Powering reductive charge shift reactions—linking fullerenes of different electron acceptor strength to secure an energy gradient†
Carmen
Villegas
a,
Juan Luis
Delgado
ab,
Pierre-Antoine
Bouit
b,
Bruno
Grimm
c,
Wolfgang
Seitz
c,
Nazario
Martín
*ab and
Dirk M.
Guldi
*c
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Ciudad Universitaria s/n, 28040, Madrid, Spain. E-mail: nazmar@quim.ucm.es; Fax: (+34) 91-394-4103; Tel: (+34) 91-394-4227
bIMDEA-Nanociencia, Facultad de Ciencias, Módulo C-IX, 3a planta, Ciudad Universitaria de Cantoblanco, 28049, Madrid, Spain
cDepartment of Chemistry and Pharmacy & Interdisciplinary Center of Molecular Materials (ICMM) Friedrich-Alexander-University Erlangen-Nuremberg Egerlandstr. 3, 91058, Erlangen, Germany. E-mail: dirk.guldi@chemie.uni-erlangen.de; Fax: (+49) 9131-85-28307
First published on 29th June 2011
Abstract
We report here on one of the rare cases that realizes a reductive charge shift from a primary electron acceptor (C60) to a secondary electron acceptor (C70) in a ZnP–C60–C70 conjugate. Initially, charge separation evolves from an excited state electron donor (ZnP). By far the most far reaching benefit from such a reductive pathway is a marked slow down of the energy wasting charge recombination when compared to the ZnP–C60 conjugate (i.e., picoseconds versus nanoseconds).
Introduction
Nature employs a myriad of interlocking principles to create sophisticated and amazing architectures, in which a formidable level of control over organizing individual building blocks and replacing them has been realized. To this end, the structure of the bacterial photosynthetic reaction centers provides important incentives for arranging electron donors and electron acceptors. Sunlight is harvested by antenna chlorophylls before it is unidirectionally funneled to the special “bacteriochlorophyll dimer” pair. Here, excited state interactions in the form of an electron transfer yield an oxidized special “bacteriochlorophyll dimer”, while the electrons are inserted into a redox chain. Importantly, a directed cascade of electron transfer reactions mediates the electrons through the well-balanced redox gradients in the redox chain and retards the charge separation.1Fullerenes are readily available and exhibit exciting characteristics.2 For example, the delocalization of charges within the giant, spherical carbon framework together with the rigid, confined structure of the π-sphere offers unique opportunities for stabilizing charged entities.3 Above all, the small reorganization energies of fullerenes in charge-transfer reactions have led to a notable breakthrough in multicomponent electron donor–acceptor systems by providing accelerated charge separation and decelerated charge recombination.4 In most of such multicomponent systems, photoexcitation is the inception to reduce the electron accepting fullerenes and to generate a hole at the electron donor. A redox gradient governs a hole transfer from the primary electron donor to the secondary electron donor, etc.5
In general, there have been only a small number of multicomponent electron donor–acceptor systems, in which a unidirectional flow of electrons evolves and, in turn, mediates electrons between different electron acceptors rather than holes between different electron donors.6 This is what the current work addresses. In particular, we present a paradigm shift by using two fullerenes that are covalently linked to each other and an excited state electron donating ZnP—see Chart 1. To ensure an unambiguous characterization of the electron transfer pathways, two different fullerenes, that is, C60 and C70, were chosen. Implementation of a redox gradient was realized by using different fullerene functionalization. In particular, a fulleropyrrolidine of C60 and a fulleropyrazoline of C70—to improve the electron acceptor character—served as primary and secondary electron acceptor, respectively.
![Structure of ZnP–C60–C70 (1), C60–C70 (2), ZnP azide (3), formyl containing 2-pyrazolino[70]fullerene (4) and ZnP–C60 (5). Compounds 1, 2, 4 and 5 were obtained as isomeric mixtures.](/image/article/2011/SC/c1sc00331c/c1sc00331c-c1.gif) | ||
| Chart 1 Structure of ZnP–C60–C70 (1), C60–C70 (2), ZnP azide (3), formyl containing 2-pyrazolino[70]fullerene (4) and ZnP–C60 (5). Compounds 1, 2, 4 and 5 were obtained as isomeric mixtures. | ||
Results and discussion
The synthesis of 1 has been carried out by means of copper catalyzed “click-chemistry” of fullerene dimer (2) with zinc porphyrine azide (3)7 in moderate yields (40%).82, on the other hand, was formed by reacting formyl containing 2-pyrazolino[70]fullerene (4)9 with 2-propargyl-glycine and C60 (57% yield).10 Finally, as a reference system, we have synthesized 5 bearing only one fragment of 1, following a similar methodology.8,11 It is worth mentioning that compounds 1, 2, and 4 were obtained as a mixture of isomers stemming from the difference site and regioselectivity in the cycloaddition reaction on the C70fullerene.121 displays in THF (Figure S1†) the absorption characteristics of C60 and C70 at 253/315 and 360/465 nm, respectively. The presence of the Soret- and Q-bands of ZnP at 426 and 557/597 nm complete the characterization of 1. A closer inspection reveals, however, that the absorption spectrum of 1 fails to resemble the sum of the individual spectra, that is, 2 and 3.
Electrochemistry
The electrochemical properties of 1–3 have been investigated by means of room temperature cyclic voltammetry/osteryoung square wave voltammetry experiments (Figure S3, Table S1†). For 3, the first oxidation process occurs at 0.52 to yield the ZnP radical cation. The ZnP dication, on the other hand is formed at 0.90 V. Reduction processes at −1.84 and −2.00 V relate to the reduction of ZnP to the corresponding radical anion and dianion, respectively. At −1.62 V, the reduction of the azide fragment set in. In principle, C60 and C70 exhibit nearly identical reduction processes.13 In 2, however, the electron withdrawing effects of the pyrazoline ring14 shifts the C70reduction processes of the fulleropyrazoline to −0.72, −1.13, −1.50, and −1.87 V. The C60reduction processes of the fulleropyrrolidine are cathodically shifted that are detectable at −0.85, −1.26, −1.64, and −2.02 V. 1 shows two oxidation processes at 0.52 and 0.89 V to lead to the ZnP radical cation and ZnP dication. On the reductive side, several redox processes are noted. Whereas the reduction of the fulleropyrazoline of C70 involves processes at −0.72, −1.13, −1.52, and −1.86 V, reduction of the fulleropyrrolidine of C60 is seen at −0.85, −1.25, −1.62, and −2.01 V generating the C70/C60radical anion, the C70/C60 dianion, the C70/C60 trianion, and the C70/C60tetraanion, respectively. The reduction of ZnP to the corresponding radical anion and dianion at −1.86 and −2.01 V, respectively, and the reduction of the triazole at −1.62 V are superimposed to the reduction processes of C60 and C70 and intensity the reductive currents in the corresponding regions.Photophysical studies
Transient absorption measurements in THF with the references were performed in the following order—ZnP (3), pyrrolidino[60]fullerene, and pyrazolino[70]fullerene (4)—before turning to the C60–C70 (2) and ZnP–C60 conjugates (5) and, finally, to the ZnP–C60–C70 conjugate (1).When photoexciting the ZnP reference (3) at 420 or 550 nm, within 2.0 ps the higher singlet excited state features of ZnP transform into those of the lowest singlet excited state. Specifically, characteristic maxima at 455, 580, and 630 nm as well as minima at 565 and 605 nm are clear attributes of the singlet–singlet absorptions of ZnP.15 On a time-scale of up to 3.0 ns, the fate of the ZnP singlet excited state is dominated by intersystem crossing—driven by a strong spin–orbit coupling—to afford the corresponding triplet excited state (2.1 ns–4.8 × 108 s−1). The latter was identified by a long-lived (100 μs–1.0 × 104 s−1) and strongly absorbing triplet–triplet transition that maximizes at 840 nm.16
Excitation of the pyrrolidino[60]fullerene and pyrazolino[70]fullerene references at 387 nm or 550 nm (not shown) populates their singlet excited states with characteristic absorption maxima at 610/920 and 610/1290 nm, respectively. Both singlet excited states undergo intersystem crossing to the corresponding triplet manifolds with characteristic lifetimes of 1.4 ns (7.0 × 108 s−1) for C60 and 0.97 ns (1.03 × 109 s−1) for C70 as well as characteristic maxima at 700 and 970 nm.17
When conjugating C60 with C70, the transient absorption features that were recorded immediately upon photoexcitation of (2) are best described as the superimposition of the C60 and the C70 singlet excited states featuring maxima at 610/920 and 610/1290 nm, respectively. Particularly interesting is the fact that the C60 singlet excited state decays in the presence of C70 with dynamics (15 ps–6.0 × 1010 s−1) that are appreciably faster than those seen in the C60 reference. Owing to the shear impossibility of oxidizing C60 or C70 in the photochemical processes, the aforementioned seems to prompt an intramolecular transduction of singlet excited state energy from C60 to C70. In fact, the sole photoproduct decays with dynamics that resembles those seen for the intersystem crossing in the C70 reference (0.97 ns–1.03 × 109 s−1). Crucial support for the energy transfer notion comes from the observation that the only triplet excited state seen at time delays of 3.0 ns and beyond was that of C70 with its fingerprint absorption at 970 nm.18 Our transient absorption measurements failed, on the other hand, to provide for 2 any appreciable evidence for the 700 nm maximum of the C60 triplet excited state.19
Next, the transient absorption changes of the ZnP–C60 conjugate (5) were recorded with several time delays after the 550 nm laser pulse and compared with those seen for the ZnP reference—see Fig. 1. At early times, these are practically identical to those of the ZnP reference, disclosing a strong maximum at 455 nm and a strong bleaching at 565 nm. However, at delay times of around 10 ps, the 455 nm absorption starts to decay and simultaneously a new transition grows in the visible, that is, a maximum around 630 nm (50 ps–2.0 × 1010 s−1), which is accompanied by a near-infrared feature, namely a 1005 nm maximum. Based on a spectral comparison with former spectroelectrochemical and pulse radiolytical investigations,20 we ascribe the earlier band to the ZnP π-radical cation (ZnP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ˙+), while the latter band corresponds to the C60 π-radical anion (C60˙−). In accordance with these results, we propose that charge separation evolves from the ZnP singlet excited state to the electron accepting fullerene to yield ZnP˙+–C60˙−. The absorption of the ZnP˙+–C60˙− state decays in the nanosecond regime (1.3 ns–7.6 × 108 s−1). Notable is that the semiflexible spacer connecting ZnP with C60 provides ample room for configurational changes in the ZnP–C60 conjugate. Based on previous studies, such changes in configuration are likely to be the inception for through space rather than through bond electron transfer interactions.21
˙+), while the latter band corresponds to the C60 π-radical anion (C60˙−). In accordance with these results, we propose that charge separation evolves from the ZnP singlet excited state to the electron accepting fullerene to yield ZnP˙+–C60˙−. The absorption of the ZnP˙+–C60˙− state decays in the nanosecond regime (1.3 ns–7.6 × 108 s−1). Notable is that the semiflexible spacer connecting ZnP with C60 provides ample room for configurational changes in the ZnP–C60 conjugate. Based on previous studies, such changes in configuration are likely to be the inception for through space rather than through bond electron transfer interactions.21
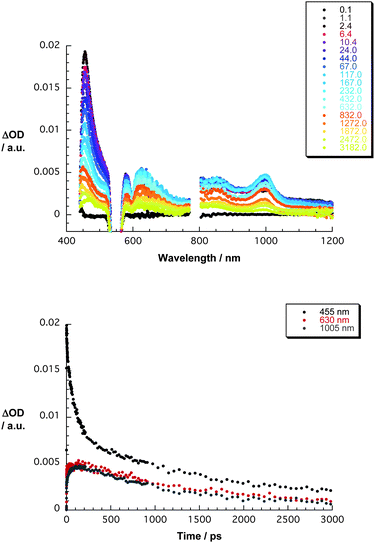 | ||
| Fig. 1 Upper part—differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (550 nm) of ZnP–C60 in THF with several time delays between 0 and 3000 ps at room temperature—see figure legend for time delays. Lower part—time-absorption profiles of the spectra shown in the upper and central parts at 455, 630, and 1005 nm monitoring the charge separation and charge recombination. | ||
Finally, we turned to the ZnP–C60–C70 conjugate (1)—see Fig. 2. Immediately upon 550 nm photoexcitation we noticed the characteristics of the ZnP singlet excited state in the visible and those of the C70 singlet excited state in the near-infrared with maxima at 455 and 1290 nm, respectively. Both are, however, metastable and transform rapidly (10 ps–1.0 × 1011 s−1) into a new transient product. To this end, evidence for the ZnP π-radical cation (ZnP˙+) evolves in the visible region—550 to 800 nm. Changes in the near-infrared region, namely at 1005 and 1315, on the other hand, relate to the involvement of the C60 π-radical anion (C60˙−) and the C70 π-radical anion (C70˙−).22,23 Important is that at time delays of around 1.0 ns, only the features of the C70 π-radical anion (C70˙−) are discernable at 1315 nm, while those of the C60 π-radical anion (C60˙−) at 1005 nm have completely disappeared. From analyzing the C60 π-radical anion (C60˙−) decay at, for example, 1005 nm, we have determined a lifetime of 135 ps (7.1 × 109 s−1). A comparison with the intrinsic lifetime of ZnP˙+–C60˙− (1.3 ns–7.6 × 108 s−1) leads us to conclude that the nature of the ZnP˙+–C60˙−–C70 decay is an exothermic and unidirectional charge shift, which involves an electron shift between the two electron accepting fullerenes. The correspondingly formed ZnP˙+–C60–C70˙− shows no appreciable decay in the 1100 to 1500 nm range on the 3.0 ns time scale. Such an impressive gain in stability is also substantiated when inspecting the visible region. Here, the ZnP π-radical cation (ZnP˙+), which absorbs between 550 and 800 nm, is stable without showing any noticeable decay. From complementary nanosecond experiments a lifetime of 100 ± 30 ns—Figure S18—has been derived. As far as the ZnP˙+–C60–C70˙− quantum yield is concerned, a multiwavelength analysis reveals 30%. One factor that may prevent higher quantum yields is the semiflexible spacer.
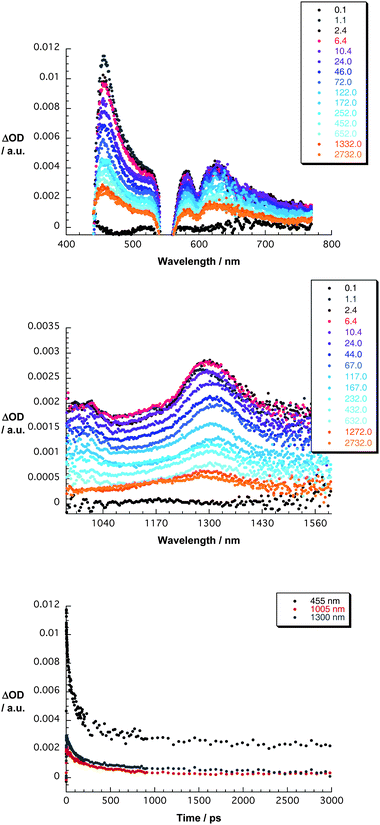 | ||
| Fig. 2 Upper part—differential absorption spectra (visible) obtained upon femtosecond flash photolysis (550 nm) of ZnP–C60–C70 in THF with several time delays between 0 and 3000 ps at room temperature —see figure legend for time delays. Central part—differential absorption spectra (extended near-infrared) obtained upon femtosecond flash photolysis (550 nm) of ZnP–C60–C70 in THF with several time delays between 0 and 3000 ps at room temperature—see figure legend for time delays. Lower part—time-absorption profiles of the spectra shown in the upper and central parts at 455, 1005, and 1300 nm monitoring the charge separation and charge shift. | ||
Conclusions
In short, a fulleropyrrolidine of C60 and a fulleropyrazoline of C70 have been integrated together with ZnP into a novel ZnP–C60–C70 conjugate towards creating a gradient of redox centers. In femtosecond flash photolysis studies we confirm fluorescence experiments that are not discussed and the occurrence of an electron transfer sequence.24 In particular, an initial charge separation yields ZnP˙+–C60˙−–C70—with a lifetime of one hundred picoseconds—and is followed by a subsequent charge shift to afford ZnP˙+–C60–C70˙−—with a lifetime of around one hundred nanoseconds. Overall, our finding constitutes one of the rare cases, in which reductive charge shift from a primary electron acceptor (C60) to a secondary electron acceptor (C70) retards charge recombination as opposed to the more common oxidative charge shift from primary to secondary donors.
Acknowledgements
Cluster of Excellence “Engineering of Advanced Materials” is gratefully acknowledged. This work has been supported by the MICINN of Spain (CT2008-00795, and Consolider-Ingenio CSD2007-00010 on Molecular Nanoscience), Comunidad de Madrid (MADRISOLAR-2, S2009/PPQ-1533) and EU (FUNMOLS FP7-212942-1). P.A.B. thanks IMDEA-Nanociencia for a postdoctoral research grant. J.L.D. thanks the MICINN of Spain for a Ramón y Cajal Fellowship co-financed by the EU Social Funds. C.V. thanks to CM for a research grant.Notes and references
- (a) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS; (b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS.
- (a) D. M. Guldi, N. Martín, ed., Fullerenes: From Synthesis to Optoelectronic Properties; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2002 Search PubMed; (b) F. Langa, J.-F. Nierengarten, ed., Fullerenes. Principles and Applications, RSC, Cambridge, UK, 2007 Search PubMed; (c) N. Martín, Chem. Commun., 2006, 2093 RSC.
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS.
- (a) G. Accorsi and N. Armaroli, J. Phys. Chem. C, 2010, 114, 1385 CrossRef CAS; (b) D. M. Guldi, G. M. Aminur Rahman, V. Sgobba and C. Ehli, Chem. Soc. Rev., 2006, 35, 471 RSC.
- As representative examples of D1–D2–A triads, see: (a) B. M. Illescas, J. Santos, M. Wielopolski, C. M. Atienza, N. Martín and D. M. Guldi, Chem. Commun., 2009, 5374 RSC; (b) H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607 CrossRef CAS; (c) P. A. Liddell, D. Kuciauskas, J. P. Sumida, B. Nash, D. Nguyen, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 1997, 119, 1400 CrossRef CAS.
- (a) C. A. Wijesinghe, M. E. El-Khouly, J. D. Blakemore, M. E. Melvin Zandler, S. Fukuzumi and F. D'Souza, Chem. Commun., 2010, 46, 3301 RSC; (b) D. Gonzalez-Rodriguez, E. Carbonell, G. de Miguel Rojas, C. Atienza Castellanos, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2010, 132, 16488 CrossRef CAS.
- M. A. Fazio, O. P. Lee and D. I. Schuster, Org. Lett., 2008, 10, 4979 CrossRef CAS.
- Compound 1 is formed in moderate yield (40%). Since typical click-chemistry reactions take place with high yields, this result could be accounted for by the competition between the cycloaddition reaction of the azide derivative on the terminal alkyne group and on the fullerene units. Nevertheless, in order to minimize this competitive process, the reaction is carried out at room temperature. See supporting information†.
- (a) J. L. Delgado, F. Cardinali, E. Espíldora, M. R. Torres, F. Langa and N. Martín, Org. Lett., 2008, 10, 3705 CrossRef CAS; (b) J. L. Delgado, F. Oswald, F. Cardinali, F. Langa and N. Martín, J. Org. Chem., 2008, 73, 3184 CrossRef CAS; (c) J. L. Delgado, E. Espíldora, M. Liedtke, A. Sperlich, D. Rauh, A. Baumann, C. Deibel, V. Dyakonov and N. Martín, Chem.– Eur. J., 2009, 15, 13474 CAS.
- (a) M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798 CrossRef CAS; (b) M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS; (c) N. Tagmatarchis and M. Prato, Synlett, 2003, 768 CAS.
- The chemical structure of 1 was confirmed by NMR, HPLC, and MS measurements (m/z calculated for C210H85N11O2Zn 2858.6 [M], found 2859.7 [M + 1]). See supporting information.† For some examples on related Zn–C60 dyads, see: (a) J. Iehl, R. Pereira de Freitas, B. Delavaux-Nicot and J.-F. Nierengarten, Chem. Commun., 2008, 2450 RSC; (b) M. A. Fazio, O. P. Lee and D. I. Schuster, Org. Lett., 2008, 10, 4979 CrossRef CAS; (c) J. Iehl, I. Osinska, R. Louis, M. Holler and J.-F. Nierengarten, Tetrahedron Lett., 2009, 50, 2245 CrossRef CAS; (d) J. Iehl, M. Vartanian, M. Holler, J.-F. Nierengarten, B. Delavaux-Nicot, J.-M. Strub, A. Van Dorsselaer, Y. Wu, J. Mohanraj, K. Yoosaf and N. Armaroli, J. Mater. Chem., 2011, 21, 1562 RSC.
- Only recently good control of the site-, regio-, diastereo- and, enantioselectivity on C70 has been reported. See: E. E. Maroto, A. de Cozar, S. Filippone, A. Martín-Domenech, M. Súarez, F. P. Cossío and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 6060 CrossRef CAS.
- (a) Q. Xie, E. Pérez-Cordero and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 3978 CrossRef CAS; (b) L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593 CrossRef CAS; (c) J.-F. Nierengarten, A. Herrmann, R. R. Tykwinski, M. Riittimann, F. Diederich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 1997, 80, 293 CrossRef CAS.
- (a) For a recent review, see: J. L. Delgado, N. Martín, P. de la Cruz, F. Langa, Chem. Soc. Rev., 10.1039/C1CS15105C; (b) J. L. Delgado, P. de la Cruz, V. López-Arza, F. Langa, D. B. Kimball, M. M. Haley, Y. Araki and O. Ito, J. Org. Chem., 2004, 69, 2661 CrossRef CAS; (c) J. L. Delgado, P. de la Cruz, V. López-Arza and F. Langa, Tetrahedron Lett., 2004, 45, 1651 CrossRef CAS; (d) F. Langa, P. de la Cruz, E. Espildora, A. de la Hoz, J. L. Bourdelande, L. Sanchez and N. Martín, J. Org. Chem., 2001, 66, 5033 CrossRef CAS.
- (a) M. Z. Hoffmann, F. Bolletta, L. Moggi and G. L. Hug, J. Phys. Chem. Ref. Data, 1989, 18, 219 CrossRef; (b) J. Rodriguez, C. Kirmaier and D. Holten, J. Am. Chem. Soc., 1989, 111, 6500 CrossRef CAS; (c) S. L. Murov, I. Carmichael, G. L. Hug, Handbook of Photochemistry; Marcel Dekker Inc.; New York, 1993 Search PubMed; (d) K. Kalyanasundaram, Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press; London, 1992 Search PubMed.
- (a) L. Pekkarimen and H. Linschitz, J. Am. Chem. Soc., 1960, 82, 2407 CrossRef; (b) W. A. Lee, M. Graetzel and K. Kalyanasundaram, Chem. Phys. Lett., 1984, 107, 308 CrossRef CAS.
- (a) N. M. Dimitrijevic and P. V. Kamat, J. Phys. Chem., 1992, 96, 4811 CrossRef CAS; (b) D. M. Guldi, P. V. Kamat, In Chemistry, Physics and Technology; K. M. Kadish, R. S. Ruoff, ed.; John Wiley and Sons: New York, 2000, p 225–282 Search PubMed; (c) D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS.
- (a) M. R. Wasielewski, M. P. O'Neil, K. R. Lykke, M. J. Pellin and D. M. Gruen, J. Am. Chem. Soc., 1991, 113, 2774 CrossRef CAS; (b) K. Tanigaki, T. W. Ebbesen and S. Kuroshima, Chem. Phys. Lett., 1991, 185, 189 CrossRef CAS; (c) H. T. Etheridge and R. B. Weisman, J. Phys. Chem., 1995, 99, 2782 CrossRef CAS; (d) T. Nojiri, A. Watanabe and O. Ito, J. Phys. Chem. A, 1998, 102, 5215 CrossRef CAS.
- S. Nonell, J. W. Arbogast and C. S. Foote, J. Phys. Chem., 1992, 96, 4169 CrossRef CAS.
- (a) Z. Gasyna, W. R. Browett and M. Stillman, Inorg. Chem., 1985, 24, 2440 CrossRef CAS; (b) D. M. Guldi, A. Hirsch, M. Scheloske, E. Dietel, A. Troisi, F. Zerbetto and M. Prato, Chem.–Eur. J., 2003, 9, 4968 CrossRef CAS.
- (a) D. I. Schuster, P. Cheng, P. D. Jarowski, D. M. Guldi, C. Luo, L. Echegoyen, S. Pyo, A. R. Holzwarth, S. E. Braslavsky, R. M. Williams and G. Klihm, J. Am. Chem. Soc., 2004, 126, 7257 CrossRef CAS; (b) A. S. D. Sandanayaka, H. Sasabe, Y. Araki, Y. Furusho, O. Ito and T. Takata, J. Phys. Chem. A, 2004, 108, 5145 CrossRef CAS; (c) D. I. Schuster, S. MacMahon, D. M. Guldi, L. Echegoyen and S. E. Braslavsky, Tetrahedron, 2006, 62, 1928 CrossRef CAS; (d) F. Wessendorf, J.-F. Gnichwitz, G. H. Sarova, K. Hager, U. Hartnagel, D. M. Guldi and A. Hirsch, J. Am. Chem. Soc., 2007, 129, 16057 CrossRef CAS; (e) K. Maurer, B. Grimm, F. Wessendorf, K. Hartnagel, D. M. Guldi and A. Hirsch, Eur. J. Org. Chem., 2010, 5010 CrossRef CAS; (f) F. Wessendorf, B. Grimm, D. M. Guldi and A. Hirsch, J. Am. Chem. Soc., 2010, 132, 10786 CrossRef CAS.
- Such a nearly simultaneous formation of the C60 π-radical anion and the C70 π-radical anion can only be rationalized on the basis that the ZnP singlet excited state reduces the adjacent C60, while the C70 singlet excited state is reduced by the ZnP singlet ground state.
- D. R. Lawson, D. L. Feldhein, C. A. Foss, P. K. Dorhout, C. M. Elliott, C. R. Martin and B. Parkinson, J. Phys. Chem., 1992, 96, 7175 CrossRef CAS.
- The ZnP centered fluorescence is in the ZnP–C60–C70 conjugate (1) quenched by a factor of more than 10.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure, and complete characterizations of all the new compounds. See DOI: 10.1039/c1sc00331c |
| This journal is © The Royal Society of Chemistry 2011 |