Catalytic asymmetric [4 + 2] additions with aliphatic nitroalkenes†
Keith J.
Bartelson
,
Ravi P.
Singh
,
Bruce M.
Foxman
and
Li
Deng
*
Department of Chemistry, Brandeis University, Waltham, MA 02454-9110, USA. E-mail: deng@brandeis.edu; Fax: +1 781 736 2516; Tel: +1 781 736 2529
First published on 18th July 2011
Abstract
We describe an unprecedented cycloaddition reaction of 2-pyrones with aliphatic nitroalkenes catalyzed by a new bifunctional cinchona alkaloid-derived catalyst bearing a bulky TIPS-ether at the 9-position. The [2.2.2] bicyclic adducts were obtained in good yield with excellent diastereo- and enantioselectivity. Carbon isotope effects were measured by 13C NMR and are indicative of a stepwise mechanism. Finally, a synthetic application is demonstrated, highlighting the utility of the cycloadducts.
Introduction
Six-membered ring structures are prevalent in natural and synthetic products. Consequently, asymmetric transformations enabling rapid and stereoselective construction of stereochemically defined six-membered rings are of particular importance in organic synthesis. The development of asymmetric [4 + 2] addition reactions, such as the Diels–Alder reaction, has largely focused on those involving carbonyl-based dienophiles.1 Nonetheless, asymmetric [4 + 2] additions with non carbonyl-based dienophiles such as α,β-unsaturated nitriles and nitroalkenes expand upon the range of chiral six-membered ring structures accessible by asymmetric [4 + 2] additions.2 For example, an asymmetric [4 + 2] addition between a diene and a nitroalkene followed by the reduction of the nitro group3 provides a rapid construction of chiral cyclohexyl amines, a class of highly valuable chiral building blocks (Scheme 1).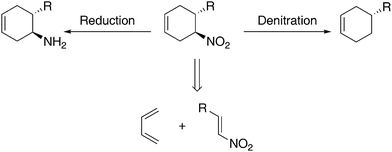 | ||
| Scheme 1 Nitrocyclohexene synthesis and common transformations. | ||
Great strides have been made toward the synthesis of functionalized chiral nitrocyclohexanes based on a strategy featuring in situ generation of activated dienes from enonesviaenamine catalysis by chiral amines. Barbas first demonstrated the concept with acyclic enones and aromatic nitroalkenes, albeit in moderate enantioselectivity (38% ee).2b Exploring this strategy with five- to seven-membered cyclic enones, Cordova obtained high diastereoselectivity and enantioselectivity (>25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 32–86% ee).2c In 2009, Xu developed an improved secondary chiral amine catalyst for this reaction, achieving excellent selectivities with cyclic enones (>25:1 dr, 83–96% ee).2d Shortly thereafter, Melchiorre reported the use of 9-amino cinchona alkaloids to achieve high selectivities with acyclic enones (2:1 to >19:1 dr and 88–99% ee).2e
:1 dr, 32–86% ee).2c In 2009, Xu developed an improved secondary chiral amine catalyst for this reaction, achieving excellent selectivities with cyclic enones (>25:1 dr, 83–96% ee).2d Shortly thereafter, Melchiorre reported the use of 9-amino cinchona alkaloids to achieve high selectivities with acyclic enones (2:1 to >19:1 dr and 88–99% ee).2e
Another successful strategy for the formation of functionalized chiral nitrocyclohexanes involves reaction cascades with nitroalkenes in which Michael addition/aldol reactions are mediated by enamine and iminium catalysis, respectively.4,5 The reacting partners of the nitroalkenes are typically easily enolizable β-ketoesters, γ,δ- unsaturated β-ketoesters, aldehydes, or malonates containing additional electrophilic functionalities.
Notably, these aforementioned asymmetric reactions have so far successfully been applied to almost exclusively aromatic nitroalkenes. To our knowledge, only a single documentation in the literature exists for the formation of a nitrocyclohexane from an aliphatic nitroalkene with synthetically useful selectivity; a tandem Michael/Henry reaction of (E)-2-(nitrovinyl)cyclohexane with 2,5-dihydroxy-3,4-dihydrofuran under the catalysis of a diphenylprolinol silyl ether, developed by Hayashi (45% yield, 99% ee).5b Consequently, the development of highly enantioselective and diastereoselective catalytic asymmetric [4 + 2] additions or ring-forming reaction cascades that are generally applicable to the synthetically important but less active aliphatic nitroalkenes remains an important but challenging task in asymmetric synthesis. In this article, we wish to report a highly diastereoselective and enantioselective [4 + 2] addition between 2-pyrones and aliphatic nitroalkenes promoted by a cinchona alkaloid-derived bifunctional catalyst.
Results and discussion
We2a,6 and others7 have shown that cinchona alkaloids with hydrogen bond donors can promote highly enantioselective and diastereoselective Diels–Alder reactions with inactive but synthetically important 3-hydroxy-2-pyrones in a stereospecific fashion. Thus, we postulated that such bifunctional catalysis may enable us to develop an asymmetric Diels–Alder reaction of 2-pyrones with alkyl nitroalkenes. Accordingly we initiated an investigation aiming at the realization of this transformation.Our investigation commenced with a catalyst screen (Fig. 1) for the model reaction of 3-hydroxy-2-pyrone 6A with (E)-1-nitro-1-pentene 7a (Table 1). In the absence of catalyst the reaction did not proceed (entry 1, Table 1), however in the presence of 5 mol% of a natural or modified cinchona alkaloid at room temperature in dichloromethane the reaction readily reached full conversion. Surprisingly, in addition to the expected adducts endo-8Aa and exo-9Aa, another stereoisomer of the [4 + 2] adduct, 10Aa, was also obtained as a minor product (Table 1). The formation of 10Aa could arise from a stepwise conjugate addition/aldol reaction cascade (vide infra).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | T/°C | t/h |
8Aa:9Aa:10Aab |
eec8 (%) |
| a Unless noted, reactions were performed with 0.10 mmol 6A and 0.20 mmol 7a in 50 μL solvent (2.0 M). b Determined by 1H NMR analysis. c Determined by chiral HPLC analysis. See ESI1 for details. d Reaction was performed with 100 μL solvent (1.0 M). | ||||||
| 1 | — | CH2Cl2 | 23 | 24 | — | — |
| 2 | 1 | CH2Cl2 | 23 | 1.5 | 34:62:4 |
9 |
| 3 | 2 | CH2Cl2 | 23 | 4.5 | 78:12:10 |
49 |
| 4 | 3a | CH2Cl2 | 23 | 4.5 | 62:30:8 |
73 |
| 5 | 3b | CH2Cl2 | 23 | 3 | 66:23:11 |
74 |
| 6 | 3c | CH2Cl2 | 23 | 1.5 | 65:29:6 |
78 |
| 7 | 4a | CH2Cl2 | 23 | 1.5 | 71:21:8 |
88 |
| 8 | 4b | CH2Cl2 | 23 | 2 | 72:19:9 |
88 |
| 9 | 4c | CH2Cl2 | 23 | 1.5 | 73:22:5 |
87 |
| 10 | 4d | CH2Cl2 | 23 | 1.5 | 72:19:9 |
87 |
| 11 | 4e | CH2Cl2 | 23 | 1.5 | 69:25:6 |
84 |
| 12 | 4f | CH2Cl2 | 23 | 1.5 | 68:27:5 |
84 |
| 13 | 4b | THF | 23 | 1.5 | 80:14:6 |
92 |
| 14 | 4b | THF | −20 | 5 | 80:12:8 |
96 |
| 15d | 4b | THF | −20 | 20 | 82:8:10 |
96 |
| 16d | 5 | THF | −20 | 24 | 69:22:9 |
61 |

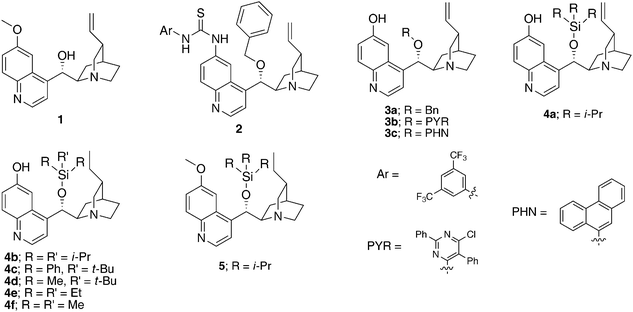 | ||
| Fig. 1 Cinchona alkaloid catalysts screened. | ||
Quinidine 1 afforded low endo-selectivity and enantioselectivity (entry 2, Table 1). Quinidine-derived 6′-thiourea catalyst 2 provided endo-adduct 8Aa as the major product but in only 49% ee (entry 3, Table 1). Interestingly, a dramatic increase in enantioselectivity was observed with 6′-OH cinchona alkaloid 3a bearing a benzyl group as the 9-substituent (entry 4 vs. 3, Table 1). However, catalyst tuning via introduction of other aromatic groups as the 9-substituent failed to improve the enantioselectivity (entry 4 vs. 5 and 6, Table 1).
Postulating that a silyl ether group at the 9-position may provide a steric environment different than that presented by aryl-based groups (benzyl, PYR, PHN), 9-silyl ether cinchona alkaloid 4a was prepared8 and utilized to catalyze the model reaction. Gratifyingly, 4a afforded significantly improved diastereo- and enantioselectivity over those by 3a–c (entry 7 vs. 6, Table 1). The corresponding dihydroquinidine-derived analogue 4b was found to perform equally well as catalyst 4a (entry 8 vs. 7, Table 1).
To probe the effect of the bulk of the silyl ether on the catalytic selectivity, reactions with silyl ether catalysts 4b-4f were investigated.8Catalysts derived from larger silyl ethers such as TIPS, TBDPS and TBS (4b, 4c and 4d, respectively) provided similarly high levels of selectivity (entries 8–10, Table 1). On the other hand, smaller silyl ether catalysts such as those derived from TMS and TES (4e and 4f) gave slightly diminished results (entries 11 and 12, Table 1). The highest enantioselectivity could be achieved with triisopropylsilyl substituted catalyst 4b. It should be noted that 6′–OH cinchona alkaloids bearing a 9-silyl ether group have not yet been reported in the literature, although both a 6′-thiourea cinchona alkaloid derivative bearing a silyl ether group9 and trimethylsilylquinine10 were reported to be effective catalysts for asymmetric desymmetrization-fragmentations and ketene-aldehyde cycloadditions, respectively.
With optimal catalyst 4b in hand the remaining reaction parameters were screened. At room temperature THF was found to be the optimal solvent, in which both the diastereoselectivity and the enantioselectivity were noticeably improved (entry 13 vs. 8, Table 1).8 Cooling the reaction to −20 °C provided additional increases in both diastereoselectivity and enantioselectivity (entry 14, Table 1). By lowering the reaction concentration to 1.0 M, the reaction proceeded with the highest levels of selectivity to give the endo-adduct 8a in 96% ee and with high endo/exo selectivity (entry 15, Table 1).
To gauge the impact of the 6′–OH group on the catalytic selectivity of 4b, a reaction with the corresponding 6′–OMe cinchona alkaloid 5 was investigated. It was found that both the diastereoselectivity and enantioselectivity deteriorated dramatically (entry 16 vs. 15, Table 1), consistent with the proposal that 6′–OH catalyst 4b promotes this reaction via bifunctional catalysis.
The substrate scope was explored with respect to both the nitroalkenes and the 2-pyrones (Table 2). Importantly, reactions proceeded cleanly in excellent diastereo- and enantioselectivity with both linear (entries 1–3, Table 2) and branched aliphatic nitroalkenes (entry 4, Table 2). The reaction also tolerates α-branched nitroalkenes, providing adducts 8Ae and 8Af with high enantioselectivity but with slightly lower diastereoselectivity (entries 5 and 6, Table 2). It is worth noting that 10 was not observed in reactions with α-branched nitroalkenes as it was with the other nitroalkenes. Both unsubstituted and substituted homobenzyl groups were viable substrates, affording adducts 8Ag and 8Ah with good diastereoselectivity and excellent enantioselectivity (entries 7 and 8, Table 2). Additionally nitroalkenes bearing a benzyl protected alcohol (7i) or a trimethylsilyl group (7j) gave excellent results (entries 9 and 10, Table 2). Finally, substituted 2-pyrones were well tolerated as chloro, bromo and methyl substitution provided cycloadducts 8Ba, 8Ca and 8Da with similarly high levels of enantio- and diastereoselectivity (entries 11–13, Table 2). Though endo-adducts 8 could be readily separated from exo-adducts 9, the separation of 8 and 10 proved unfeasible. In all cases, except where 10 was not produced, a mixture of 8 and 10 was obtained with the ratio of 8:10 ranging from 89:11 to 94:6 as determined by NMR analysis (Table 2).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | R | R′ | Product | T/°C | t/h |
8:9:10b |
Yield (8 + 10)c(%) |
8:10b |
eed8 (%) |
| a Unless noted, reactions were performed with 0.20 mmol 6 and 0.40 mmol 7 in 200 μL THF (1.0 M). b Determined by 1H NMR analysis. c Isolated yield of 8 + 10. d Determined by chiral HPLC analysis. e Absolute configuration determined by X-ray analysis of a single crystal of 8Ah (see ESI1). f Inseparable mixture of 8Ca, 9Ca and 10Ca. | |||||||||
| 1 | H |

|
8Aa | −20 | 20 | 82:8:10 |
82 | 92:8 |
96 |
| 2 | H |

|
8Ab | −20 | 28 | 81:9:10 |
77 | 91:9 |
95 |
| 3 | H |

|
8Ac | 0 | 21 | 85:8:7 |
75 | 94:6 |
95 |
| 4 | H |

|
8Ad | −20 | 28 | 80:9:11 |
81 | 92:8 |
97 |
| 5 | H |

|
8Ae | 0 | 46 | 77:23:0 |
74 | 100:0 |
96 |
| 6 | H |

|
8Af | 0 | 54 | 78:22:0 |
81 | 100:0 |
96 |
| 7 | H |

|
8Ag | −20 | 20 | 74:15:11 |
79 | 90:10 |
96 |
| 8 | H |

|
8Ah | −20 | 20 | 74:18:8 |
68 | 92:8 |
95e |
| 9 | H |

|
8Ai | 0 | 20 | 79:11:10 |
78 | 89:11 |
96 |
| 10 | H |

|
8Aj | −20 | 4.5 | 90:3:7 |
77 | 93:7 |
95 |
| 11 | Cl |

|
8Ba | 0 | 28 | 89:1:10 |
86 | 92:8 |
96 |
| 12 | Br |

|
8Ca | 0 | 17 | 80:12:8 |
56f | 91:9 |
96 |
| 13 | Me |

|
8Da | 0 | 18 | 90:0:10 |
75 | 90:10 |
98 |

A synthetic application of this new asymmetric [4 + 2] addition with aliphatic nitroalkenes is demonstrated in the first enantioselective synthesis of the sphingosine analogue 15, which displays antiparasitic activities (L. Amazonensis IC50 = 0.26 μM and T. CruziIC50 = 0.19 μM) that exceed those of the current clinically used drugs pentamidine (L. Amazonensis IC50 = 29.4 μM) and benznidazol (T. CruziIC50 = 7.4 μM).11 These parasites lead to leishmaniasis and trypanosomiasis, diseases that manifest themselves through skin deforming lesions and can lead to death if left untreated. To our knowledge, only one racemic synthesis of 15 has been reported in the literature by del Olmo.11 As shown in Scheme 2, our enantioselective synthesis began with the reduction of a mixture of 8Ac and 10Ac (94:6 ratio) to the corresponding amines, which, without purification, were subjected to Boc2O. The resulting N-Boc amine 11, upon chromatographic purification, was obtained as a pure stereoisomer in 70% yield from 8Ac. A retro Diels–Alder extrusion of CO2 from 11 furnished the chiral enone 12, which was reduced in a highly diastereoselective manner to the corresponding allylic alcohol 13. Lithium aluminum(tri(tert- butoxy)hydride was found to be essential for the high diastereoselectivity as other reducing agents gave significantly poorer results. Subsequent hydrogenation of crude 13 afforded the desired N-Boc amino alcohol 14 as a single diastereomer. Standard deprotection of the Boc-protected amine afforded the desired amino alcohol 15, the structure of which was confirmed by comparison to an authentic sample prepared via del Olmo's route.8
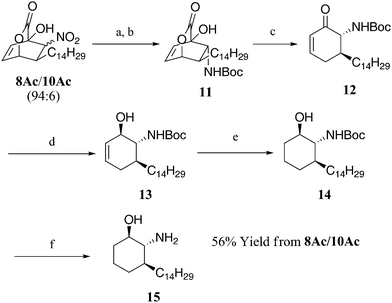 | ||
| Scheme 2 Enantioselective synthesis of 15. Reagents and conditions: (a) Sn powder, 1.0 M HCl, THF, 20 °C for 1 h then 60 °C for 1 h; (b) Boc2O, H2O, THF, 20 °C, 16 h, 70% (two steps), 96% ee; (c) toluene, 150 °C, 2.5 h, 89%, 96% ee; (d) Li(t-BuO)3AlH, THF, 0 °C, 0.5 h; (e) Pd/C, H2, THF, 20 °C, 4 h, 94% (two steps); (f) TFA, CH2Cl2, 20 °C, 4 h, 96%. THF = tetrahydrofuran, TFA = trifluoroacetic acid. | ||
In order to determine whether the cycloadducts were formed through a concerted pathway or a stepwise Michael addition/Henry reaction cascade, we investigated the carbon isotope effects for the [4 + 2] reaction between pyrone 6A and nitroalkene 7a using Singleton's method at natural abundance (Fig. 2).12 If the [4 + 2] reaction were to proceed via a concerted Diels–Alder pathway, carbon isotope effects would be expected at both the α- and β-positions of nitroalkene 7a and only products 8Aa and 9Aa could be formed, meaning that 10Aa could conceivably arise via epimerization (pathway A, Fig. 2).13 On the other hand, if the reaction were to proceed though a stepwise Michael addition/Henry reaction cascade, a carbon isotope effect would be expected at only one of the alkene carbons of 7a, depending on which step in the cascade is the rate-determining step, and 8Aa, 9Aa, and 10Aa could be formed (pathway B, Fig. 2).
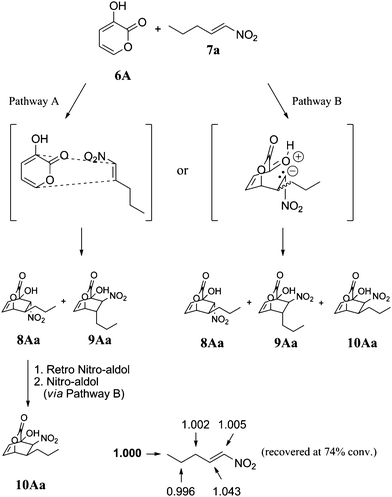 | ||
| Fig. 2 Carbon isotope effects (R/R0) calculated for nitroalkene 7a. The methyl carbon (value in bold) was taken as the internal standard. | ||
When the reaction was stopped at 74% conversion, the 13C ratio of each carbon in the recovered nitroalkene to the same carbon in virgin nitroalkene was measured using quantitative 13C NMR. As shown in Fig. 2, the only appreciable carbon isotope effect was observed at the β-position of the nitroalkene [13C(recovered)/13C(virgin) = 1.043, average of three runs], thus indicating that the [4 + 2] cycloaddition proceeds through a stepwise mechanism to form adducts 8–10.8
Conclusions
In conclusion, we have developed a new bifunctional cinchona alkaloid-derived catalyst that promotes an unprecedented asymmetric organocatalytic [4 + 2] cycloaddition reaction between 2-pyrones and aliphatic nitroalkenes. A carbon kinetic isotope effect study proves that the reaction does not occur through a concerted pathway but is instead stepwise. To our knowledge, this is the first example of an asymmetric organocatalytic [4 + 2] cycloaddition reaction successfully employing aliphatic nitroalkenes as dienophile partners. The synthetic potential of this methodology for the asymmetric synthesis of six-membered cyclic amines was illustrated in the development of a concise enantioselective synthesis of the antiparasitic compound 15.Acknowledgements
We are grateful for the generous financial support of the National Institutes of Health (GM-61591).Notes and references
- For reviews of asymmetric Diels–Alder reactions, see:
(a) E. J. Corey, Angew. Chem., Int. Ed., 2002, 41, 1650 CrossRef CAS
; (b) A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef
- For asymmetric Diels–Alder reactions with α,β-unsaturated nitriles, see:
(a) Y. Wang, H. Li, Y.-Q. Wang, Y. Liu, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2007, 129, 6364 CrossRef CAS
-
N. Ono, The Nitro Group in Organic Synthesis, John Wiley & Sons, Chichester, UK, 2000, ch. 8 Search PubMed
- For cascade reactions involving nitroalkenes in the synthesis of nitrocyclohexenes catalyzed by proline derived catalysts:
(a) D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861 CrossRef CAS
- For cascade reactions involving nitroalkenes in the synthesis of nitrocyclohexanes catalyzed by cyclic diamine derived catalysts, see:
(a) Y. Hoashi, T. Yabuta and Y. Takemoto, Tetrahedron Lett., 2004, 45, 9185 CrossRef CAS
- R. P. Singh, K. Bartelson, Y. Wang, H. Su, X. Lu and L. Deng, J. Am. Chem. Soc., 2008, 130, 2422 CrossRef CAS
- For reviews of Diels–Alder reactions with 2-pyrones, see:
(a) K. Afarinkia, V. Vinader, T. D. Nelson and G. H. Posner, Tetrahedron, 1992, 48, 9111 CrossRef CAS
- See ESI† for details.
- G. Dickmeiss, V. De Sio, J. Udmark, T. B. Poulsen, V. Marcos and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 6650 CrossRef CAS
- C. Zhu, X. Shen and S. G. Nelson, J. Am. Chem. Soc., 2004, 126, 5352 CrossRef CAS
- O. Rebollo, E. del Olmo, G. Ruiz, J. L. López-Pérez, A. Giménez and A. San Feliciano, Bioorg. Med. Chem. Lett., 2008, 18, 184 CrossRef CAS
-
(a) D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357 CrossRef CAS
-
(a) D. A. Singleton, B. E. Schulmeier, C. Hang, A. A. Thomas, S.-W. Leung and S. R. Merrigan, Tetrahedron, 2001, 57, 5149 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures, analytical data, and 1H/13C spectra. CCDC reference number 827937. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00326g |
| This journal is © The Royal Society of Chemistry 2011 |