Methods for converting cysteine to dehydroalanine on peptides and proteins†
Justin M.
Chalker
a,
Smita B.
Gunnoo
a,
Omar
Boutureira
a,
Stefanie C.
Gerstberger
a,
Marta
Fernández-González
a,
Gonçalo J. L.
Bernardes
a,
Laura
Griffin
b,
Hanna
Hailu
b,
Christopher J.
Schofield
a and
Benjamin G.
Davis
*a
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Road, Oxford, OX1 3TA, United Kingdom. E-mail: ben.davis@chem.ox.ac.uk; Fax: +44 (0) 1865 285002; Tel: +44 (0) 1865 275652
bUCB Celltech, 208 Bath Road, Slough, Berkshire SL1 3WE, United Kingdom
First published on 7th June 2011
Abstract
Dehydroalanine is a synthetic precursor to a wide array of protein modifications. We describe multiple methods for the chemical conversion of cysteine to dehydroalanine on peptides and proteins. The scope and limitations of these methods were investigated with attention paid to side reactions, scale, and aqueous- and bio-compatibility. The most general method investigated—a bis-alkylation–elimination of cysteine to dehydroalanine—was applied successfully to multiple proteins and enabled the site-selective synthesis of a glycosylated antibody.
Introduction
Dehydroalanine: a precursor to post-translational modifications
Dehydroalanine (Dha, 1, Scheme 1) is an amino acid residue of both biological and synthetic interest. In nature, dehydroalanine is found in lanthionine-containing antibiotic peptides—lantibiotics—where it is formed by the enzymatic dehydration of serine.1–3Dehydroalanine imparts a conformational constraint on peptides4 and is an electrophilic center for reactions with nucleophiles. Intramolecular additions to dehydroalanine occur naturally, as in the biosynthesis of lantibiotics such as Nisin (Scheme 1A).1–3Dehydroalanine also occurs naturally in proteins. For example, tyrosine is converted to dehydroalanine in thyroglobulin during thyroid hormone biosynthesis.5,6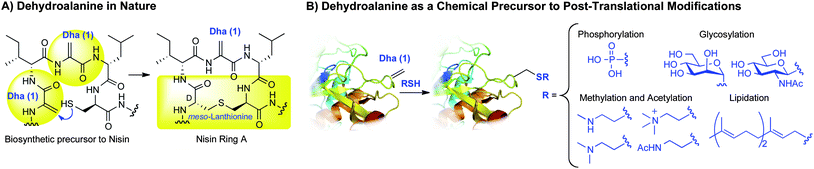 | ||
| Scheme 1 Dehydroalanine is a precursor to modified peptides and proteins. | ||
Synthetically, dehydroalanine is a useful chemical precursor to a range of post-translational modifications (PTMs)7,8 and their analogues by the conjugate addition of a thiol (Scheme 1B).9–14Protein modifications accessible viadehydroalanine comprise some of the most prevalent post-translational modifications in nature: phosphorylation,15,16glycosylation,17–19methylation,20acetylation,21,22 and lipidation.23 Though the addition of thiols to dehydroalanine can lead to epimeric products, this does not necessarily preclude their use in biology where one or both of the epimers can serve as enzyme substrates or enable binding of other biomacromolecules. For instance, S-linked glycoproteins derived from Dha are acceptors for glycosylation by endoglycosidase A.24 The sulfur-based methyl- and acetyl lysine analogs (Scheme 1B) have also been validated as lysine mimics in several biological contexts.14,25,26Phosphocysteine has been proposed as a mimic for phosphoserine13 and is a natural protein modification,27 as is farnesyl cysteine.23 Both modifications are accessible through Dha (Scheme 1B).13
The importance of post-translational modifications in biomolecular function is well established,7,8 but a detailed understanding of structure–function relationships of PTMs is often lacking. Studies of post-translationally modified proteins would therefore benefit from ready access to modified proteins such as those in Scheme 1B.28 Native chemical ligation can supply post-translationally modified proteins and useful analogs with the atomic precision conferred by total chemical synthesis. However, this strategy is often technically challenging—especially when multiple ligations are required.29 Amber codon suppression is another powerful technology that has provided proteins bearing post-translational modifications.30–34 This method, however, requires the evolution of a unique aminoacyl-tRNA synthetase for each modified amino acid, and in some cases requires several additional chemical steps to access the final protein.35–37 Moreover, for many residues bearing PTMs (e.g.trimethyllysine, phosphoserine, glycosylserines, and prenylated cysteines), no synthetases have been reported for their direct incorporation during translation. Dehydroalanine, in contrast, can be directly converted to several PTMs by an operationally simple conjugate addition of a thiol (Scheme 1B).
This route to modified proteins, however, is predicated on the efficient and reliable incorporation of dehydroalanine at the site of modification. Several chemical and biochemical methods for the incorporation of dehydroalanine into peptides and proteins have been described, yet each strategy has limitations. We discuss the merits and drawbacks of these methods below. This assessment helped frame our strategy for developing more general methods for the incorporation of dehydroalanine into protein substrates.
Previous reports for installing dehydroalanine in proteins
Site-selective incorporation of dehydroalanine into proteins has received the attention of chemists for nearly half a century. These efforts are summarized in Scheme 2. Initial reports from Koshland and co-workers described the conversion of the nucleophilic serine of chymotrypsin to dehydroalanine in an effort to investigate the role of the serine in catalysis.38,39 After selective sulfonylation at the nucleophilic serine, base-mediated elimination provided dehydroalanine (Scheme 2A). Koshland et al. proposed that the electrophilic properties of dehydroalanine might enable “chemical mutations” by the conjugate addition of nucleophiles and demonstrated such additions to confirm dehydroalanine formation.39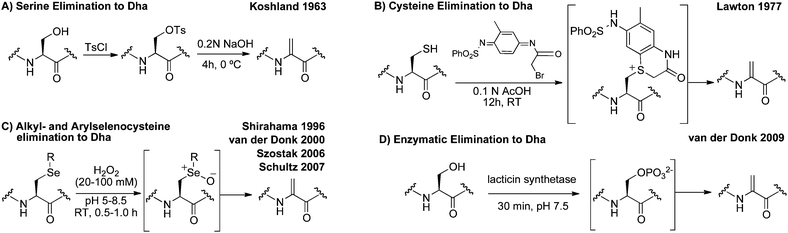 | ||
| Scheme 2 Representative methods for the incorporation of dehydroalanine into peptides and proteins. | ||
Koshland's pioneering work exploited the enhanced nucleophilicity of the active site serine in chymotrypsin. More general methods for incorporating Dha have utilized the nucleophilic properties of the cysteinyl thiol.40 Holmes and Lawton, for instance, used the conversion of cysteine to dehydroalanine as a method for mapping cysteine residues since peptide-backbone hydrolysis occurs at dehydroalanine when heated (80 °C) under acidic conditions.41Cysteinyl residues were dialkylated and the resulting sulfonium salts eliminated to dehydroalanine after prolonged incubation in aqueous acetic acid (Scheme 2B).
Oxidative elimination of S-alkyl cysteinyl residues is another strategy for dehydroalanine synthesis; however, elimination of S-alkyl cysteinyl sulfoxides typically requires temperatures incompatible with protein substrates.42 Oxidative elimination of phenylselenocysteine and related derivatives, in contrast, can be achieved via the selenoxide at room temperature and has provided access to dehydroalanine in peptides43–45 and proteins12,14 (Scheme 2C). Methods that exploit the promiscuity of enzymes in lantibiotic biosynthesis have also been reported for the conversion of serine to Dha (Scheme 2D).46
Despite this long-standing interest in dehydroalanine, the inherent limitations of the methods in Scheme 2 have precluded their wider use in peptide and protein modification. None of these methods enable general, chemo- and site-selective incorporation of dehydroalanine. For instance, base-induced elimination may require a pH incompatible with many substrates (Scheme 2A).38,39 Lawton's method (Scheme 2B), requires a significant amount of organic solvent to dissolve the appropriate reagent which can cause protein precipitation.41 Oxidative eliminations of alkyl-cysteines and alkyl-selenocysteines typically employ oxidants that react with other residues such as methionine (Scheme 2C).14,45 Additionally, while lacticin synthetase (Scheme 2D) is relatively promiscuous, enzymatic routes to Dha are currently limited to peptides with specific leader sequences.46
Proposed methods for the conversion of cysteine to dehydroalanine
In this report, we disclose an assessment of methods for the conversion of cysteine to dehydroalanine. Cysteine is a convenient target for protein modification because of its strong nucleophilicity and ease of incorporation into proteins using standard biochemical techniques.40 We consider and compare four complementary modes of reactivity for the conversion of cysteine to dehydroalanine: the reduction–elimination of cysteine disulfides, base-mediated elimination of cysteine disulfides and related derivatives, oxidative elimination of cysteine, and the bis-alkylation-elimination of cysteine (Scheme 3).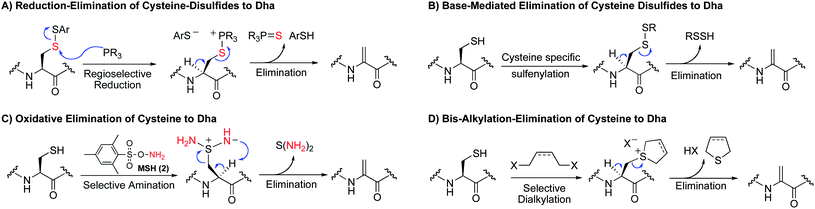 | ||
| Scheme 3 Four complementary modes of elimination of cysteine to dehydroalanine. | ||
The reduction–elimination of cysteine disulfides (Scheme 3A) is designed on the basis of reports on the reaction of electron rich phosphines with disulfides. Phosphine attack upon disulfides is an endothermic process47 and proceeds through a late transition state.48,49 It should therefore proceed in such a way that the thiol with the lowest pKa will be the leaving group. The logical consequence is that by making a mixed disulfide from an electron-withdrawing thiol and cysteine, attack on the sulfur of cysteine should be favored. The resulting phosphonium salt can then undergo elimination to give dehydroalanine. Harpp and Gleason observed similar eliminations from β-keto disulfides using electron rich phosphines,50 Xian and co-workers observed the formation of dehydroalanine from cystine when treated with tris(dimethylamino)phosphine (HMPT),51 and we have observed such eliminations from cysteine-carbohydrate disulfides using HMPT.52
While we anticipated that the intermediate thiophosphonium salt would eliminate more rapidly than the disulfide alone (Scheme 3A), we did not rule out the possibility of base-mediated elimination of the disulfide itself. Thus, after specific conversion to an electron-deficient disulfide, base-induced elimination is another route to dehydroalanine considered here (Scheme 3B).
A third method for the conversion of cysteine to Dha is the oxidative elimination of cysteine to dehydroalanine using O-mesitylenesulfonylhydroxylamine (MSH, 2, Scheme 3C).13 This reaction is nearly instantaneous between 0 °C and room temperature—a rate that compares favorably to the corresponding sulfoxide elimination of S-alkyl cysteinyl residues. In our initial report of this reaction, we applied this transformation to a single-cysteine mutant of a model protease, selectively converting cysteine to dehydroalanine.13 While side reactions were not observed on this model protease, MSH is a reactive oxidizing and aminating reagent53–55 and the full scope and limitations of this method are unreported.
A fourth method considered for the synthesis of Dha is the bis-alkylation–elimination of cysteine (Scheme 3D). This method is inspired in part by Holmes' and Lawton's method for cysteine mapping (Figure 2B).41 We also noted that this transformation is observed in both murine and human metabolism of 1,4-dihalobutanes and Busulfan (1,4-bis(methansulfonyl)-butandiol) where the cysteinyl residue of glutathione was converted to dehydroalanine.56–61 In these latter cases, the occurrence of these transformations under physiological conditions bodes well for application in biology as a synthetic tool for protein modification and in vivo chemistry.
During these investigations, careful attention was paid to side reactions, substrate scope, synthetic limitations, and aqueous compatibility. After exploratory work on amino acid and peptide model systems, these methods were evaluated on several model proteins, including the protease subtilisin from Bacillus lentus and the single-domain antibody cAb-Lys3. We disclose the results of these studies here. Finally, the most general method for the conversion of cysteine to dehydroalanine was applied to several proteins and used in the site-selective synthesis of a glycosylated antibody.
Results and discussion
Conversion of cysteine to dehydroalanine on amino acid and peptide substrates
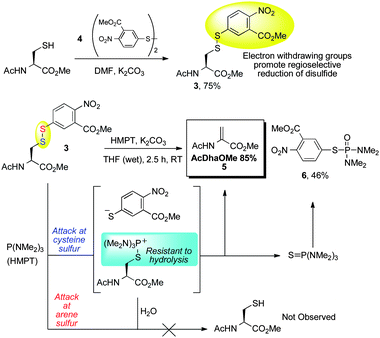 | ||
| Scheme 4 Model reduction–elimination of cysteine disulfide. | ||
With 3 in hand, the reduction–elimination was tested. Upon addition of 2 equivalents of HMPT, the reaction mixture turned red, consistent with disulfide attack and release of the aryl thiolate. The major product of the reaction was the desired AcDhaOMe (5, 85%); phosphoramide 6 was also isolated in 46% yield. The formation of 6 is consistent with a mechanism in which the aryl thiolate leaving group attacks either the phosphine(thio)oxide (Scheme 4) or HMPT itself.50 Importantly, AcCysOMe was not observed, indicating that HMPT attack at the arene sulfur of 3 is disfavored and that the hydrolysis of the cysteine phosphonium intermediate is slow relative to elimination (Scheme 4). It should be pointed out that carbonate is not a strong enough base to eliminate the disulfide directly (3 was synthesized in the presence of carbonate); elimination therefore occurs after attack of the disulfide by HMPT.
With the desired reduction–elimination validated using model substrate 3, we then investigated a one-pot procedure whereby cysteine is first treated with Ellman's reagent (as the free acid, rather than ester 4) and then the resulting disulfide is reduced and eliminated using HMPT. DMF was used as the solvent and triethylamine as the base. Gratifyingly, Dha was generated efficiently from the dipeptide 7 in 83% yield (Scheme 5). While we anticipated that the electron-rich HMPT was the most suitable phosphine for the transformation in Schemes 4 and 5, we tested several other phosphorous nucleophiles since HMPT and the product of oxidation (HMPA) are toxic. The results of this screening are shown in Scheme 5. Unfortunately, though not unexpectedly, the use of other less electron-rich phosphines gave inferior yields and resulted mainly in reduction and regeneration of 7.63
 | ||
| Scheme 5 One-pot reduction–elimination of cysteine to dehydroalanine. | ||
The successful use of disulfides as precursors to dehydroalanine is promising since they can easily be installed on proteins in a cysteine-specific manner.40 The reduction–elimination strategy, however, is not the only method to convert cysteine-disulfides to dehydroalanine. In some cases, disulfides of cysteine can be eliminated directly to dehydroalanine.64–68 We considered this method next.
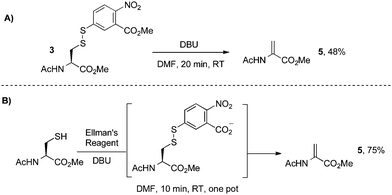 | ||
| Scheme 6 Direct elimination of cysteine disulfides with DBU. | ||
This DBU-mediated elimination is direct, simple, and scalable. However, it is known that disulfides can also be converted to the corresponding sulfinic acids in the presence of hydroxide, especially labile disulfides such as those derived from Ellman's reagent.64 For this reason, the elimination of Ellman disulfides with base is likely limited to organic solvents and therefore not suitable for protein modification (vide infra). To resolve this issue, we considered other derivatives of cysteine that could undergo a similar elimination and better tolerate aqueous systems. In particular, we anticipated that Mukaiyama's reagent69 was well-suited for this transformation given its high reactivity towards sulfur nucleophiles.70
Indeed, Mukaiyama's reagent reacted efficiently with cysteine. In DMF, the adduct of cysteine and Mukaiyama's reagent reacted to give dehydroalanine within 5 min in the presence of DBU (Scheme 7). The yield, however, was moderate and did not improve with extended reaction time. Treating AcCysOMe with DBU in the absence of Mukaiyama's reagent led to only trace AcDhaOMe (5).63
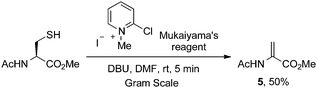 | ||
| Scheme 7 Mukaiyama's reagent in the arylation and elimination of cysteine to dehydroalanine. | ||
Unfortunately, while the use of Mukaiyama's reagent and base to convert cysteine to dehydroalanine is easy and scalable, it also requires a high pH (>10). Triethylamine is not basic enough to promote this elimination and application to protein substrates is likely limited to those tolerant of high pH (vide infra). We then turned to methods more likely to proceed at lower pH: oxidative elimination using MSH and the bis-alkylation–elimination strategy.
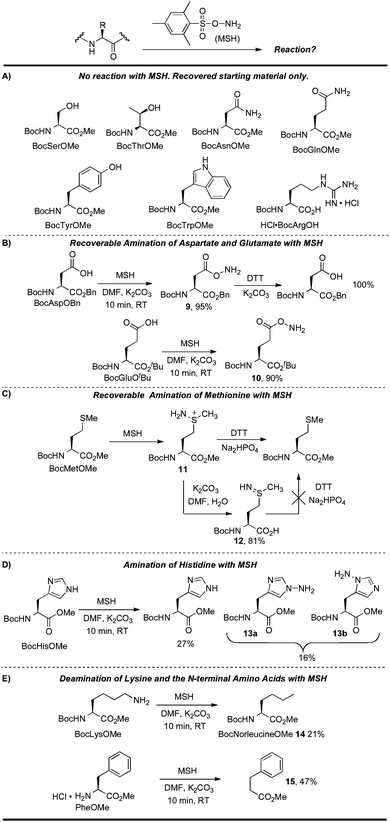 | ||
| Scheme 8 Reactions of MSH with functionalized amino acids. | ||
No reaction was observed for the serine, threonine, asparagine, glutamine, tyrosine, and tryptophan derivatives in Scheme 8A under standard conditions. BocArgOH also did not react with MSH, likely because the basic guanidine side chain is protected in its protonated form. The free C-terminus of BocArgOH was also recovered without incident under these conditions. Carboxy groups can, however, be aminated by MSH under basic conditions, as seen in the reaction with aspartate and glutamate side chains. Efficient carboxy-amination was observed in these cases to provide 9 and 10 in high yield. While the rapid reaction of MSH with aspartate and glutamate revealed compatibility issues with these nucleophilic residues, the products of these aminations were easily reduced back to their respective carboxylic acid. For instance, treating 9 with dithiothreitol (DTT) led to rapid and quantitative reduction to BocAspOBn (Scheme 8B).
Methionine is also aminated by MSH, but the native thioether can again be regenerated using DTT. Therefore, this side reaction does not preclude use on peptides or proteins containing methionine because the resulting intermediate sulfiliminium salt (11) can be reduced back to the original amino acid residue (Scheme 8C).71 Other reducing agents such as phosphines, zinc metal, sodium dithionite, and sodium ascorbate were not effective in this reduction.63
To clarify the role of the putative sulfiliminium 11 and discount other pathways for Met recovery, we examined other possible intermediates. For example, it was not clear whether DTT could reduce methionine sulfilimine 12 (as opposed to the sulfiliminium salt 11). 12 was therefore isolated and reacted with DTT under basic conditions. It is notable that quite basic conditions (K2CO3, pH > 11) are required to generate 12 and it is unlikely that such an intermediate is formed during protein modification since a much lower pH is used in this reaction for protein modification (pH 8.0 or lower).13 Even after 15 h, DTT did not reduce 12 to the thioether (Scheme 8C). This result suggests that the reducible intermediate is not sulfilimine 12 but another intermediate such as 11. Finally, it is worth noting that double amination at sulfur was not observed in the synthesis of 12, even in the presence of excess MSH.63 This result suggests that for each methionine in a given substrate, it is likely that no more than a single amination will be observed at sulfur under conditions typical for protein modification. It is also notable that during these studies, no vinyl glycine or dehydrobutyrine was observed, indicating that elimination of 11 and 12 does not occur under these conditions.
Histidine and lysine also reacted with MSH (Scheme 8D and 8E). BocHisOMe gave a regioisomeric mixture of aminated products (13a, 13b), consistent with previous studies that describe the amination of nitrogen heterocycles with MSH and related aminating reagents.53,72 Poor mass balance for this reaction suggests that some material is lost on workup, perhaps as the water-soluble imidazolium product of di-N-amination of the His sidechain. This proposal is consistent with a lower recovery with increasing equivalents of MSH.63Lysine gave a mixture of products after reaction with MSH. Deaminated lysine (norleucine derivative 14) was the major product isolated from this mixture, albeit in relatively low yield (21%) (Scheme 8E). This result was initially counterintuitive: MSH, an aminating and oxidizing agent, effectively de-aminated and reduced lysine. This reaction, however, likely proceeds through the hydrazine—the product of amination at the ε-amino group—before further oxidation to the mono-substituted diimide and ultimate loss of nitrogen.73 The course suggested for this reaction is depicted in Scheme 9 and is based on an analogous “hydrodeamination”74 using hydroxylamine-O-sulfonic acid.72
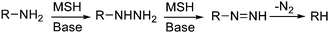 | ||
| Scheme 9 “Hydrodeamination” using MSH. | ||
To investigate N-terminal deamination, PheOMe was treated with excess MSH. Deamination of the α-amino group was indeed observed and 15 was isolated in 47% yield (Scheme 8E). To further explore the limitations of an exposed N-terminus, model peptide 16 was synthesized and MSH was used to convert the cysteine to dehydroalanine (Scheme 10). While the desired peptide 17 could be isolated in useful yield, a significant side product was observed that had a mass that corresponded to loss of both H2S and NH2, consistent with concomitant deamination and Dha formation to give 18.
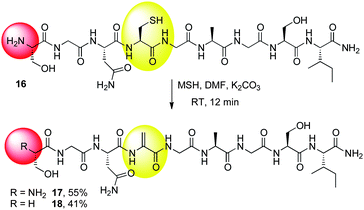 | ||
| Scheme 10 N-terminal deamination using MSH on a model peptide. | ||
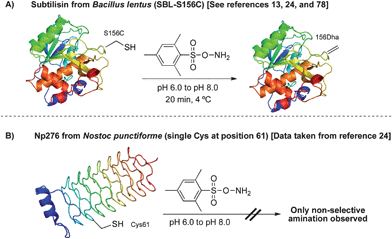
From this assessment of MSH reactivity, it appears that the predominant side reactions occur at nucleophilic amino acid residues (Asp, Glu, Met, Lys, and His) and at the N-terminal amino group. For Asp, Glu, and Met, the product of amination can be converted back to the native side chain by reduction with DTT. Additionally, careful control of pH could minimize amination of lysine and histidine. MSH-mediated elimination of cysteine to dehydroalanine can proceed at a pH as low as 6 and lysine and histidine might be protected in their protonated forms.13
Therefore, while these side reactions can and do occur, they do not preclude selective reaction at cysteine. The selectivity that allowed successful conversion of cysteine to dehydroalanine on our model protein (subtilisin from Bacillus lentus) was likely due to a combination of factors such as the high nucleophilicity of cysteine, the pH, and differential accessibility of side chains.13 These factors should be taken into consideration for use in peptide and protein modification. With a goal of avoiding these side-reactions entirely, we next pursued a potentially more selective transformation: the bis-alkylation–elimination of cysteine to dehydroalanine.
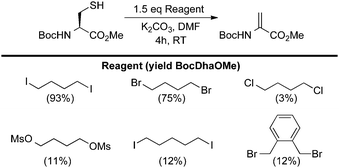 | ||
| Scheme 11 Reagents for the bis-alkylation–elimination of cysteine to dehydroalanine. | ||
From the results in Scheme 11, several aspects of the bis-alkylation–elimination reaction are revealed. First, the 1,4-dibromo- and 1,4-diiodobutanes are the most efficient of the reagents screened to S-alkylate, cyclize, and eliminate. 1,4-Dichlorobutane is slow to alkylate and mostly unreacted BocCysOMe was recovered. Despite prior observations56–61 that had guided our reasoning (vide supra), Busulfan (1,4-bis(methansulfonyl)-butandiol) was also relatively inefficient under these reaction conditions, generating BocDhaOMe in only 11% yield. 1,5-Diiodopentane provided only 12% of the desired elimination product under the same conditions. This result suggested that formation of the six-member sulfonium ion is slower than the corresponding 5-membered ring formed with 1,4-diiodobutane or that the resulting cyclic sulfonium is less labile (or both). Finally, α,α′-dibromo-o-xylene generated only 12% Dha. In this example, the isolation of dimerized product 19 suggested that intermediate cyclization is slow (Scheme 12A). When a larger excess of α,α′-dibromo-o-xylene was used (5 equivalents) to minimize the formation of 19, the major product was instead the alkylated, uncyclized benzyl bromide 20 which proved sufficiently stable to be isolated and characterized (Scheme 12B).63 Heating 20 at 37 °C for 4 h in the presence of base led to modest yields of dehydroalanine (40%), providing further evidence for its intermediacy and relatively inefficient cyclization-elimination (Scheme 12C). Together, these results suggested that a saturated 1,4-dialkylating reagent would best enable efficient formation of the cyclic tetrahydrothiophenium intermediate, the immediate precursor to Dha.
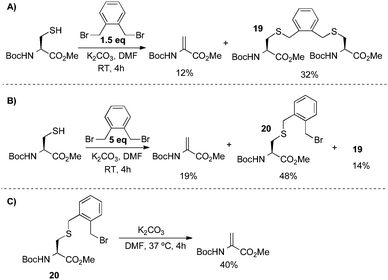 | ||
| Scheme 12 Bis-alkylation–elimination with α,α′-dibromo-o-xylene. | ||
The reconnaissance work depicted in Schemes 11 and 12 guided the design of water-soluble reagents with the requisite 1,4-dihalobutane moiety. The syntheses of these reagents (21 and 22) are outlined in Scheme 13.
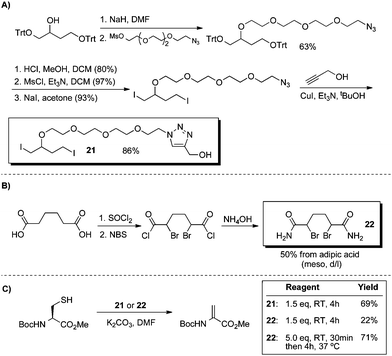 | ||
| Scheme 13 Synthesis of water-soluble 1,4-dihalobutane derivatives and their use in the conversion of cysteine to dehydroalanine. | ||
Reagent 21 contains a hydrophilic tetraethyleneglycol unit tethered to a 1,4-diiodobutane core. Reagent 22 is a bisamide of the 1,4-dibromobutane core and was inspired in part by Kajihara's observations that diethyl meso-2,5-dibromoadipate converted cysteine to dehydroalanine on peptide substrates.75 To compare 21 and 22 directly to the parent reagents in Scheme 11, the same model reaction was studied in DMF (Scheme 13C). Using di-iodide 21, we obtained useful yields of Dha at room temperature. Bis-amide 22 initially afforded only a low yield of elimination product (22%), but with complete consumption of cysteine. It appeared that the cyclization was slow for 22; when cysteine was first alkylated at room temperature and then incubated for 4 h at 37 °C, a 71% yield of Dha was obtained (Scheme 13).76
With a preliminary assessment of four modes of elimination of cysteine to dehydroalanine on model substrates in hand, their application to protein substrates was pursued next.
Conversion of cysteine to dehydroalanine on protein substrates
All protein reactions were monitored by LC-MS. The reaction conversions reported are calculated from the relative peak height of the deconvoluted mass spectrum. For a modification at a single site, we have previously shown that relative MS peak height correlates well with the relative amount of protein measured by independent methods.77 For the substrates and chromatographic conditions employed, all protein material generally co-elutes in a single peak in the total ion chromatogram (TIC). In a typical analysis, the mass spectra for all protein material contained in this peak are combined and the resulting ion series is then deconvoluted using a maximum entropy algorithm. A representative analysis showing the TIC, combined ion series, and deconvoluted spectra can be found on page 55 of the supplementary information.†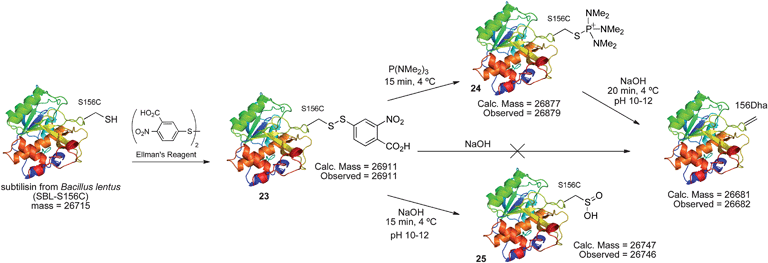 | ||
| Scheme 14 Elimination of cysteine disulfides to Dha in protein model SBL. | ||
The disulfide 23 derived from Ellman's reagent was not stable to nucleophilic attack by hydroxide and could not be directly eliminated to dehydroalanine. However, adduct 26, formed from reaction of cysteine with Mukaiyama's reagent, can be eliminated under such conditions. Upon treatment with 1 M NaOH (pH ∼11–12), rapid elimination to dehydroalanine was observed by LC-MS. The product protein did not react with Ellman's reagent, indicating that all cysteine was consumed in the reaction sequence. The presence of dehydroalanine was further corroborated by the conjugate addition of 2-mercaptoethanol.63 The overall reaction sequence is depicted in Scheme 15.
 | ||
| Scheme 15 Elimination of cysteine to dehydroalanine after reaction with Mukaiyama's reagent. | ||
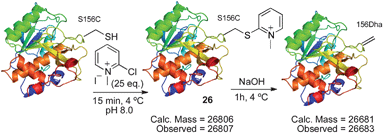
While this 2-step, one-pot method for the conversion of cysteine to dehydroalanine is also restricted to substrates tolerant of high pH, it represents a selective and operationally simple method for installing dehydroalanine. Pursuing methods for dehydroalanine synthesis at a milder pH, we turned to oxidative and bis-alkylation–elimination.
 | ||
| Scheme 16 MSH in the oxidative elimination of cysteine to dehydroalanine on protein substrates. | ||
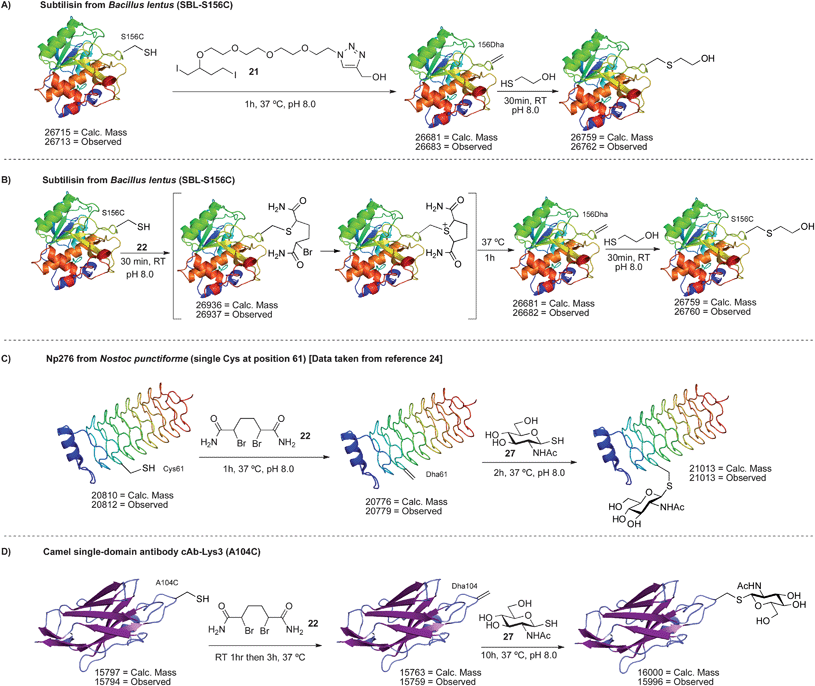 | ||
| Scheme 17 Bis-alkylation–elimination of cysteine to dehydroalanine on protein surfaces. | ||
A similar reactivity was observed with 22 on SBL-S156C. After 30 min at room temperature, the alkylated uncyclized intermediate was detected by LC-MS (Scheme 17B). This outcome was consistent with the model reactions in Scheme 13C. To induce cyclization and elimination, the reaction was simply incubated for one hour at 37 °C. Full conversion to dehydroalanine was observed (Scheme 17B). Conversion of cysteine to dehydroalanine was confirmed by Ellman's assay and addition of 2-mercaptoethanol.63
The reaction of di-bromide 22 with the single cysteine mutant of Np276 was examined next (Scheme 17C). Using 22, the selective conversion of cysteine to dehydroalanine was indeed possible, overcoming the non-selective amination observed with MSH. Reaction at cysteine was once again verified by Ellman's assay and the formation of dehydroalanine was demonstrated by the addition of various thiols. For instance, the dehydroalanine residue was subsequently converted to N-acetyl-glucosamine cysteine (GlcNAc cysteine).24,63 The synthesis of this S-linked glycoprotein was critical in our concurrent investigation of endoglycosidase A in glycoprotein synthesis.24
As a more demanding test for reagent 22, a mutant of the camel single-domain antibody cAb-Lys3 was used as a substrate (cAb-Lys3-A104C, Scheme 17D).79,80 This antibody mutant contains a single reactive cysteine at position 104 and two disulfide bonds located in the core of the antibody. The correct folding of the A104C mutant was inferred by an enzyme-linked immunosorbent assay (ELISA) that showed binding to hen egg white lysozyme, the antigen to which cAb-Lys3 was raised. Circular dichroism (CD) spectroscopy also showed a secondary structure comparable to the wild type antibody.63 Upon reaction of the A104C mutant with 22, efficient conversion to dehydroalanine was observed by LC-MS (Scheme 17D). The protein product did not react with Ellman's reagent, demonstrating all free cysteine was consumed in the reaction, and the formation of Dha was corroborated by the addition of 2-mercaptoethanol.63 In control experiments, it was shown that the wild type antibody did not react with 22, Ellman's reagent, or 2-mercaptoethanol.63 These results indicate that the reaction observed for the mutant antibody occurs at cysteine 104. It is notable that in this example, no organic solvent was used and 22 was simply added as a solid—an important feature for fragile proteins such as antibodies that may not tolerate organic solvent. It should also be highlighted that this reaction sequence is a rare example of regioselective cysteine modification81–83 and demonstrates that the incorporation and modification of a single cysteine ‘tag’ is possible in proteins that contain multiple natural cysteine residues.84,85 Of course, such selectivity is less likely when the protein contains multiple reduced cysteines, rather than disulfides, but regioselective modification can still be achieved if these cysteine residues have different solvent accessibility.81
Finally, the dehydroalanine on cAb-Lys3 was converted to a GlcNAc cysteine residue by the addition of the glycosyl thiol of N-acetylglucosamine (27). S-Linked glycopeptides are found naturally86–88 and we propose their use as analogs of the more commonly found O-linked glycoproteins.89 Moreover, antibody glycosylation often improves their pharmacokinetic and biophysical properties.90,91 Not surprisingly, much attention has been devoted to the synthesis of homogeneously glycosylated antibodies in recent years.92–96 We note here that conjugate addition to dehydroalanine is one of the few methods for the site-specific synthesis of S-linked glycoproteins and the results in Scheme 17 attest to the facility of 21 and 22 in accessing these macromolecules.97,98
Conclusions
We have disclosed an assessment of multiple, complementary methods for the conversion of cysteine to dehydroalanine—a useful ‘tag’ in the ‘tag-and-modify’ approach to protein modification.84,85 Side reactions, selectivity issues, and substrate scope have been described in detail. These mechanistically distinct approaches all allow complete conversion of Cys to Dha in proteins, though each method varies in scope.The most general of these methods—the bis-alkylation–elimination with reagent 22—was applied successfully to three model proteins, including both an enzyme and an antibody. Reagent 22 is stable, simple to prepare, and easy to handle. The elimination of cysteine to dehydroalanine using 22 is also straightforward. The selectivity of 22 can be compared to haloacetamides—a class of reagents long used for selective alkylation of cysteine.40 To demonstrate the scope and utility of 22, dehydroalanine was incorporated into the camel single-domain antibody cAb-Lys3, allowing subsequent conversion to GlcNAc cysteine. This is a rare example of the site-selective chemical glycosylation of an antibody.90–96
More generally, dehydroalanine itself is a target of synthesis (e.g. in the synthesis of lantibiotics) and also a precursor to many other post-translational modifications (Scheme 1). For many of these modifications there are currently no known methods for their synthesis using recombinant expression techniques. The syntheses of dehydroalanine described in this report should enable access to many of these modified proteins.
Acknowledgements
The authors acknowledge their sources of generous support. J.M.C. is a Rhodes Scholar and National Science Foundation Graduate Research Fellow. We also thank the following sources of funding: UCB Celltech for a CASE Award (S.B.G.); The European Commission (Marie Curie IEF, O.B.); Xunta de Galicia, Spain (M.F.G.); FCT, Portugal (G.J.L.B.). B.G.D. is a recipient of a Royal Society Wolfson Research Merit Award and is supported by an EPSRC Life Sciences Interface Platform Grant. We also thank Dr John Porter and Dr Alastair Lawson (UCB Celltech) for helpful discussions.Notes and references
- J. M. Willey and W. A. van der Donk, Annu. Rev. Microbiol., 2007, 61, 477–501 CrossRef CAS.
- R. W. Jack and G. Jung, Curr. Opin. Chem. Biol., 2000, 4, 310–317 CrossRef CAS.
- G. Jung, Angew. Chem., Int. Ed. Engl., 1991, 30, 1051–1068 CrossRef.
- D. E. Palmer, C. Pattaroni, K. Nunami, R. K. Chadha, M. Goodman, T. Wakamiya, K. Fukase, S. Horimoto, M. Kitazawa, H. Fujita, A. Kubo and T. Shiba, J. Am. Chem. Soc., 1992, 114, 5634–5642 CrossRef CAS.
- J.-M. Gavaret, J. Nunez and H. J. Cahnmann, J. Biol. Chem., 1980, 255, 5281–5285 Search PubMed.
- J.-M. Gavaret, H. J. Cahnmann and J. Nunez, J. Biol. Chem., 1981, 256, 9167–9173 CAS.
- C. T. Walsh, S. Garneau-Tsodikova and G. J. Gatto, Jr, Angew. Chem., Int. Ed., 2005, 44, 7342–7372 CrossRef CAS.
- C. T. Walsh, Posttranslational Modification of Proteins: Expanding Nature's Inventory, Roberts and Company Publishers, Englewood, CO, 2006 Search PubMed.
- Y. Zhu and W. A. van der Donk, Org. Lett., 2001, 3, 1189–1192 CrossRef CAS.
- D. P. Galonic, W. A. van der Donk and D. Y. Gin, Chem.–Eur. J., 2003, 9, 5997–6006 CrossRef CAS.
- M. R. Levengood and W. A. van der Donk, Nat. Protoc., 2007, 1, 3001–3010 Search PubMed.
- J. Wang, S. M. Schiller and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 6849–6851 CrossRef CAS.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 6399–6401 CrossRef CAS.
- P. Cohen, Nat. Cell Biol., 2002, 4, E127–E130 CrossRef CAS.
- M. K. Tarrant and P. A. Cole, Annu. Rev. Biochem., 2009, 78, 797–825 Search PubMed.
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS.
- K. Ohtsubo and J. D. Marth, Cell, 2006, 126, 855–867 CrossRef CAS.
- A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, ed., Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, 2009 Search PubMed.
- C. Martin and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2005, 6, 838–849 CrossRef CAS.
- A. Kimura, K. Matsubara and M. Horikoshi, J. Biochem., 2005, 138, 647–662 Search PubMed.
- S. C. Kim, R. Sprung, Y. Chen, Y. Xu, H. Ball, J. Pei, T. Cheng, Y. Kho, H. Xiao, L. Xiao, N. V. Grishin, M. White, X.-J. Yang and Y. Zhao, Mol. Cell, 2006, 23, 607–618 CrossRef CAS.
- F. L. Zhang and P. J. Casey, Annu. Rev. Biochem., 1996, 65, 241–269 CrossRef CAS.
- M. Fernández-González, O. Boutureira, G. J. L. Bernardes, J. M. Chalker, M. A. Young, J. C. Errey and B. G. Davis, Chem. Sci., 2010, 1, 709–715 RSC.
- M. D. Simon, F. Chu, L. R. Racki, C. C. de la Cruz, A. L. Burlingame, B. Panning, G. J. Narlikar and K. M. Shokat, Cell, 2007, 128, 1003–1012 CrossRef CAS.
- C. Chatterjee and T. W. Muir, J. Biol. Chem., 2010, 285, 11045–11050 CrossRef CAS.
- T. C. Stadtman, BioFactors, 1994, 4, 181–185 Search PubMed.
- D. P. Gamblin, S. I. van Kasteren, J. M. Chalker and B. G. Davis, FEBS J., 2008, 275, 1949–1959 CrossRef CAS.
- P. E. Dawson and S. B. H. Kent, Annu. Rev. Biochem., 2003, 69, 923–960 Search PubMed.
- H. Neumann, S. Y. Peak-Chew and J. W. Chin, Nat. Chem. Biol., 2008, 4, 232–234 CrossRef CAS.
- D. P. Nguyen, M. M. Garcia Alai, P. B. Kapadnis, H. Neumann and J. W. Chin, J. Am. Chem. Soc., 2009, 131, 14194–14195 CrossRef CAS.
- H.-w. Ai, J. W. Lee and P. G. Schultz, Chem. Commun., 2010, 46, 5506–5508 RSC.
- D. Groff, P. R. Chen, F. B. Peters and P. G. Schultz, ChemBioChem, 2010, 11, 1066–1068 CrossRef CAS.
- Y.-S. Wang, B. Wu, Z. Wang, Y. Huang, W. Wan, W. K. Russell, P.-J. Pai, Y. N. Moe, D. H. Russell and W. R. Liu, Mol. BioSyst., 2010, 6, 1557–1560 RSC.
- X. Li, T. Fekner, J. J. Ottesen and M. K. Chan, Angew. Chem., Int. Ed., 2009, 48, 9184–9187 CrossRef CAS.
- S. Virdee, Y. Ye, D. P. Nguyen, D. Komander and J. W. Chin, Nat. Chem. Biol., 2010, 6, 750–757 CrossRef CAS.
- D. P. Nguyen, M. M. Garcia Alai, S. Virdee and J. W. Chin, Chem. Biol., 2010, 17, 1072–1076 Search PubMed.
- D. H. Strumeyer, W. N. White and D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 931–935 Search PubMed.
- H. Weiner, W. N. White, D. G. Hoare and D. E. Koshland, J. Am. Chem. Soc., 1966, 88, 3851–3859 Search PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.–Asian J., 2009, 4, 630–640 CrossRef.
- T. J. Holmes and R. G. Lawton, J. Am. Chem. Soc., 1977, 99, 1984–1986 Search PubMed.
- D. H. Rich, J. Tam, P. Mathiaparanam, J. A. Grant and C. Mabuni, J. Chem. Soc., Chem. Commun., 1974, 897–898 RSC.
- K. Hashimoto, M. Sakai, T. Okuno and H. Shirahama, Chem. Commun., 1996, 1139–1140 RSC.
- N. M. Okeley, Y. Zhu and W. A. van der Donk, Org. Lett., 2000, 2, 3603–3606 CrossRef CAS.
- F. P. Seebeck and J. W. Szostak, J. Am. Chem. Soc., 2006, 128, 7150–7151 CrossRef CAS.
- Y. O. You, M. R. Levengood, L. A. F. Ihnken, A. K. Knowlton and W. A. van der Donk, ACS Chem. Biol., 2009, 4, 379–385 CrossRef CAS.
- D. N. Harpp and J. G. Gleason, J. Am. Chem. Soc., 1971, 93, 2437–2445 CrossRef CAS.
- J. E. Leffler, Science, 1953, 117, 340–341 CrossRef CAS.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334–338 CrossRef CAS.
- D. N. Harpp and J. G. Gleason, J. Org. Chem., 1970, 35, 3259–3263 CrossRef CAS.
- H. Wang, J. Zhang and M. Xian, J. Am. Chem. Soc., 2009, 131, 13238–13239 Search PubMed.
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS.
- Y. Tamura, J. Minamikawa and M. Ikeda, Synthesis, 1977, 1–17 CrossRef CAS.
- C. R. Johnson, R. A. Kirchhoff and H. G. Corkins, J. Org. Chem., 1974, 39, 2458–2459 Search PubMed.
- J. Mendiola, J. A. Rincón, C. Mateos, J. F. Soriano, Ó. de Frutos, J. K. Niemeier and E. M. Davis, Org. Process Res. Dev., 2009, 13, 263–267 Search PubMed.
- J. J. Roberts and G. P. Warwick, Biochem. Pharmacol., 1961, 6, 205–216 Search PubMed.
- W. Onkenhout, W. M. van Loon, W. Buijs, A. van der Gen and N. P. Vermeulen, Drug Metab. Dispos., 1986, 14, 608–612 Search PubMed.
- D. H. Marchand, R. P. Remmel and M. M. Abdel-Monem, Drug Metab. Dispos., 1988, 16, 85–92 Search PubMed.
- M. Czerwinski, J. P. Gibbs and J. T. Slattery, Drug Metab. Dispos., 1996, 24, 1015–1019 Search PubMed.
- A. J. L. Cooper, I. R. Younis, Z. V. Niatsetskaya, B. F. Krasnikov, J. T. Pinto, W. P. Petros and P. S. Callery, Drug Metab. Dispos., 2008, 36, 1546–1552 Search PubMed.
- I. R. Younis, M. Elliott, C. J. Peer, A. J. L. Cooper, J. T. Pinto, G. W. Konat, M. Kraszpulski, W. P. Petros and P. S. Callery, J. Pharmacol. Exp. Ther., 2008, 327, 770–776 Search PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS.
- See Electronic Supplementary Information for full details†.
- J. W. Donovan and T. M. White, Biochemistry, 1971, 10, 32–38 Search PubMed.
- A. S. Nashef, D. T. Osuga, H. S. Lee, A. I. Ahmed, J. R. Whitaker and R. E. Feeney, J. Agric. Food Chem., 1977, 25, 245–251 Search PubMed.
- G. Federici, S. Dupre, R. M. Matarese, S. P. Solinas and D. Cavallini, Int. J. Pept. Protein Res., 1977, 10, 185–189 Search PubMed.
- E. Helmerhorst and G. B. Stokes, Biochemistry, 1983, 22, 69–75 Search PubMed.
- A. K. Galande, J. O. Trent and A. F. Spatola, Biopolymers, 2003, 71, 534–551 CrossRef CAS.
- T. Mukaiyama, M. Usui, E. Shimada and K. Saigo, Chem. Lett., 1975, 1045–1048 CAS.
- D. Crich and I. Sharma, Angew. Chem. Int. Ed., 2009, 48, 2355–2358 CAS.
- T. L. Gilchrist and C. J. Moody, Chem. Rev., 1977, 77, 409–435 CrossRef CAS.
- R. G. Wallace, Aldrichim. Acta, 1980, 13, 3–11 Search PubMed.
- E. M. Kosower, Acc. Chem. Res., 1971, 4, 193–198 CrossRef CAS.
- G. A. Doldouras and J. Kollonitsch, J. Am. Chem. Soc., 1978, 100, 341–342 CrossRef CAS.
- R. Okamoto, S. Souma and Y. Kajihara, J. Org. Chem., 2009, 74, 2494–2501 CrossRef CAS.
- Since 22 was synthesized as a mixture of meso and d/l, alkyation and cyclization will result in diastereomeric intermediates before converging to the dehydroalanine product. Though the rate of bis-alkylation and elimination of these diastereomers may differ, the good yields in model systems and high conversions on protein substrates suggest that any difference in these rates is not detrimental to the synthesis of dehydroalanine.
- Y. A. Lin, J. M. Chalker and B. G. Davis, J. Am. Chem. Soc., 2010, 132, 16805–16811 CrossRef CAS.
- Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642–9643 CrossRef CAS.
- S. Muyldermans, Rev. Mol. Biotechnol., 2001, 74, 277–302 Search PubMed.
- M. M. Harmsen and H. J. De Haard, Appl. Microbiol. Biotechnol., 2007, 77, 13–22 Search PubMed.
- B. Bader, K. Kuhn, D. J. Owen, H. Waldmann, A. Wittinghofer and J. Kuhlmann, Nature, 2000, 403, 223–226 CrossRef CAS.
- D. Macmillan, R. M. Bill, K. A. Sage, D. Fern and S. L. Flitsch, Chem. Biol., 2001, 8, 133–145 CrossRef CAS.
- J. J. Smith, D. W. Conrad, M. J. Cuneo and H. W. Hellinga, Protein Sci., 2005, 14, 64–73 CAS.
- B. G. Davis, Pure Appl. Chem., 2009, 81, 285–298 CrossRef CAS.
- J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011 DOI:10.1021/ar200056q , In press.
- C. J. Lote and J. B. Weiss, FEBS Lett., 1971, 16, 81–85 Search PubMed.
- J. B. Weiss, C. J. Lote and H. Bobinski, Nat. New Biol., 1971, 234, 25–26 Search PubMed.
- T. J. Oman, J. M. Boettcher, H. Wang, X. N. Okalibe and W. A. van der Donk, Nat. Chem. Biol., 2011, 7, 78–80 Search PubMed.
- N. E. Zachara and G. W. Hart, Chem. Rev., 2002, 102, 431–438 CrossRef CAS.
- R. Jefferis, Trends Pharmacol. Sci., 2009, 30, 356–362 Search PubMed.
- R. Jefferis, in Post-translational Modification of Protein Biopharmaceuticals, ed. G. Walsh, Wiley-VCH Verlag GmbH & Co., Weinheim, Germany, 2009, pp. 79–108 Search PubMed.
- G. M. Watt, J. Lund, M. Levens, V. S. Kumar Kolli, R. Jefferis and G.-J. Boons, Chem. Biol., 2003, 10, 807–814 CrossRef CAS.
- H. Li, N. Sethuraman, T. A. Stadheim, D. Zha, B. Prinz, N. Ballew, P. Bobrowicz, B.-K. Choi, W. J. Cook, M. Cukan, N. R. Houston-Cummings, R. Davidson, B. Gong, S. R. Hamilton, J. P. Hoopes, Y. Jiang, N. Kim, R. Mansfield, J. H. Nett, S. Rios, R. Strawbridge, S. Wildt and T. U. Gerngross, Nat. Biotechnol., 2006, 24, 210–215 CrossRef CAS.
- Y. Wei, C. Li, W. Huang, B. Li, S. Strome and L.-X. Wang, Biochemistry, 2008, 47, 10294–10304 CrossRef CAS.
- T. I. Potgieter, M. Cukan, J. E. Drummond, N. R. Houston-Cummings, Y. Jiang, F. Li, H. Lynaugh, M. Mallem, T. W. McKelvey, T. Mitchell, A. Nylen, A. Rittenhour, T. A. Stadheim, D. Zha and M. d'Anjou, J. Biotechnol., 2009, 139, 318–325 Search PubMed.
- Y. Mimura, R. Jefferis, Y. Mimura-Kimura, J. Abrahams and P. M. Rudd, in Therapeutic Monoclonal Antibodies: From Bench to Clinic, ed. Z. An, John Wiley & Sons, Inc., Hoboken, NJ, 2009, pp. 67–89 Search PubMed.
- B. G. Davis, Chem. Rev., 2002, 102, 579–601 CrossRef CAS.
- D. P. Gamblin, E. M. Scanlan and B. G. Davis, Chem. Rev., 2009, 109, 131–163 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, 1H and 13C NMR spectra for all novel compounds, HPLC and MS data for peptide substrates, and ESI-MS for all protein reactions. See DOI: 10.1039/c1sc00185j |
| This journal is © The Royal Society of Chemistry 2011 |