, 4.25; N, 26.2%. Mp 180 °C. ES−-MS (m/z): 260.2 [53%] C11H11N5O3 − H+, 306.0 (100%) C11H11N5O3 + Cl−,520.8 [25%] 2(C11H11N5O3) − H+, 566.2 (12%) 2(C11H11N5O3) + Cl−. FT-IR: (ν, cm−1) 3346 (m), 3042 (m), 3010 (w), 2936 (w), 1707 (s), 1611 (m), 1583 (m), 1544 (s), 1523 (s).
Crystallography
Crystal data for tert-butyl-5-amino-3-methyl-1H-pyrazole-1-carboxylate: C9H15N3O2, M = 197.24, colourless prism, 0.24 × 0.19 × 0.13 mm3, monoclinic, space groupP21/c (no. 14), a = 9.8180(3), b = 9.6472(3), c = 11.6088(4) Å, β = 106.9080(10)°, V = 1052.01(6) Å3, Z = 4, Dc = 1.245 g cm−3, F000 = 424, Smart-6K, MoKα radiation, λ = 0.71073 Å, T = 120(2) k, 2θmax = 61.0°, 9721 reflections collected, 3199 unique (Rint = 0.0403). Final GooF = 1.024, R1 = 0.0479, wR2 = 0.1228, R indices based on 2223 reflections with I > 2σ(I) (refinement on F2), 135 parameters, 4 restraints. Lp and absorption corrections applied, µ = 0.090 mm−1.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 993 reflections collected, 3450 unique (Rint = 0.0388). Final GooF = 1.031, R1 = 0.0486, wR2 = 0.1254, R indices based on 2428 reflections with I > 2σ(I) (refinement on F2), 173 parameters, 0 restraints. Lp and absorption corrections applied, µ = 0.114 mm−1.
366 reflections collected, 9934 unique (Rint = 0.0401). Final GooF = 1.053, R1 = 0.0523, wR2 = 0.1414, R indices based on 7653 reflections with I > 2σ(I) (refinement on F2), 503 parameters, 14 restraints. Lp and absorption corrections applied, µ = 0.192 mm−1.
993 reflections collected, 3450 unique (Rint = 0.0388). Final GooF = 1.031, R1 = 0.0486, wR2 = 0.1254, R indices based on 2428 reflections with I > 2σ(I) (refinement on F2), 173 parameters, 0 restraints. Lp and absorption corrections applied, µ = 0.114 mm−1.
366 reflections collected, 9934 unique (Rint = 0.0401). Final GooF = 1.053, R1 = 0.0523, wR2 = 0.1414, R indices based on 7653 reflections with I > 2σ(I) (refinement on F2), 503 parameters, 14 restraints. Lp and absorption corrections applied, µ = 0.192 mm−1.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 7.7509(4), b = 8.2203(5), c = 13.2848(7) Å, α = 94.481(2), β = 104.643(2), γ = 110.579(2)°, V = 753.54(7) Å3, Z = 2, Dc = 1.618 g cm−3, F000 = 376, Smart-6K, MoKα radiation, λ = 0.71073 Å, T = 120(2) k, 2θmax = 61.1°, 12989 reflections collected, 4594 unique (Rint = 0.0481). Final GooF = 1.025, R1 = 0.0585, wR2 = 0.1402, R indices based on 2963 reflections with I > 2σ(I) (refinement on F2), 235 parameters, 2 restraints. Lp and absorption corrections applied, µ = 0.153 mm−1.
(no. 2), a = 7.5596(4), b = 12.6366(6), c = 13.7797(6) Å, α = 100.6610(10), β = 94.926(2), γ = 96.370(2)°, V = 1277.98(11) Å3, Z = 2, Dc = 1.586 g cm−3, F000 = 628, Smart-6K, MoKα radiation, λ = 0.71073 Å, T = 120(2) k, 2θmax = 56.0°, 17616 reflections collected, 6149 unique (Rint = 0.0494). Final GooF = 1.011, R1 = 0.0587, wR2 = 0.1474, R indices based on 3877 reflections with I > 2σ(I) (refinement on F2), 390 parameters, 0 restraints. Lp and absorption corrections applied, µ = 0.136 mm−1.
666 reflections collected, 9546 unique (Rint = 0.0692). Final GooF = 1.008, R1 = 0.0613, wR2 = 0.1458, R indices based on 5157 reflections with I > 2σ(I) (refinement on F2), 457 parameters, 0 restraints. Lp and absorption corrections applied, µ = 0.126 mm−1.
(no. 2), a = 7.557(8), b = 14.027(16), c = 15.689(17) Å, α = 70.74(2), β = 77.71(2), γ = 87.03(2)°, V = 1534(3) Å3, Z = 2, Dc = 1.393 g cm−3, F000 = 668, Smart-6K, MoKα radiation, λ = 0.71073 Å, T = 120(2) k, 2θmax = 50.0°, 15248 reflections collected, 5366 unique (Rint = 0.2267). Final GooF = 1.812, R1 = 0.2848, wR2 = 0.5981, R indices based on 2474 reflections with I > 2σ(I) (refinement on F2), 383 parameters, 280 restraints. Lp and absorption corrections applied, µ = 0.273 mm−1. While the crystallographic data are of low precision on this sample due to the extremely poor crystal quality the overall nature of the structure is apparent. As a result the limited structural data available are provided for completeness. The model obtained is sufficient to give evidence for the structural details that are of interest.
(no. 2), a = 7.7509(4), b = 8.2203(5), c = 13.2848(7) Å, α = 94.481(2), β = 104.643(2), γ = 110.579(2)°, V = 753.54(7) Å3, Z = 2, Dc = 1.618 g cm−3, F000 = 376, Smart-6K, MoKα radiation, λ = 0.71073 Å, T = 120(2) k, 2θmax = 61.1°, 12989 reflections collected, 4594 unique (Rint = 0.0481). Final GooF = 1.025, R1 = 0.0585, wR2 = 0.1402, R indices based on 2963 reflections with I > 2σ(I) (refinement on F2), 235 parameters, 2 restraints. Lp and absorption corrections applied, µ = 0.153 mm−1.
(no. 2), a = 7.5596(4), b = 12.6366(6), c = 13.7797(6) Å, α = 100.6610(10), β = 94.926(2), γ = 96.370(2)°, V = 1277.98(11) Å3, Z = 2, Dc = 1.586 g cm−3, F000 = 628, Smart-6K, MoKα radiation, λ = 0.71073 Å, T = 120(2) k, 2θmax = 56.0°, 17616 reflections collected, 6149 unique (Rint = 0.0494). Final GooF = 1.011, R1 = 0.0587, wR2 = 0.1474, R indices based on 3877 reflections with I > 2σ(I) (refinement on F2), 390 parameters, 0 restraints. Lp and absorption corrections applied, µ = 0.136 mm−1.
666 reflections collected, 9546 unique (Rint = 0.0692). Final GooF = 1.008, R1 = 0.0613, wR2 = 0.1458, R indices based on 5157 reflections with I > 2σ(I) (refinement on F2), 457 parameters, 0 restraints. Lp and absorption corrections applied, µ = 0.126 mm−1.
(no. 2), a = 7.557(8), b = 14.027(16), c = 15.689(17) Å, α = 70.74(2), β = 77.71(2), γ = 87.03(2)°, V = 1534(3) Å3, Z = 2, Dc = 1.393 g cm−3, F000 = 668, Smart-6K, MoKα radiation, λ = 0.71073 Å, T = 120(2) k, 2θmax = 50.0°, 15248 reflections collected, 5366 unique (Rint = 0.2267). Final GooF = 1.812, R1 = 0.2848, wR2 = 0.5981, R indices based on 2474 reflections with I > 2σ(I) (refinement on F2), 383 parameters, 280 restraints. Lp and absorption corrections applied, µ = 0.273 mm−1. While the crystallographic data are of low precision on this sample due to the extremely poor crystal quality the overall nature of the structure is apparent. As a result the limited structural data available are provided for completeness. The model obtained is sufficient to give evidence for the structural details that are of interest.
Acknowledgements
We thank Dr Nigel Clarke for assistance in interpreting the rheological results and Dr Budhika Mendis for assistance in collecting the TEM data. GOL would like to thank the Commonwealth Scholarship Commission and the Herchel Smith Fellowship Fund for financial support.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699 RSC.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489 CrossRef CAS.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237 CrossRef CAS.
- D. K. Smith, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH, Weinheim, 2008, p. 111 Search PubMed.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. L. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS.
- L. A. Estroff, L. Addadi, S. Weiner and A. D. Hamilton, Org. Biomol. Chem., 2004, 2, 137 RSC.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263 CrossRef.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315 CrossRef CAS.
- B. Escuder, F. Rodríguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044 RSC.
- F. Fages, Angew. Chem., Int. Ed. Engl., 2006, 45, 1680 CrossRef CAS.
- H. Maeda, Chem.–Eur. J., 2008, 14, 11274 CrossRef.
- G. O. Lloyd and J. W. Steed, Nat. Chem., 2009, 1, 437 CrossRef CAS.
- R. Oda, I. Huc and S. J. Candau, Angew. Chem., Int. Ed., 1998, 37, 2689 CrossRef CAS.
- H. Henstock, J. Am. Chem. Soc., 1939, 61, 670 CrossRef CAS.
- D. J. Abdallah and R. G. Weiss, Chem. Mater., 2000, 12, 406 CrossRef CAS.
- D. R. Trivedi and P. Dastidar, Cryst. Growth Des., 2006, 6, 2114 CrossRef CAS.
- A. Ballabh, D. R. Trivedi and P. Dastidar, Chem. Mater., 2006, 18, 3795 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Angew. Chem., Int. Ed., 2000, 39, 3447 CrossRef CAS.
- F. Fages, F. Vögtle and M. Žinic, Top. Curr. Chem., 2005, 256, 77 CAS.
- M. de Loos, J. van Esch, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 613 CrossRef CAS.
- P. J. Smith, M. V. Reddington and C. S. Wilcox, Tetrahedron Lett., 1992, 33, 6085 CrossRef CAS.
- S. Nishizawa, P. Buhlmann, M. Iwao and Y. Umezawa, Tetrahedron Lett., 1995, 36, 6483 CrossRef CAS.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179 CrossRef CAS.
- F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680 CrossRef CAS.
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Langmuir, 2009, 25, 8451 CrossRef CAS.
- D. K. Kumar, D. A. Jose, A. Das and P. Dastidar, Chem. Commun., 2005, 4059 RSC.
- A. Westcott, C. J. Sumby, R. D. Walshaw and M. J. Hardie, New J. Chem., 2009, 33, 902 RSC.
- H. J. Kim, E. Y. Jung, L. Y. Jin and M. Lee, Macromolecules, 2008, 41, 6066 CrossRef CAS.
- S. I. Kawano, N. Fujita, K. J. C. van Bommel and S. Shinkai, Chem. Lett., 2003, 12 CrossRef CAS.
- H. F. Anwar and M. H. Elnagdi, ARKIVOC, 2009, 198 CAS.
- H. Graubaum, J. Prakt. Chem., 1993, 335, 585 CrossRef CAS.
- C. M. Pask, K. D. Camm, C. A. Kilner and M. A. Halcrow, Tetrahedron Lett., 2006, 47, 2531 CrossRef CAS.
- P. Orsini, G. Traquandi, P. Sansonna and P. Pevarello, Tetrahedron Lett., 2005, 46, 933 CrossRef CAS.
- W. Seelen, M. Schafer and A. Ernst, Tetrahedron Lett., 2003, 44, 4491 CrossRef CAS.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684 RSC.
- D. M. Blow and A. Rich, J. Am. Chem. Soc., 1960, 82, 3566 CrossRef CAS.
- W. M. Muller, U. Muller, G. Mieden-Gundert, F. Vogtle, M. Lescanne, K. Heuze, A. D'Aleo and F. Fages, Eur. J. Org. Chem., 2002, 2891 CrossRef CAS.
- Z. M. Yang, H. W. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208 RSC.
- M. Suzuki, M. Yumoto, H. Shirai and K. Hanabusa, Org. Biomol. Chem., 2005, 3, 3073 RSC.
- D. C. Lee, K. K. McGrath and K. Jang, Chem. Commun., 2008, 3636 RSC.
- F. Rodriguez-Llansola, B. Escuder and J. F. Miravet, Org. Biomol. Chem., 2009, 7, 3091 RSC.
- J. J. Panda, A. Mishra, A. Basu and V. S. Chauhan, Biomacromolecules, 2008, 9, 2244 CrossRef CAS.
- D. S. Tsekova, B. Escuder and J. F. Miravet, Cryst. Growth Des., 2008, 8, 11 CrossRef CAS.
- T. H. Kim, J. Seo, S. J. Lee, S. S. Lee, J. Kim and J. H. Jung, Chem. Mater., 2007, 19, 5815 CrossRef CAS.
- J. W. Chung, B. K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750 CrossRef CAS.
- I. Hwang, W. S. Jeon, H. J. Kim, D. Kim, H. Kim, N. Selvapalam, N. Fujita, S. Shinkai and K. Kim, Angew. Chem., Int. Ed., 2007, 46, 210 CrossRef CAS.
- D. G. Velazquez, D. D. Diaz, A. G. Ravelo and J. J. M. Tellado, J. Am. Chem. Soc., 2008, 130, 7967 CrossRef.
- H. Komatsu, S. Matsumoto, S. Tamaru, K. Kaneko, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5580 CrossRef CAS.
- A. Srivastava, S. Ghorai, A. Bhattacharjya and S. Bhattacharya, J. Org. Chem., 2005, 70, 6574 CrossRef.
- A. Shome, S. Debnath and P. K. Das, Langmuir, 2008, 24, 4280 CrossRef CAS.
- T. Kar, S. Debnath, D. Das, A. Shome and P. K. Das, Langmuir, 2009, 25, 8639 CrossRef CAS.
- S. Kiyonaka, S. L. Zhou and I. Hamachi, Supramol. Chem., 2003, 15, 521 CrossRef CAS.
- S. L. Zhou, S. Matsumoto, H. D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem.–Eur. J., 2005, 11, 1130 CrossRef CAS.
- K. J. C. van Bommel, C. van der Pol, I. Muizebelt, A. Friggeri, A. Heeres, A. Meetsma, B. L. Feringa and J. van Esch, Angew. Chem., Int. Ed., 2004, 43, 1663 CrossRef CAS.
- S. Kiyonaka, S. Shinkai and H. Hamachi, Chem.–Eur. J., 2003, 9, 976 CrossRef CAS.
- M. A. Halcrow, Dalton Trans., 2009, 2059 RSC.
- O. Roubeau, A. Colin, W. Schmitt and R. Clerac, Angew. Chem., Int. Ed., 2004, 43, 3283 CrossRef CAS.
- Y. J. Wang, L. M. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884 CrossRef.
- K. Hanabusa, K. Hiratsuka, M. Kimura and H. Shirai, Chem. Mater., 1999, 11, 649 CrossRef CAS.
- J. Z. Wang, K. P. Loh, Z. Wang, Y. L. Yan, Y. L. Zhong, Q. H. Xu and P. C. Ho, Angew. Chem., Int. Ed., 2009, 48, 6282 CrossRef CAS.
- Y. L. Wang, W. Li and L. Wu, Langmuir, 2009, 25, 13194 CrossRef CAS.
- S. Bag, P. N. Trikalitis, P. J. Chupas, G. S. Armatas and M. G. Kanatzidis, Science, 2007, 317, 490 CrossRef CAS.
- P. Terech, D. Pasquier, V. Bordas and C. Rossat, Langmuir, 2000, 16, 4485 CrossRef CAS.
- J. L. Li, X. Y. Liu, R. Y. Wang and J. Y. Xiong, J. Phys. Chem. B, 2005, 109, 24231 CrossRef CAS.
- J. Brinksma, B. L. Feringa, R. M. Kellogg, R. Vreeker and J. van Esch, Langmuir, 2000, 16, 9249 CrossRef CAS.
- P. Terech and S. Friol, Tetrahedron, 2007, 63, 7366 CrossRef CAS.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679 CrossRef.
- P. Terech, C. Rossat and F. Volino, J. Colloid Interface Sci., 2000, 227, 363 CrossRef CAS.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Commun., 2008, 2644 RSC.
- R. G. Larson, The Structure and Rheology of Complex Fluids, Oxford University Press, Oxford, 1999 Search PubMed.
- N. M. Sangeetha, S. Bhat, A. R. Choudhury, U. Maitra and P. Terech, J. Phys. Chem. B, 2004, 108, 16056 CrossRef CAS.
- W. H. Shih, W. Y. Shih, S. I. Kim, J. Liu and I. A. Aksay, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 4772 CAS.
- L. J. Gibson and M. F. Ashby, Proc. R. Soc. London, Ser. A, 1982, 382, 43 CrossRef CAS.
- G. A. Buxton and N. Clarke, Phys. Rev. Lett., 2007, 98, 238103 CrossRef.
- J. T. O'Brien, J. S. Prell, M. F. Bush and E. R. Williams, J. Am. Chem. Soc., 2010, 132, 8248 CrossRef CAS.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley–Blackwell, Chichester, 2nd edn, 2009 Search PubMed.
- M. G. Cacace, E. M. Landan and J. J. Ramsden, Q. Rev. Biophys., 1997, 30, 241 CrossRef CAS.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699 RSC.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615 CrossRef CAS.
- A. H. H. Elghandour, M. M. M. Ramiz, M. K. A. Ibrahim and M. R. H. Elmoghayar, Org. Prep. Proced. Int., 1989, 21, 479 CrossRef CAS.
- P. S. Corbin, S. C. Zimmerman, P. A. Thiessen, N. A. Hawryluk and T. J. Murray, J. Am. Chem. Soc., 2001, 123, 10475 CrossRef CAS.
- M. Tiliakos, P. Cordopatis, A. Terzis, C. P. Raptopoulou, S. P. Perlepes and E. Manessi-Zoupa, Polyhedron, 2001, 20, 2203 CrossRef CAS.
- V. Fares, A. Flamini and P. Pasetto, J. Chem. Soc., Perkin Trans. 1, 2000, 4520 RSC.
- P. S. Corbin and S. C. Zimmerman, J. Am. Chem. Soc., 2000, 122, 3779 CrossRef CAS.
- J. T. Lenthall, K. M. Anderson, S. J. Smith and J. W. Steed, Cryst. Growth Des., 2007, 7, 1858 CrossRef CAS.
- D. R. Kelman, K. A. Claborn, W. Kaminsky, K. I. Goldberg, D. T. Li and D. X. West, J. Mol. Struct., 2003, 654, 145 CrossRef CAS.
- L. S. Reddy, S. Basavoju, V. R. Vangala and A. Nangia, Cryst. Growth Des., 2006, 6, 161 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS.
- G. R. Desiraju, CrystEngComm, 2003, 5, 466 RSC.
- J. D. Dunitz, CrystEngComm, 2003, 5, 506 RSC.
- C. B. Aakeroy and D. J. Salmon, CrystEngComm, 2005, 7, 439 RSC.
- M. J. Zaworotko, Cryst. Growth Des., 2007, 7, 4 CrossRef CAS.
- A. D. Bond, CrystEngComm, 2007, 9, 833 RSC.
- J. W. Steed, CrystEngComm, 2003, 5, 169 RSC.
- D. R. Turner, E. C. Spencer, J. A. K. Howard, D. A. Tocher and J. W. Steed, Chem. Commun., 2004, 1352 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- J. L. Atwood and L. J. Barbour, Cryst. Growth Des., 2003, 3, 3 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of the synthesis and analytical data for 1. Additional rheology, TEM, EELS, SEM and electron diffraction data for gel samples. PXRD pattern of dried gel samples. Crystallographic data in CIF or other electronic format with further figures of structures. CCDC reference numbers CCDC 783190–783196. See DOI: 10.1039/c0sm00594k |
| This journal is © The Royal Society of Chemistry 2011 |