Kinetics and thermodynamics in surface-confined molecular self-assembly
Rico
Gutzler
*,
Luis
Cardenas
and
Federico
Rosei
*
Institut National de la Recherche Scientifique, Université du Québec, 1650 boulevard Lionel-Boulet, Varennes, QC J3X 1S2, Canada. E-mail: gutzler@emt.inrs.ca; rosei@emt.inrs.ca
First published on 3rd October 2011
Abstract
Two-dimensional molecular self-assembly at the liquid/solid interface is a widely employed approach in surface science to pattern surfaces at the nanometre scale. A multitude of supramolecular structures can be realized depending on parameters such as the functionalization of the molecular building blocks, the temperature at which self-assembly takes place, the type of solvent and solute concentration. How these and other parameters influence the kinetics and thermodynamics of the self-assembly process is the subject of this review.
Introduction
Molecular self-assembly is a widely used strategy to pattern and functionalize surfaces at the nanometre scale with applications such as functional nanomaterials and organic electronics.1–3 The physisorption of organic molecules onto surfaces and their arrangement into ordered monolayers has been extensively investigated, leading to a myriad of different supramolecular structures with different molecular building blocks, both at the liquid/solid interface and the vacuum/solid interface.4–15 Despite numerous investigations following the pioneering work of Foster & Frommer16 and Rabe & Buchholz,17,18 it is not yet possible to deliberately engineer molecular monolayers or to predict the structure of such a monolayer for a given molecular building unit. To tackle these shortcomings, it is important to understand the mechanisms that underlie surface-confined molecular self-assembly. Polymorphism – the formation of two or more distinct crystalline monolayer structures – is one facet of two-dimensional molecular self-assembly that lacks a detailed understanding, although many parameters have been identified that can be used to control polymorphism and to switch between several different polymorphs. A nonexclusive list of these parameters includes the type of solvent,19–24 concentration of the molecular building block,25–27 molecular interactions,28–34 size and structure of the adsorbing molecule,30,35–37 type of surface38–42 and temperature.43–48 Care must be taken to discriminate between polymorphism in three-dimensional and two-dimensional crystals, the latter type being relevant in self-assembled organic monolayers. For example, concentration-dependent polymorphism in 3D cannot occur due to thermodynamic reasons, while it is now a commonly accepted feature in 2D crystallization, as pointed out by Matzger and coworkers.49At the liquid/solid interface, interactions between molecular building blocks, solvent molecules and the surface are crucial in defining the structure of a monolayer. These interactions constitute one aspect of a complex thermodynamic description of the self-assembly process, which necessarily also includes parameters such as temperature, entropy, or chemical potentials.13 Attempts have been made to find suitable thermodynamic models that semi-quantitatively describe isolated experimental results,37,47,48,50,51 but, until now, a conclusive and more global thermodynamic description is still lacking. Furthermore, as in any reaction, kinetic factors influence supramolecular self-assembly. Several studies show that kinetically stabilized phases can form at a surface, which over time transform into thermodynamically more stable polymorphs.49,52,53 Some of the parameters that govern kinetics and thermodynamics of molecular self-assembly are elucidated here in view of their importance for the self-assembly process.
The complexity of a thorough description of supramolecular self-assembly at surfaces stems from the intricate adsorption process itself. Once the solution is applied to a clean surface, commonly highly oriented pyrolytic graphite (HOPG), the dissolved molecular building units (analyte) start to adsorb on the substrate. They are free to move on the surface and eventually bind to other adsorbed molecules or desorb back into solution. Stable monolayers can be formed when a sufficiently large number of molecules on the surface assemble into large crystalline domains. However, a molecule adsorbed in a monolayer is not necessarily fixed in its position and may desorb back into the bulk solution. The monolayer is thus in a dynamic equilibrium with the solution and constant adsorption/desorption is likely to happen on timescales relevant for experiments.
Parameters such as adsorption/desorption rates, analyte mobility in solution and on the surface and the nucleation rates of different polymorphs can influence the kinetics of the two-dimensional crystallization. Solvation enthalpy, adsorption enthalpy and entropy changes between the dissolved state and adsorbed state of the analyte describe the energetic contribution of surface-confined molecular self-assembly and hence merge into a thermodynamic description. A microscopic picture of the adsorption/surface diffusion processes can be offered for the growth of surface nanostructures from adsorbed species.1 If the deposition rate, i.e. the rate at which molecules adsorb on the surface, is given by F and the surface diffusivity of the adsorbed molecule is given by D, the ratio D/F represents a measure of the average distance a molecule has to travel on the surface before it encounters other molecules, thus forming a nucleus for monolayer growth. A large value of D/F implies quick diffusion and slow adsorption. Equilibrium structures are the consequence, as each molecule has enough time to settle in an energetic minimum. Small values of D/F, on the other hand, result from fast adsorption and little time for diffusion. This regime would stabilize kinetically favoured structures if a metastable energetic minimum is attainable faster than the thermodynamic global minimum. How quick molecules adsorb and how fast they move on the surface defines whether the formed structure is only metastable or if it already is in its thermodynamically most preferred state.
Molecular monolayers can be observed and characterized by scanning probe techniques. The scanning tunnelling microscope (STM) has proven to be a particularly versatile tool for imaging physisorbed molecules.54–56 It allows imaging of thin molecular layers adsorbed on electrically conductive surfaces with submolecular resolution in real space and with a temporal resolution at the second-scale. Unlike diffraction techniques, the STM does not provide space-averaged information, but allows us to image spatially non-periodic structures and defects, such as grain boundaries, and simplifies the discrimination of coexisting polymorphs within a molecular layer. For instance, slow phase transitions from one polymorph into another composed of the same molecular building block can be observed in real time by STM,49,52,53 yielding important information on phase transition kinetics. Studying the dynamic behaviour of the domains of co-adsorbed molecular building blocks facilitates conclusions about the relative energetic stability of polymorphs.36,57 Likewise, grain boundary dynamics recorded by STM allows a qualitative insight into molecular interactions within a monolayer.58–61 The STM is thus an excellent tool to study the evolution of molecular monolayers and to characterize (meta)stable polymorphs.
Kinetic and thermodynamic control over 2D molecular self-assembly
Thermodynamics is concerned with energy differences between static states of a system, while kinetics includes rates of change and thus time-dependent phenomena. Transitions from one polymorph into another, under constant conditions or as a response to external stimuli, are such time-dependent processes the rate of which is determined by an activation energy. Competitive co-adsorption of different analytes and resulting dynamic changes in the relative surface coverage of each molecule is another example that is determined by kinetic factors, such as nucleation rates. Both examples will be discussed in the following sections.Kinetics and the importance of equilibration time
Successive phase transitions from one initial polymorph into a second one, followed by a transition into a final, third polymorph were observed by STM for the molecule hexakis(n-dodecyl)-peri-hexabenzocoronene (HBC-C12, Fig. 1a).52 This molecule adsorbs readily on top of an alkane (n-C50H102) monolayer adsorbed on HOPG into an oblique structure from a n-tetradecane solution (α phase, Fig. 1b). One hour after starting the experiment, only this phase is observable. Within the next hour, the oblique phase transforms into another, different polymorph (β phase, Fig. 1c). Once the transition is completed, the final polymorph (γ phase, Fig. 1d) starts to form, which is the only remaining phase after eight hours. Solvent evaporation was suppressed by using a liquid cell so that the concentration of the solute remained constant. This ensures that the transitions are not induced by an artificial increase in concentration due to solvent evaporation. The authors conclude that the phases α and β are metastable, that γ is the thermodynamically most stable polymorph and that the kinetic barrier between two phases is of the order of the thermal energy (see Fig. 1e for a potential energy diagram). The α phase is the least densely packed structure of HBC-C12, while the γ phase is the most densely packed. For this system, there is a trend towards dense-packed polymorphs as the thermodynamically stable structure; a trend that is likewise observed in the self-assembly of other molecules. Maximizing the number of molecules adsorbed on the surface minimizes energy by exchanging solvation enthalpy with adsorption enthalpy and drives self-assembly towards densely packed monolayers.![(a) The chemical structure of hexakis(n-dodecyl)-peri-hexabenzocoronene. (b) A STM topograph and molecular model of the self-assembled monolayer of HBC-C12 on n-C50H102 in the oblique α-phase. The α-phase transforms spontaneously into the β-phase (c), and subsequently into the γ-phase (d). A potential energy diagram of the transitions α → β → γ as a function of time is shown in (e). The barrier heights separating the minima of different polymorphs need to be of the order of kBT to facilitate the phase transitions. See ref. [52]. Reproduced and adapted with permission from the American Chemical Society.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f1.gif) | ||
| Fig. 1 (a) The chemical structure of hexakis(n-dodecyl)-peri-hexabenzocoronene. (b) A STM topograph and molecular model of the self-assembled monolayer of HBC-C12 on n-C50H102 in the oblique α-phase. The α-phase transforms spontaneously into the β-phase (c), and subsequently into the γ-phase (d). A potential energy diagram of the transitions α → β → γ as a function of time is shown in (e). The barrier heights separating the minima of different polymorphs need to be of the order of kBT to facilitate the phase transitions. See ref. [52]. Reproduced and adapted with permission from the American Chemical Society. | ||
Time-dependent phase transitions were also observed for octadecylcarbamic acid tetradecyl ester, a linear molecule, when assembling from phenyloctane solution onto HOPG.53 Two different polymorphs form at the surface, one of which slowly grows at the expense of the other. The authors interpret this conversion in view of Ostwald's rule of stages. This rule describes the initial formation of a kinetically favoured metastable crystal structure and its subsequent transformation into the thermodynamically most stable crystal structure. The polymorph with higher free energy is kinetically more accessible, but changes over time into the other more stable polymorph. Ostwald's rule of stages seems to be applicable not only to this study, but also gives an adequate description of the phase transformations in the HBC-C12 monolayers in the study discussed above and likewise explains the following temperature induced phase transitions.
Temperature
The same molecule assembling on Au(111) from n-tetradecane solution shows a time-dependent transition from an initial polymorph (Fig. 2a) to a second polymorph (Fig. 2b), which is completed after one hour.43 Two further transitions can be induced by progressively heating the sample to 50 °C, where at 30 °C a third phase (Fig. 2c) appears and above 35 °C, a fourth, final phase is discernible (Fig. 2d). Above 50 °C, no new phase transitions are observable. The phase transitions are not reversible, i.e., after cooling the annealed surface down to room temperature, imaging still yields the high-temperature phase. Again, care was taken to minimize solvent evaporation by using an environmental chamber saturated with n-tetradecane. The first structure that forms is not stable and rapidly transforms into the second polymorph, implying that it is kinetically favoured but thermodynamically unstable. The third phase is more stable than the second phase, but requires additional thermal energy to overcome a kinetic barrier. The same holds true for the transition from the third to the final, fourth phase. Phases two and three are metastable with respect to the fourth phase, which is supported by the non-reversibility of the transition. It is apparent from both studies on HBC-C12 that kinetic barriers that separate energetically different polymorphs can be surmounted by either waiting for a certain amount of time, or by supplying thermal energy to accelerate the transition process.![STM images of the four phases observed for HBC-C12 on Au(111) with models of the molecular structure. (a) The initial first metastable phase, which evolves rapidly into the second phase (b). Heating to 30 °C transforms the second phase into the third phase (c). Above 35 °C, the final and thermodynamically most stable phase appears (d). See ref. [43]. Reproduced and adapted with permission from the American Chemical Society.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f2.gif) | ||
| Fig. 2 STM images of the four phases observed for HBC-C12 on Au(111) with models of the molecular structure. (a) The initial first metastable phase, which evolves rapidly into the second phase (b). Heating to 30 °C transforms the second phase into the third phase (c). Above 35 °C, the final and thermodynamically most stable phase appears (d). See ref. [43]. Reproduced and adapted with permission from the American Chemical Society. | ||
Concentration
Structural modifications in molecular monolayers can be induced by changing the environment in which self-assembly takes place. Matzger and coworkers changed in situ the concentration of the solution from which the analyte, an amide with an aliphatic chain (Fig. 3a), physisorbs on graphite.49 The polymorph that is stable at high concentrations (phase II, Fig. 3b) is destabilized by the addition of pure solvent and re-assembles first into a kinetic phase (phase IV, Fig. 3c), and subsequently into the thermodynamically preferred phase (phase V, Fig. 3d). The addition of pure solvent to the solution effectively reduces the concentration of the analyte in solution, thereby changing the adsorption–desorption equilibrium. The densely packed phase II is destabilized by the addition of solvent and transforms into the polymorph that is stable at the new concentration. Although this polymorph corresponds to phase V, phase IV is formed as an intermediate polymorph, since it is kinetically more easily accessible than phase V. After equilibration times of the order of several minutes, the initially observed coexistence of all three phases disappears and phase V is the only remaining polymorph. Fig. 3e displays a reaction coordinate diagram corresponding to the transition from phase II through the intermediate phase IV and eventually settling into the most stable polymorph phase V. It depicts the relative energetic stability of each polymorph for the concentration at which the transition is observed. Since at no concentration phase IV is observed as a stable polymorph, it is concluded that it is a kinetic form and not thermodynamically stable.![(a) The chemical structure of the investigated amide. (b) The STM image and computed model of the close-packed phase II assembled from phenyloctane. (c) The porous structure IV that forms upon lowering the concentration of the analyte in solution by adding pure solvent. (d) Subsequent to the formation of phase IV, phase V assembles and remains as the thermodynamic stable polymorph. The transition is schematically shown in a reaction diagram (e), which shows phase IV as a kinetic metastable state in the transition from phase II to the energetically more favorable phase V. See ref. [49]. Reproduced and adapted with permission from the American Chemical Society.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f3.gif) | ||
| Fig. 3 (a) The chemical structure of the investigated amide. (b) The STM image and computed model of the close-packed phase II assembled from phenyloctane. (c) The porous structure IV that forms upon lowering the concentration of the analyte in solution by adding pure solvent. (d) Subsequent to the formation of phase IV, phase V assembles and remains as the thermodynamic stable polymorph. The transition is schematically shown in a reaction diagram (e), which shows phase IV as a kinetic metastable state in the transition from phase II to the energetically more favorable phase V. See ref. [49]. Reproduced and adapted with permission from the American Chemical Society. | ||
Competitive coadsorption, nucleation rates and Ostwald ripening
The importance of kinetics can also be observed in competitive adsorption of two different analytes from binary mixture solutions. This differs from the above studies, which describe the adsorption of one type of molecule on the surface and the transition of monolayers made of this building block only. Two or more molecular building blocks, on the other hand, can co-adsorb to form either mixed monolayers where the constituents co-assemble to form one polymorph,29,50,62–69 or adsorb to form separate, pure domains of polymorphs consisting of only one type of molecule.57,70,71 The latter case, for example, is observable for a binary mixture of the liquid crystal solvent 4′-octyl-4-biphenylcarbonitrile (8CB, Fig. 4a) and n-tetracontane (C40H82).57 Both molecules co-assemble on HOPG, albeit with different nucleation rates. This leads to the ordering of each component into pure domains consisting of one type of molecule. While during the initial stages the largest part of the surface is covered with domains of C40H82 and little coverage is due to 8CB, the 8CB domains grow in time at the expense of the C40H82 domains (cf. Figs 4b–e). After one to two days, the surface is almost completely covered by 8CB. It is also observed that C40H82 grows in many small domains, while 8CB assembles into large domains. The conclusion drawn from the time-dependent relative coverage is that the 8CB polymorph is thermodynamically more stable than the C40H82 polymorph, since it prevails in the long run. The authors argue that the different sizes of the domains indicate the importance of the nucleation rates and growth rates of each polymorph. Their proposed analytical model defines a ratio r = RG/RN between the growth rate, RG, and nucleation rate, RN, for both molecular types. A large ratio r for a certain molecular type leads to the formation of well-ordered domains that extend over large areas, as observed for 8CB. On the contrary, a small ratio r, when domains nucleate quickly but are slow in growth, yields many small domains, which describes the assembly of C40H82. Since in the beginning C40H82 forms many small domains while 8CB almost forms no domains, it can be inferred that RN(C40H82) > RN(8CB). Nevertheless, in the long run, the molecule with the larger growth rate should eventually cover most of the surface, just as was observed in this study where 8CB eventually displaces the C40H82 domains. A qualitative interpretation of the data in view of nucleation rates and relative thermodynamic stability explains the transition from predominant C40H82 coverage to near complete 8CB coverage. These findings can almost certainly be generalized to the other discussed studies: a fast nucleating polymorph assembles quickly on the surface, resulting in a high coverage. A different, slower nucleating polymorph, which is otherwise energetically more stable, assembles with a time lag. But since this polymorph is thermodynamically more favourable, it will eventually outgrow the fast nucleating polymorph.![(a) The chemical structure of 4′-octyl-4-biphenylcarbonitrile. (b) A monolayer of co-adsorbed 8CB and n-tetracontane that form separate pure domains. (c–e) Surface coverage of 8CB increases at the expense of n-tetracontane and small 8CB domains coalesce into larger domains. Topographs were recorded at 1.7 min time intervals. Reproduced and adapted from ref. [57] with permission from the American Chemical Society.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f4.gif) | ||
| Fig. 4 (a) The chemical structure of 4′-octyl-4-biphenylcarbonitrile. (b) A monolayer of co-adsorbed 8CB and n-tetracontane that form separate pure domains. (c–e) Surface coverage of 8CB increases at the expense of n-tetracontane and small 8CB domains coalesce into larger domains. Topographs were recorded at 1.7 min time intervals. Reproduced and adapted from ref. [57] with permission from the American Chemical Society. | ||
Similar kinetic transitions are observed for multicomponent systems.36 At the heptanoic acid/graphite interface for example, a binary mixture of trimesic acid (TMA, Fig. 5a) and 1-undecanol (Fig. 5b) shows the initial formation of pure TMA domains (Fig.5c, triangular domain in the centre of each STM image). Over time, these domains gradually dissolve and cede to a monolayer structure comprised of both TMA and 1-undecanol, which surrounds the pure domain. This bicomponent self-assembled network seems to be thermodynamically more stable than the pure TMA domain, arguably due to a higher adsorption enthalpy of the long-chain alcohol compared to TMA. Apart from the open-pore network, also not otherwise observable close-packed structures of TMA could be stabilized within the TMA/alcohol network.72 In view of these results, the kinetically preferred pure TMA structure might be favoured in the short term over the mixed structure due to a higher nucleation rate.
![The chemical structure of (a) TMA and (b) 1-undecanol. (c) STM images showing the growth of the tapes of TMA/1-undecanol SAMNs at the expense of the flower structure consisting of pure TMA. See ref. [36]. Reproduced and adapted with permission from the American Chemical Society.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f5.gif) | ||
| Fig. 5 The chemical structure of (a) TMA and (b) 1-undecanol. (c) STM images showing the growth of the tapes of TMA/1-undecanol SAMNs at the expense of the flower structure consisting of pure TMA. See ref. [36]. Reproduced and adapted with permission from the American Chemical Society. | ||
Another noteworthy observation on the dynamic behaviour of separated homomolecular domains based on kinetics is Ostwald ripening, which describes the growth of large domains at the expense of small domains.59–61,73,74 The driving force is thermodynamic in origin and is due to an evolution towards a minimum in line energy, i.e., minimizing the energy of the boundary of a domain. Molecules at the boundary are commonly less strongly bonded than in the inside of a domain due to a lower number of binding partners. Minimizing the number of molecules at the boundary with respect to the number of molecules inside a domain equals minimizing the overall energy. Since small domains have a larger ratio of molecules at the boundary, with respect to molecules in the inside, than large domains and these outer molecules detach faster than molecules from the inside due to their lower binding energy, small domains shrink while larger domains grow. The kinetic process itself – the detachment of molecules from small domains and their inclusion into larger domains – depends on the rate at which the detachment/attachment occurs. Kinetics allows an understanding of dynamic processes that occur when molecules self-assemble at the liquid/solid interface. But although time-dependent phenomena are encountered in such experiments, they do not constitute the majority of reported observations.
Thermodynamics
Stable self-assembled monolayers are studied quite often and are usually assumed to be thermodynamically controlled. The whole system comprised of surface, solution and self-assembled interfacial monolayer can be characterized in terms of the Gibbs free energy G. A minimum in this state function corresponds to the system being in thermodynamic equilibrium, i.e., the self-assembled molecular layer is in equilibrium with the solution phase. Two descriptions can be encountered in the literature for changes in the Gibbs free energy, one dependent on chemical potentials μi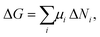 | (1a) |
| ΔG = ΔH − TΔS, | (1b) |
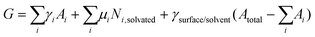 | (2) |
Two different extremes in this model are apparent. The first one corresponds to high-concentration solutions with a much larger number of molecules dissolved compared to adsorbed molecules. In this case, after the formation of a monolayer covering the whole surface, the last term in eqn (2) goes to zero. The Gibbs free energy is then minimized by a balance of the two remaining terms, defined by the chemical potential in solution μi and the surface free energy γi. Minimizing the surface free energy is achieved through the maximization of the number of molecules on the surface and a maximization of interactions of the molecule per unit area. This scenario is the one encountered most often in the literature. The second case corresponds to low concentrations of the analyte in solution. Under these conditions, the number of molecules in solution above the surface might contain fewer molecules than necessary to cover the entire substrate with a monolayer of a given polymorph. If all molecules adsorb on the surface, the second term in eqn (2) equals zero and the Gibbs free energy depends on a balance between the first and the third term, characterized by the surface free energy of the molecular layer γi and the surface free energy of the solvent/surface interface γsurface/solvent. The requirement to minimize the surface free energy yields the formation of polymorphs in which each analyte molecule maximizes its interaction in the monolayer. While in the high-concentration regime the interaction energy per unit area is an adequate measure to compare the relative stability of different polymorphs, in the low-concentration regime the molar adsorption energy is more useful. Few experiments have been reported for the latter case of low analyte concentrations.65 Samorì's group used eqn (2) to rationalize the formation of bimolecular layers from different building blocks37 and a similar interpretation regarding the adsorption energy per molecule at low analyte concentrations versus adsorption energy per unit area at high analyte concentrations was discussed by Matzger and co-workers.49,70
Concentration in a monocomponent system
The thermodynamic equilibrium of the solution/interface system can be changed by tuning parameters, such as concentration, temperature and type of solvent. De Feyter's group investigated the concentration dependence of monolayer formation using alkoxylated dehydrobenzo[12]annulenes (DBAs, Fig. 6a) at the 1,2,4-trichlorobenzene/HOPG interface.25 When assembling from solutions with high DBA concentration, the molecules form a densely packed linear motif (Fig. 6c). At low concentrations, a porous honeycomb network is formed (Fig. 6d) and both polymorphs coexist for intermediate concentrations. The relative coverage of the surface of each polymorph can thus be controlled by a deliberate choice of concentration (cf. Fig. 6b). A thermodynamic model was derived from a formula similar to eqn (2) that describes how the relative coverage correlates to concentration and temperature. Starting from the assumption of an equilibrium between the two polymorphs and the molecules in solution and assuming full coverage of the surface with molecules, a measure of the relative coverage can be expressed as | (3) |
![(a) The chemical structure of alkoxylated dehydrobenzo[12] annulenes. (b) Surface coverage θ dependence of the honeycomb structure on DBA concentration. Low concentrations favor the honeycomb polymorph. (c) STM image and molecular model of the linear structure. (d) STM image and molecular model of the honeycomb structure. See ref. [25]. Reproduced and adapted with permission from Wiley-VCH Verlag GmbH & Co. KGaA.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f6.gif) | ||
| Fig. 6 (a) The chemical structure of alkoxylated dehydrobenzo[12] annulenes. (b) Surface coverage θ dependence of the honeycomb structure on DBA concentration. Low concentrations favor the honeycomb polymorph. (c) STM image and molecular model of the linear structure. (d) STM image and molecular model of the honeycomb structure. See ref. [25]. Reproduced and adapted with permission from Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Y h is the coverage of the surface with the honeycomb structure, Yl the part of the surface covered with the linear structure, and h and l are the numbers of molecules per unit area in the respective pattern. μ0,solution, μ0,h and μ0,l are the chemical potentials of the molecules in solution, in the honeycomb and in the linear structure, respectively. As observed by experiment, this formula describes how lowering the concentration c results in increase of the ratio of hexagonal versus linear structure Yh/Yl (for l > h). The two variables concentration c and temperature T can, in principle, be used to tune the relative coverage. Eqn (3) comprises important parameters that influence the thermodynamic equilibrium and thus govern molecular self-assembly, which depends on the analyte and the solvent. The type of solvent enters the equation through the chemical potential μ0,solution, which is related to the solvation enthalpy, and μ0,h, μ0,l are characteristic for different analytes and their adsorption geometry on the surface, being related to adsorption enthalpy. The two differences (μ0,solution − μ0,h) and (μ0,solution − μ0,l) in the exponential term delineate how relative chemical potentials of each polymorph influence equilibrium conditions. Since the derivation of eqn (3) is general and is independent of the choice of analyte, solvent, or surface, it should hold in principle for all systems in which one component can assemble into two different polymorphs. The observation that the concentration can be used to control which polymorph assembles on a surface is general and has been confirmed by various other studies.24–27,47,50,51,75,76 Generally, low concentrations result in the formation of low-density structures.
Charra et al. elaborated on this model to account for their observation of a clear-cut phase transition where only one of two polymorphs is observed, but not both at the same time.48 Just like DBA in the previous study described, the molecule TSB3,5 (Fig. 7a) assembles from phenyloctane into one dense and one porous structure (Figs 7b and c). By adjusting the deposition and annealing temperatures, the composition of the monolayer could be tuned. After deposition of TSB3,5 at slightly below room temperature and annealing for one hour at 60 °C, large domains of the thermodynamically more stable densely packed structure are observed. In a control experiment, where the solution was applied to the substrate held at 60 °C and allowing equilibration for one hour, large domains of the porous structures are observed. The authors explain this finding as a result of different nucleation and growth rates of the two structures and argue that a kinetic blockade inhibits the transformation from the thermodynamically less stable to the more stable structure, even at elevated temperatures. Based on this finding, the influence of concentration on monolayer formation was studied. The samples were allowed to equilibrate at high temperature (55 °C) to reach thermodynamic equilibrium and subsequently the fraction of the surface covered with each of the two structures was measured. In contrast to the previous study by De Feyter and coworkers, the ratio did not gradually change with concentration but shows a clear phase transition at a critical concentration. Concentrations higher than the critical concentration favoured the formation of only the densely packed structure, while below the critical concentration only the porous structure is observed (Fig. 7d). Eqn (3) was modified accordingly to include this phase transition by taking into account intermolecular interactions within the monolayer. This is done by assuming that not single molecules in solution and in different polymorphs are in thermodynamic equilibrium with each other, but whole domains. This leads to the modified equation
 | (4) |
![(a) Chemical structures of TSB (R is an alkyl chain of 6, 8, 10, 12, or 14 carbon atoms). (b) STM topograph of the densely packed structure. (c) STM topograph of the porous structure. (d) The fraction of the surface covered by the porous structure as a function of concentration (red triangles) and model for different values of N (straight lines). See ref. [48]. Reproduced and adapted with permission from the American Institute of Physics.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f7.gif) | ||
| Fig. 7 (a) Chemical structures of TSB (R is an alkyl chain of 6, 8, 10, 12, or 14 carbon atoms). (b) STM topograph of the densely packed structure. (c) STM topograph of the porous structure. (d) The fraction of the surface covered by the porous structure as a function of concentration (red triangles) and model for different values of N (straight lines). See ref. [48]. Reproduced and adapted with permission from the American Institute of Physics. | ||
The newly introduced dimensionless parameter N, which enters the equation through a redefinition of the chemical potential for 2D aggregates of aggregation number N, accounts for molecular interactions that are responsible for the formation of domains rather than disordered structures. Eqn (4) reduces to eqn (3) for the case N = 1, i.e., non-interacting particles. This model now reproduces well the observed sharp transition between the two polymorphs for large numbers of N (cf. Fig. 7d). The combination of a kinetic interpretation of the nucleation and growth process, as well as the thermodynamic model that explains well the behaviour in equilibrium allow for the assembly of very large and defect free domains of either one of the two polymorphs.
Concentration in a multicomponent system
While the above treatments describe well the assembly of one component into two separate polymorphs, the assembly of two different analytes that co-adsorb into the same polymorph has been investigated by Lackinger and coworkers.50 The two carboxylic acid molecules trimesic acid (TMA, Fig. 8a) and 1,3,5-tris(4-carboxyphenyl) benzene (BTB, Fig. 8b) were shown to assemble into six different polymorphs at the solvent/graphite interface (Fig. 8c). At high TMA/BTB ratios of concentration in solution, two pure TMA polymorphs form, while in the contrary case at low TMA/BTB ratios, one pure BTB polymorph prevails. At intermediate ratios, three further structures form with a composition of TMA and BTB that is governed by the relative amount of TMA and BTB molecule in solution.![The chemical structures of (a) TMA and (b) BTB. (c) STM topographs and molecular models of the six different polymorphs A–F that assemble at different concentrations. (d) A phase diagram of the polymorphs A–F. The concentration of TMA is represented by the abscissa, the concentration of BTB by the ordinate. See ref. [50]. Reproduced and adapted with permission from the American Chemical Society.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f8.gif) | ||
| Fig. 8 The chemical structures of (a) TMA and (b) BTB. (c) STM topographs and molecular models of the six different polymorphs A–F that assemble at different concentrations. (d) A phase diagram of the polymorphs A–F. The concentration of TMA is represented by the abscissa, the concentration of BTB by the ordinate. See ref. [50]. Reproduced and adapted with permission from the American Chemical Society. | ||
Again, a thermodynamic model based on chemical potentials μi and number of molecules per unit area in a certain phase σi with i = TMA,BTB was used to rationalize these findings:
| G = [σTMA (structure)μTMA (cTMA) + σBTB(structure)μBTB(cBTB) + esolventρcavity(structure)]A | (5) |
The area of the covered surface is A, while ρcavity is the relative cavity area portion and esolvent is an additional free energy per unit area. To derive this formula, the authors approximated the number of molecules in solution as constant and unaffected by the adsorption molecules on the surface. This is justified by the much larger number of molecules in solution compared to the number of molecules at the surface. The second term in eqn (2) thus becomes constant in the calculation of differences in the Gibbs free energy. In eqn (5), apart from the area density σi (number of molecules per unit area, see l and h in the above model) in a given structure and the chemical potential μi in this structure, an additional third term is introduced to takes into account possible solvent co-adsorption in the cavities of the porous structures. This term can thus be related to the third term in eqn (2). Although often no solvent or guest co-adsorption inside cavities of two-dimensional monolayers can be resolved by STM, likely due to short resident times of the molecules in the cavity and their high mobility, co-adsorption is considered to be important in the stabilization of the monolayers. Examples where guest coadsorption in cavities induce changes in the monolayer structure and stabilize otherwise unstable polymorphs underpin the importance of this assumption31,77–81 and corroborate the empirical inclusion of the third term. Eqn (5) was used to calculate a phase diagram for TMA/BTB adsorption, which agrees well with experiment (Fig. 8d). The dependence of the monolayer structure on the relative concentration of TMAversusBTB is adequately described by the model and accurately predicts the transition from pure TMA polymorphs to the pure BTB, with three intermediate ones in which both molecules co-assemble when the relative concentration of TMAversusBTB is decreased.
Temperature
The experimental verification of the above discussed models focused primarily on the effect of analyte concentration in solution. However, it is apparent from, for example, eqn (3) that not only concentration but also temperature can be used to control the self-assembly of molecules. This parameter was addressed by Lackinger and co-workers using BTB (Fig. 9a) as the analyte.47BTB assembles from saturated fatty acid solutions into two different polymorphs at room temperature; a dense-packed row structure when heptanoic acid is used as solvent (similar to Fig. 9c), and a hexagonal porous structure when nonanoic acid is used (Fig. 9b). Octanoic acid yields both polymorphs in coexistence. Increasing the temperature at which the molecules adsorb from octanoic acid to about 43 °C resulted in the assembly of the row structure only. This transition is reversible and, after cooling the sample to room temperature, the hexagonal structure reappeared. A similar behavior was found for nonanoic acid, for which the transition from hexagonal to row structure takes place at about 55 °C (see Figs 9b–g for several heat/cool cycles). Both polymorphs, row and hexagonal structure, are assumed to be thermodynamically stable under given conditions and the reversibility of the transition evidences that the low-temperature hexagonal structure is not merely a kinetically trapped polymorph. This stands in contrast to the non-reversible temperature-driven phase transitions observed by Fichou et al.43 (vide supra), where kinetically stable polymorphs are transformed into thermodynamically stable ones. The thermodynamic model that describes the reversible transition depends on a detailed examination of enthalpic and entropic contributions to the Gibbs free energy in the form of eqn (1b). Molecules that assemble into a static structure on a surface may lower their enthalpy by trading solvation enthalpy for adsorption enthalpy. While this decrease in enthalpy favours the formation of monolayers by minimizing the Gibbs free energy, the loss in entropy upon adsorption acts against it. A molecule in solution possesses translational and rotational degrees of freedom, which are lost, in part, when the molecule sits in a fixed position on a surface. The reduction of degrees of freedom implies a decrease in entropy, which consequently raises the free energy of the system. The loss in entropy at a given temperature has to be compensated for by an adequate reduction in enthalpy for monolayer formation to spontaneously occur. The authors approximate changes in enthalpy through molecular mechanics calculations, a method commonly employed along with ab initio calculations to compute the relative energetic stability of different polymorphs.27,37,82 Changes in entropy are computed using formulas such as the Sackur–Tetrode equation. This approach allows for the quantitative estimation of differences in the Gibbs free energy for the two phases of BTB and yields the right trend for the phase transition. The transition can only be fully understood using this model if solvent coadsorption is taken into account, which also explains to some extent the solvent-dependent critical temperature at which the transition takes place. Furthermore, analyte concentration in solution was shown to affect the transition and no phase transition at elevated temperatures could be observed in a diluted solution of BTB in nonanoic acid. The model based on detailed estimates of enthalpic and entropic factors to the Gibbs free energy offers an alternative way to the models based on eqn (1a), although both approaches should give the same results. However, quantitative exact considerations have not been reported so far and the modelling of thermodynamics of adsorption in supramolecular chemistry still calls for further research and refinement of models.![(a) The chemical structure of BTB. (b–g) Successively recorded STM images of BTB monolayers at the nonanoic acid/graphite interface at various temperatures. (b) The hexagonal porous structure at room temperature. (c) The row structure at about 55 °C. A new cooling to room temperature results in the hexagonal polymorph (d). Repeated heating and cooling cycles are depicted in (e) to (g). See ref. [47]. Reproduced and adapted with permission from the American Chemical Society.](/image/article/2011/SC/c1sc00531f/c1sc00531f-f9.gif) | ||
| Fig. 9 (a) The chemical structure of BTB. (b–g) Successively recorded STM images of BTB monolayers at the nonanoic acid/graphite interface at various temperatures. (b) The hexagonal porous structure at room temperature. (c) The row structure at about 55 °C. A new cooling to room temperature results in the hexagonal polymorph (d). Repeated heating and cooling cycles are depicted in (e) to (g). See ref. [47]. Reproduced and adapted with permission from the American Chemical Society. | ||
Conclusions
Interpretations based on the kinetics and thermodynamics of molecular self-assembly at surfaces have been discussed in several studies and help researchers to understand experimental results. Analytic thermodynamic models are being developed to improve our understanding of supramolecular chemistry in two dimensions. These models might lay the foundation for a more rational approach towards the design of experiments. Time and temperature-dependent irreversible phase transitions have been identified as a transformation of kinetically favourable to thermodynamically stable polymorphs. Nucleation and growth rates have been proposed to be the underlying factors responsible for these observations and naturally lead to a tentative adaptation of Ostwald's rule of stages for two-dimensional supramolecular monolayer growth. Competitive coadsorption of different molecules that assemble into separate domains can be kinetically controlled, as exemplified by the transition from mixed monolayers in which domains of both polymorphs are present to monolayers that almost exclusively contain only one kind of molecule. The fundamental driving force for all of these transitions that are kinetically controlled is the minimization of free energy and can thus be described on thermodynamic grounds. Several models, from purely qualitative to semi-quantitative considerations, are emerging in the literature. They share a detailed description of the analyte in solution and on the surface in terms of chemical potentials, enthalpy and entropy. Several experiments on concentration- and temperature-dependent polymorphism were successfully interpreted on the basis of these models, which could function as a starting point for future refinement. Many parameters that govern molecular self-assembly are still lacking a thorough theoretical and even experimental treatment, such as, for example, the type of solvent and accompanying factors, like solvation enthalpy for thermodynamic considerations, or viscosity and diffusion rates for kinetic considerations. A different, complementary approach towards the understanding of molecular self-assembly is based on Monte Carlo Methods.83–86 The information gained from this computational technique allows the prediction of which polymorph forms under certain conditions. Furthermore, it might help to understand processes such as nucleation and growth, which are too fast to be observed by STM. Two other prominent examples where a thermodynamic and kinetic insight can describe and improve experimental results are random networks/2D glassy systems87–89 and the synthesis of ordered surface-confined 2D polymers.90–92Experimental planning can benefit from a deeper understanding of the underlying mechanisms that control molecular self-assembly and deliberate two-dimensional crystal engineering will be one step closer once kinetics and thermodynamics are fathomed and controlled.
Acknowledgements
Our work is supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) through a Discovery Grant, as well as the Fonds Quebecois sur la Recherche en Nature et Technologies (FQRNT) through a Team Grant and the Ministere du Developpement Economique, de l′Innovation et de l′Exportation (MDEIE) through an international collaboration Grant. F. R. is grateful to the Canada Research Chairs program for partial salary support.Notes and references
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671 CrossRef CAS.
- J. A. A. W. Elemans and S. De Feyter, Soft Matter, 2009, 5, 721 RSC.
- A. Ciesielski, C.-A. Palma, M. Bonini and P. Samorì, Adv. Mater., 2010, 22, 3506 CrossRef CAS.
- M. Surin and P. Samorì, Small, 2007, 3, 190 CrossRef CAS.
- S. Stepanow, N. Lin and J. V. Barth, J. Phys.: Condens. Matter, 2008, 20, 184002 CrossRef.
- T. Kudernac, S. B. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402 RSC.
- A. Kühnle, Curr. Opin. Colloid Interface Sci., 2009, 14, 157 CrossRef.
- M. Lackinger and W. M. Heckl, Langmuir, 2009, 25, 11307 CrossRef CAS.
- Y. L. Yang and C. Wang, Chem. Soc. Rev., 2009, 38, 2576 RSC.
- Y. L. Yang and C. Wang, Curr. Opin. Colloid Interface Sci., 2009, 14, 135 CrossRef CAS.
- L. Bartels, Nat. Chem., 2010, 2, 87 CrossRef CAS.
- S. Uemura, R. Tanoue, N. Yilmaz, A. Ohira and M. Kunitake, Materials, 2010, 3, 4252 CrossRef CAS.
- C.-A. Palma, M. Bonini, T. Breiner and P. Samorì, Adv. Mater., 2009, 21, 1383 CrossRef CAS.
- A. G. Slater, P. H. Beton and N. R. Champness, Chem. Sci., 2011, 2, 1440 RSC.
- L. Cardenas, J. Lipton-Duffin and F. Rosei, Jpn. J. Appl. Phys., 2011, 50, 08LA02 CrossRef.
- J. S. Foster and J. E. Frommer, Nature, 1988, 333, 542–545 CrossRef.
- J. P. Rabe and S. Buchholz, Phys. Rev. Lett., 1991, 66, 2096 CrossRef CAS.
- J. P. Rabe and S. Buchholz, Science, 1991, 253, 424 CAS.
- F. Jäckel, M. Ai, J. S. Wu, K. Müllen and J. P. Rabe, J. Am. Chem. Soc., 2005, 127, 14580 CrossRef.
- X. Shao, X. C. Luo, X. Q. Hu and K. Wu, J. Phys. Chem. B, 2006, 110, 1288 CrossRef CAS.
- L. Kampschulte, M. Lackinger, A. K. Maier, R. S. K. Kishore, S. Griessl, M. Schmittel and W. M. Heckl, J. Phys. Chem. B, 2006, 110, 10829 CrossRef CAS.
- W. Mamdouh, H. Uji-i, J. S. Ladislaw, A. E. Dulcey, V. Percec, F. C. De Schryver and S. De Feyter, J. Am. Chem. Soc., 2006, 128, 317 CrossRef CAS.
- Y. B. Li, Z. Ma, G. C. Qi, Y. L. Yang, Q. D. Zeng, X. L. Fan, C. Wang and W. Huang, J. Phys. Chem. C, 2008, 112, 8649 CAS.
- X. R. Miao, L. Xu, Z. M. Li and W. L. Deng, J. Phys. Chem. C, 2011, 115, 3358 CAS.
- S. Lei, K. Tahara, F. C. De Schryver, M. Van der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., 2008, 120, 3006 CrossRef.
- K. Tahara, S. Okuhata, J. Adisoejoso, S. B. Lei, T. Fujita, S. De Feyter and Y. Tobe, J. Am. Chem. Soc., 2009, 131, 17583 CrossRef CAS.
- C. Meier, M. Roos, D. Künzel, A. Breitruck, H. E. Hoster, K. Landfester, A. Gross, R. J. Behm and U. Ziener, J. Phys. Chem. C, 2010, 114, 1268 CAS.
- S. B. Lei, C. Wang, S. X. Yin, H. N. Wang, F. Xi, H. W. Liu, B. Xu, L. J. Wan and C. L. Bai, J. Phys. Chem. B, 2001, 105, 10838 CrossRef CAS.
- L. Kampschulte, S. Griessl, W. M. Heckl and M. Lackinger, J. Phys. Chem. B, 2005, 109, 14074 CrossRef CAS.
- K. Tahara, S. Furukawa, H. Uji-i, T. Uchino, T. Ichikawa, J. Zhang, W. Mamdouh, M. Sonoda, F. C. De Schryver, S. De Feyter and Y. Tobe, J. Am. Chem. Soc., 2006, 128, 16613 CrossRef CAS.
- S. Lei, M. Surin, K. Tahara, J. Adisoejoso, R. Lazzaroni, Y. Tobe and S. De Feyter, Nano Lett., 2008, 8, 2541 CrossRef CAS.
- S.-L. Lee, N.-T. Lin, W.-C. Liao, C.-h. Chen, H.-C. Yang and T.-Y. Luh, Chem.–Eur. J., 2009, 15, 11594 CrossRef CAS.
- R. Gutzler, S. Lappe, K. Mahata, M. Schmittel, W. M. Heckl and M. Lackinger, Chem. Commun., 2009, 680 RSC.
- X. Zhang, T. Chen, H. J. Yan, D. Wang, Q. H. Fan, L. J. Wan, K. Ghosh, H. B. Yang and P. J. Stang, ACS Nano, 2010, 4, 5685 CrossRef CAS.
- M. Lackinger, S. Griessl, T. Markert, F. Jamitzky and W. M. Heckl, J. Phys. Chem. B, 2004, 108, 13652 CrossRef CAS.
- K. G. Nath, O. Ivasenko, J. M. MacLeod, J. A. Miwa, J. D. Wuest, A. Nanci, D. F. Perepichka and F. Rosei, J. Phys. Chem. C, 2007, 111, 16996 CAS.
- C.-A. Palma, J. Björk, M. Bonini, M. S. Dyer, A. Llanes-Pallas, D. Bonifazi, M. Persson and P. Samorì, J. Am. Chem. Soc., 2009, 131, 13062 CrossRef CAS.
- S. Cincotti and J. P. Rabe, Appl. Phys. Lett., 1993, 62, 3531 CrossRef CAS.
- S. Cincotti and J. P. Rabe, Supramol. Sci., 1994, 1, 7 CrossRef CAS.
- L. C. Giancarlo, H. B. Fang, S. M. Rubin, A. A. Bront and G. W. Flynn, J. Phys. Chem. B, 1998, 102, 10255 CrossRef CAS.
- Z. X. Xie, X. Xu, B. W. Mao and K. Tanaka, Langmuir, 2002, 18, 3113 CrossRef CAS.
- Z. Q. Zou and F. Chen, J. Appl. Phys., 2008, 103, 094304 CrossRef.
- C. Marie, F. Silly, L. Tortech, K. Müllen and D. Fichou, ACS Nano, 2010, 4, 1288 CrossRef CAS.
- L. Askadskaya and J. P. Rabe, Phys. Rev. Lett., 1992, 69, 1395 CrossRef CAS.
- X. H. Kong, K. Deng, Y. L. Yang, Q. D. Zeng and C. Wang, J. Phys. Chem. C, 2007, 111, 9235 CAS.
- W. A. English and K. W. Hipps, J. Phys. Chem. C, 2008, 112, 2026 CAS.
- R. Gutzler, T. Sirtl, J. F. Dienstmaier, K. Mahata, W. M. Heckl, M. Schmittel and M. Lackinger, J. Am. Chem. Soc., 2010, 132, 5084 CrossRef CAS.
- A. Bellec, C. Arrigoni, G. Schull, L. Douillard, C. Fiorini-Debuisschert, F. Mathevet, D. Kreher, A. J. Attias and F. Charra, J. Chem. Phys., 2011, 134, 124702 CrossRef.
- S. Ahn and A. J. Matzger, J. Am. Chem. Soc., 2010, 132, 11364 CrossRef CAS.
- L. Kampschulte, T. L. Werblowsky, R. S. K. Kishore, M. Schmittel, W. M. Heckl and M. Lackinger, J. Am. Chem. Soc., 2008, 130, 8502 CrossRef CAS.
- S. B. Lei, K. Tahara, F. C. De Schryver, M. Van der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2008, 47, 2964 CrossRef CAS.
- L. Piot, A. Marchenko, J. S. Wu, K. Müllen and D. Fichou, J. Am. Chem. Soc., 2005, 127, 16245 CrossRef CAS.
- K. Kim, K. E. Plass and A. J. Matzger, Langmuir, 2003, 19, 7149 CrossRef CAS.
- S. De Feyter and F. C. De Schryver, J. Phys. Chem. B, 2005, 109, 4290 CrossRef CAS.
- F. Rosei, M. Schunack, Y. Naitoh, P. Jiang, A. Gourdon, E. Laegsgaard, I. Stensgaard, C. Joachim and F. Besenbacher, Prog. Surf. Sci., 2003, 71, 95 CrossRef CAS.
- J. V. Barth, Annu. Rev. Phys. Chem., 2007, 58, 375 CrossRef CAS.
- R. T. Baker, J. D. Mougous, A. Brackley and D. L. Patrick, Langmuir, 1999, 15, 4884 CrossRef CAS.
- A. Stabel, R. Heinz, J. P. Rabe, G. Wegner, F. C. De Schryver, D. Corens, W. Dehaen and C. Sueling, J. Phys. Chem., 1995, 99, 8690 CrossRef CAS.
- P. Samorì, K. Müllen and H. P. Rabe, Adv. Mater., 2004, 16, 1761 CrossRef.
- M. Lackinger, S. Griessl, L. Kampschulte, F. Jamitzky and W. M. Heckl, Small, 2005, 1, 532 CrossRef CAS.
- G. M. Florio, J. E. Klare, M. O. Pasamba, T. L. Werblowsky, M. Hyers, B. J. Berne, M. S. Hybertsen, C. Nuckolls and G. W. Flynn, Langmuir, 2006, 22, 10003 CrossRef CAS.
- K. G. Nath, O. Ivasenko, J. A. Miwa, H. Dang, J. D. Wuest, A. Nanci, D. F. Perepichka and F. Rosei, J. Am. Chem. Soc., 2006, 128, 4212 CrossRef CAS.
- S. L. Xu, M. D. Dong, E. Rauls, R. Otero, T. R. Linderoth and F. Besenbacher, Nano Lett., 2006, 6, 1434 CrossRef CAS.
- W. Mamdouh, R. E. A. Kelly, M. D. Dong, L. N. Kantorovich and F. Besenbacher, J. Am. Chem. Soc., 2008, 130, 695 CrossRef CAS.
- C.-A. Palma, M. Bonini, A. Llanes-Pallas, T. Breiner, M. Prato, D. Bonifazi and P. Samorì, Chem. Commun., 2008, 5289 RSC.
- M. Li, Y. L. Yang, K. Q. Zhao, Q. D. Zeng and C. Wang, J. Phys. Chem. C, 2008, 112, 10141 CAS.
- Y. B. Li, Z. Ma, K. Deng, S. B. Lei, Q. D. Zeng, X. L. Fan, S. De Feyter, W. Huang and C. Wang, Chem.–Eur. J., 2009, 15, 5418 CrossRef CAS.
- H. Walch, A. K. Maier, W. M. Heckl and M. Lackinger, J. Phys. Chem. C, 2009, 113, 1014 CAS.
- S. Xu, S. Yin and S. Xu, Appl. Phys. A: Mater. Sci. Process., 2010, 99, 99 CrossRef CAS.
- K. Kim, K. E. Plass and A. J. Matzger, J. Am. Chem. Soc., 2005, 127, 4879 CrossRef CAS.
- S. B. Lei, K. Tahara, Y. Tobe and S. De Feyter, Chem. Commun., 2010, 46, 9125 RSC.
- J. M. MacLeod, O. Ivasenko, D. F. Perepichka and F. Rosei, Nanotechnology, 2007, 18, 424031 CrossRef CAS.
- M. Bonini, L. Zalewski, T. Breiner, F. Dötz, M. Kastler, V. Schädler, M. Surin, R. Lazzaroni and P. Samorì, Small, 2009, 5, 1521 CrossRef CAS.
- R. Heinz, A. Stabel, F. C. De Schryver and J. P. Rabe, J. Phys. Chem., 1995, 99, 505 CrossRef CAS.
- J. Adisoejoso, K. Tahara, S. Okuhata, S. Lei, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2009, 48, 7353 CrossRef CAS.
- S. B. Lei, K. Tahara, J. Adisoejoso, T. Balandina, Y. Tobe and S. De Feyter, CrystEngComm, 2010, 12, 3369–3381 RSC.
- S. Furukawa, K. Tahara, F. C. De Schryver, M. Van der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2007, 46, 2831 CrossRef CAS.
- D. Bleger, D. Kreher, F. Mathevet, A. J. Attias, G. Schull, A. Huard, L. Douillard, C. Fiorini-Debuischert and F. Charra, Angew. Chem., Int. Ed., 2007, 46, 7404 CrossRef CAS.
- M. Blunt, X. Lin, M. C. Giménez-López, M. Schröder, N. R. Champness and P. H. Beton, Chem. Commun., 2008, 2304 RSC.
- X. R. Miao, L. Xu, Y. J. Li, Z. M. Li, J. Zhou and W. L. Deng, Chem. Commun., 2010, 46, 8830 RSC.
- M. O. Blunt, J. C. Russell, M. C. Giménez-López, N. Taleb, X. Lin, M. Schröder, N. R. Champness and P. H. Beton, Nat. Chem., 2011, 3, 74 CrossRef CAS.
- J. M. MacLeod, O. Ivasenko, C. Y. Fu, T. Taerum, F. Rosei and D. F. Perepichka, J. Am. Chem. Soc., 2009, 131, 16844 CrossRef CAS.
- F. Silly, U. K. Weber, A. Q. Shaw, V. M. Burlakov, M. R. Castell, G. A. D. Briggs and D. G. Pettifor, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 201408 CrossRef.
- U. K. Weber, V. M. Burlakov, L. M. A. Perdigão, R. H. J. Fawcett, P. H. Beton, N. R. Champness, J. H. Jefferson, G. A. D. Briggs and D. G. Pettifor, Phys. Rev. Lett., 2008, 100, 156101 CrossRef CAS.
- C. Rohr, M. Balbás Gambra, K. Gruber, E. C. Constable, E. Frey, T. Franosch and B. A. Hermann, Nano Lett., 2010, 10, 833 CrossRef CAS.
- S. Lei, K. Tahara, K. Müllen, P. Szabelski, Y. Tobe and S. De Feyter, ACS Nano, 2011, 5, 4145 CrossRef CAS.
- M. O. Blunt, J. C. Russell, M. C. Giménez-López, J. P. Garrahan, X. Lin, M. Schröder, N. R. Champness and P. H. Beton, Science, 2008, 322, 1077 CrossRef CAS.
- R. Otero, M. Lukas, R. E. A. Kelly, W. Xu, E. Laegsgaard, I. Stensgaard, L. N. Kantorovich and F. Besenbacher, Science, 2008, 319, 312 CrossRef CAS.
- M. Marschall, J. Reichert, A. Weber-Bargioni, K. Seufert, W. Auwarter, S. Klyatskaya, G. Zoppellaro, M. Ruben and J. V. Barth, Nat. Chem., 2010, 2, 131 CrossRef CAS.
- D. F. Perepichka and F. Rosei, Science, 2009, 323, 216–217 CrossRef CAS.
- R. Tanoue, R. Higuchi, N. Enoki, Y. Miyasato, S. Uemura, N. Kimizuka, A. Z. Stieg, J. K. Gimzewski and M. Kunitake, ACS Nano, 2011, 5, 3923 CrossRef CAS.
- M. Bieri, M. T. Nguyen, O. Gröning, J. M. Cai, M. Treier, K. Ait -Mansour, P. Ruffieux, C. A. Pignedoli, D. Passerone, M. Kastler, K. Müllen and R. Fasel, J. Am. Chem. Soc., 2010, 132, 16669 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |