Insight into the construction of metal–organic polyhedra: metal–organic cubes as a case study†
Mohamed H.
Alkordi
ab,
Jonathan L.
Belof
b,
Edwin
Rivera
b,
Lukasz
Wojtas
b and
Mohamed
Eddaoudi
*ab
aAdvanced Membranes and Porous Materials Center, 4700 King Abdullah University of Science and Technology (KAUST), Thuwal, KSA. E-mail: Mohamed.Eddaoudi@kaust.edu.sa
bDepartment of Chemistry, University of South Florida, Tampa, Florida 33620-5250, USA
First published on 20th June 2011
Abstract
Systematic studies were conducted to gain a better understanding of the metal–organic cubes (MOCs) directed assembly and their crystallization under predetermined reaction conditions, i.e. charge and size of metal ions, solvent type, counter anions, pH, and temperature. Four novel metal–organic materials are constructed via solvothermal reactions of different metal ions and 2,2′-(1H-imidazole-4,5-diyl)di-1,4,5,6-tetrahydropyrimidine, namely [Co8(C11N6H15)12]Cl12·4H2O (1), [Ni4(C11N6H15)4](NO3)4·4DMF (2), {Cd(C11N6H15)(NO3)·DMF}n (3), and [In8(C11N6H15)12](NO3)12·4H2O (4). In addition, syntheses and crystal structures for compounds 1(a–f), constructed under deliberately modified reaction conditions of 1, are reported. In compounds 1(a–f), the CoIII-based cationic MOCs crystallize in various packing arrangements in the presence of different counter-ions. Discrete MOCs retain their structural integrity, when crystalline solid was dissolved in water, under various pH (2.03–8.07) and temperatures (298–333 K), as confirmed by solution NMR studies. The assembly of the discrete MOC, from its basic molecular building blocks under mild reaction conditions, is demonstrated and monitored through solution NMR and UV-vis studies.
Introduction
Metal–organic materials (MOMs) are regarded as a unique class of functional solid-state materials that can offer prospective answers to a number of current demanding applications in gas separation and storage, drug release, catalysis, small molecule sensing, ion exchange, and CO2 capture.1 Such attributes are directly correlated to the intrinsic properties of MOMs including, but not limited to, hybrid composition, tunable pore size and shape, and facile access to large surface areas.2 Ability to construct MOMs with desired properties and functionality, made-to-order solid state materials, has been recognized and attributed to advancements in the molecular building block (MBB) based assembly.3–5The MBBs are selected and designed to contain the proper shape, geometry, and functionality required for the construction of a MOM with a targeted topology. MOMs modularity and pre-assembly functionalization are directly correlated to the ability of consistently generating specific inorganic MBBs in situ. Therefore, it is vital to establish proper reaction conditions leading to the regular formation of the appropriate inorganic MBB. It is evident that judicial choice of pre-programmed MBBs, as well as careful design of the reaction conditions, necessary to afford such MBBs in situ, are two inseparable elements of design in any rational strategy for construction of functional MOMs. Nevertheless, only a few systematic studies aiming to understand the impact of reaction variables on the nature of isolated MOMs have appeared in the open literature.6 Herein, we report a systematic study pertaining to the assembly of metal–organic cubes (MOCs), constructed through single-metal-ion MBB approach, under variable reaction conditions.
Metal–organic polyhedra (MOPs),7,8 and MOCs in particular, with peripheral functionalities can further be employed as supermolecular building blocks (SBBs) with built-in structural information, decorating and expanding the vertices of targeted n-connected nets (n ≥ 8), towards construction of metal–organic frameworks (MOFs).9 This approach has been recently employed by our group to construct novel zeolite-like metal–organic frameworks (ZMOFs) based on 8-connected SBBs, MOCs.10
Reaction of 2,2′-(1H-imidazole-4,5-diyl)di-1,4,5,6-tetrahydro-pyrimidine, H-L1, and CoCl2·6H2O resulted in a crystalline solid containing discrete, cationic, CoIII-based MOCs, (1). This system is suitable, as an ideal case study, to assess effects of reaction conditions on the directed assembly and crystallization process of MOCs. This is mainly due to several unique attributes of the investigated MOC (1): (i) accessible crystal structure of 1, (ii) solubility of 1 in water and polar solvents yielding solvated MOCs, (iii) presence of multiple protons in L1, proved as useful probes in solution NMR structural characterization, (iv) pronounced effects of paramagnetic CoII ions in NMR spectra of coordinated L1, and (v) presence of absorption bands characteristic of Co-L1 complex(es) formation.
Experimental details
All reagents were commercial grade used without further purification. Syntheses of H-L1 and compound 1 are included, for further experimental details and syntheses procedures for compounds 1(a–f) and 2–4, see ESI.†2,2′-(1H-imidazole-4,5-diyl)di-1,4,5,6-tetrahydro-pyrimidine, H-L1
The organic ligand, H-L1, is prepared based on a modified reported procedure.11 A mixture of 1H-imidazole-4,5-dicarbonitrile (10 mmol) and excess of 1,3-diaminopropane in presence of sulfur (10 mmol) was refluxed for 1 h then vacuum dried. The solid obtained was suspended in water then filtered and dried at 80 °C to afford H-L1, as a white solid formulated as C11H16N6, (2.13 g, 9.2 mmol, 92%), Scheme 1. The purity of the ligand was confirmed by solution 1H NMR (D2O, 399.78 MHz): δ = 7.47 (s, 1H), 3.5 (t, J = 5.8 Hz, 8H), 1.97 (quin, J = 5.7 Hz, 4H). | ||
| Scheme 1 Synthesis of H-L1. | ||
[Co8(C11N6H15)12]Cl12·4H2O, 1
In a capped 25 mL scintillation vial, reaction of H-L1 (0.034 g, 0.15 mmol) and CoCl2·6H2O (0.0237 g, 0.1 mmol) in a mixture of N,N′-dimethylformamide (DMF) and water, 1 mL each, at 115 °C for 12 h resulted in red polyhedral crystals (0.04 g, 85.4% yield based on CoCl2) formulated as [Co8(C11N6H15)12]Cl12·4H2O using single-crystal X-ray diffraction structural analysis, Fig. 1.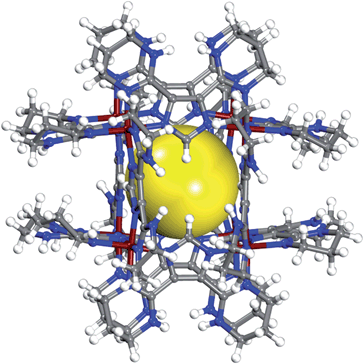 | ||
| Fig. 1 Crystal structure of the metal–organic cube (MOC) in 1, the twelve chloride counterions are omitted for clarity. Yellow sphere represent the largest sphere with diameter ∼4 Å that can fit inside the MOC excluding the vdW radii of inward atoms. C (gray), N (blue), H (white), Co (red). | ||
Results and discussion
X-ray crystal structures analyses
In all the reported structures, each mono-deprotonated L1 molecule coordinates to the metal ions in a bis-bidentate fashion and can be regarded as a linear linker bridging two metal ions. In L1, two nitrogen atoms of the 1,4,5,6-tetrahydro-pyrimidin-2-yl (thp) ring are pyridine-type (N3′) while the other two are pyrrole-type atoms (N1′). Its coordination to metal ions occurs only through the N3′ atoms of the thp rings, leaving the N1′ atoms available as hydrogen bond donors towards Cl−, Br−, (NO3)−, (SO4)2−, or (BF4)− counterions present in compounds 1, 1(a–f). For crystal structure description of compounds 1(a–f) and 2–4, see ESI.†Single crystal X-ray structure analysis of 1
In the crystal structure of 1, Fig. 1, discrete MOCs, crystallize in a cubic space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , are held together through a network of multiple weak C–H⋯Cl hydrogen bond interactions. In 1, the Co–N bond lengths of 1.911(3)–1.949(3) Å (thp), and 1.890(3)–1.906(3) Å (imidazolate) are in good agreement with bond lengths found in similar CoIII complexes, as determined from analysis of Cambridge Structural Database. The pyrrole-type nitrogen atoms of the thp rings, on the peripheral of the MOCs, are hydrogen bonded to chloride ions. Residual electron density in the Fourier difference map near N1′ atoms were assigned to hydrogen atoms, further supporting the proposed crystallographic model of mono deprotonated, imidazolate based ligand molecules. Each positively charged MOC (+12) is surrounded by twelve chloride counterions. Six chloride ions decorate the faces of a MOC, where each chloride ion is hydrogen bonded to three of the four nitrogen atoms on one face of the MOC (N1′⋯Cl distances of 3.096–3.185 Å). In addition, each of these chloride ions is hydrogen bonded to two hydrogen atoms (H⋯Cl distances of 2.697–2.739 Å, normalized data) on two C5′ atoms of a neighbouring cube. Five additional chloride ions, residing near the edges of the MOC and geometrically disordered over six positions, are simultaneously hydrogen bonded to one N1′ atom (N1′⋯Cl distance of 3.241 Å) and to two hydrogen atoms on the surface of a neighboring MOC (C–H⋯Cl distances of 2.697–2.788 Å, normalized data). The remaining chloride ion occupies the cavity inside the MOC. On each of the six faces of a MOC, two parallel imidazolate rings are separated by a centroid-to-centroid distance of 5.507 Å. In the crystal structure of 1, geometrical disorder between two sites for the methylene carbon atoms C5′ in the thp rings is observed.
, are held together through a network of multiple weak C–H⋯Cl hydrogen bond interactions. In 1, the Co–N bond lengths of 1.911(3)–1.949(3) Å (thp), and 1.890(3)–1.906(3) Å (imidazolate) are in good agreement with bond lengths found in similar CoIII complexes, as determined from analysis of Cambridge Structural Database. The pyrrole-type nitrogen atoms of the thp rings, on the peripheral of the MOCs, are hydrogen bonded to chloride ions. Residual electron density in the Fourier difference map near N1′ atoms were assigned to hydrogen atoms, further supporting the proposed crystallographic model of mono deprotonated, imidazolate based ligand molecules. Each positively charged MOC (+12) is surrounded by twelve chloride counterions. Six chloride ions decorate the faces of a MOC, where each chloride ion is hydrogen bonded to three of the four nitrogen atoms on one face of the MOC (N1′⋯Cl distances of 3.096–3.185 Å). In addition, each of these chloride ions is hydrogen bonded to two hydrogen atoms (H⋯Cl distances of 2.697–2.739 Å, normalized data) on two C5′ atoms of a neighbouring cube. Five additional chloride ions, residing near the edges of the MOC and geometrically disordered over six positions, are simultaneously hydrogen bonded to one N1′ atom (N1′⋯Cl distance of 3.241 Å) and to two hydrogen atoms on the surface of a neighboring MOC (C–H⋯Cl distances of 2.697–2.788 Å, normalized data). The remaining chloride ion occupies the cavity inside the MOC. On each of the six faces of a MOC, two parallel imidazolate rings are separated by a centroid-to-centroid distance of 5.507 Å. In the crystal structure of 1, geometrical disorder between two sites for the methylene carbon atoms C5′ in the thp rings is observed.
MALDI-TOF MS experiments
The MALDI-TOF mass spectra of 1, Fig. S1,† show a molecular ion peak of singly-charged species with wide isotope distribution and a maximum relative intensity at m/z = 3238.353 (calcd 3238.008) assigned to [Co8(C11N6H14)12]+. In addition to the observed molecular ion peak, additional molecular fragments were detected: m/z = 1966.323 (calcd 1966.632) [Co5(C11N6H16)2 (C11N6H14)5(H2O)3]+, 1677.198 (calcd 1677.449) [Co5(C11N6H16)(C11N6H14)5]+, 1444.994 (calcd 1445.305) [Co5(C11N6H14)5]+, 1157.737 (calcd 1157.250) [Co4 (C11N6H14)4]+, 811.339 (calcd 811.266) [Co2(C11N6H14) (C11N6H15)2]+, and 522.164 (calcd 522.204) [Co(C11N6H15)2]+. The plausible molecular structures matching the observed m/z ratios for singly-charged molecular ion and the observed fragments are schematically presented in Fig. 2. The observed molecular ion peak can be attributed to ionized MOC generated upon liberation of twelve HCl molecules.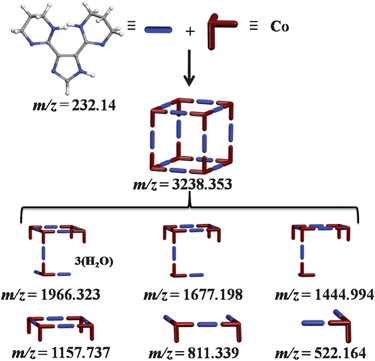 | ||
| Fig. 2 Schematic representation of the MOC and its dominant fragments observed in the MALDI-TOF mass spectra of 1. | ||
Nuclear magnetic resonance experiments
The ligand, H-L1, is an imidazole ring substituted by two 1,4,5,6-tetrahydropyrimidine (thp) rings. Each thp ring contains an amidine functional group and six methylene hydrogen atoms. Therefore, the characteristic features of such functionalities are expected to be observed in the NMR spectra of H-L1. For imidazole, even at high pH aqueous solution, it is common to observe rapid prototropic tautomerization involving the ring's nitrogen atoms N1(3) resulting in an observed average chemical environment for the C4(5) atoms.12 Furthermore, amidine functional groups, known to undergo rapid prototropic tautomerization, are expected to result in an average chemical shift for the C4′(6′) atoms of the thp rings. In addition to prototropic tautomerization, protonation of the basic nitrogen atoms of amidine and/or imidazole, especially in aqueous solution, is expected to form amidinium and/or imidazolium ions, respectively.Therefore, it is expected that either protonation or rapid tautomerization involving the basic nitrogen functionalities present in H-L1, or a combination thereof, in aqueous solution, will result in observed average chemical shifts for C4(5) of imidazole and the C4′(6′) of thp rings, Scheme 2.
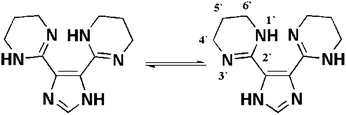 | ||
| Scheme 2 Numbering and prototropic tautomerization in H-L1. | ||
Indeed, H-L1, in either D2O or DMSO-d6 solution, exhibits a simplified 1H NMR pattern (DMSO-d6, 399.78 MHz): δ = 7.15 (s, 1 H), 3.35 (t, J = 5.65 Hz, 8 H), 1.75 ppm (quin, J = 5.70 Hz, 4 H), Fig. S2.† This indicates absence of geminal coupling between homotopic methylene protons, which could be ascribed to increased symmetry in the thp ring, a mirror plane through C2′ and C5′, due to a fast tautomerization and/or protonation of the basic amidine nitrogen atoms, vide supra. Additionally, the 13C NMR spectrum of H-L1 (DMSO-d6, 100.53 MHz): δ = 153.46, 144.41, 132.59, 39.67, 20.16 ppm, clearly indicates chemical shift equivalence for C4′(6′) atoms of the thp rings (δ = 39.67 ppm) and the C4(5) atoms of the imidazole ring (δ = 132.59 ppm). The D2O 1H NMR spectrum, pD = 11.2, of H-L1 (D2O, 399.78 MHz): δ = 7.47 (s, 1 H), 3.5 (t, J = 5.7 Hz, 8 H), 1.97 ppm (quin, J = 5.8 Hz, 4 H) referenced to the signal of DSS at 0 ppm, Fig. 3, exhibits an identical pattern to that acquired in DMSO-d6 but with noticeable small shifts toward lower shielding which could be ascribed to formation of the conjugate acid(s) through protonation (deuteration) in aqueous solution of H-L1, as expected for such basic functionalities.
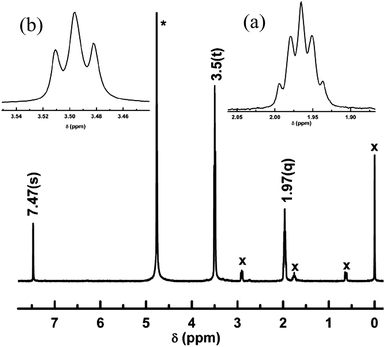 | ||
| Fig. 3 1H NMR spectrum of H-L1 in D2O acquired at 298 K with inserts of magnified (a) quintet at 1.97 ppm and (b) triplet at 3.5 ppm. * = Solvent peak, x = DSS peaks, used as an internal standard. | ||
Accordingly, it is anticipated that coordination of the H-L1 through N3′ to a Co ion will prohibit tautomerization and/or protonation, leading to a reduced symmetry of thp rings. Furthermore, tris-chelation upon coordination of three ligand molecules to a single Co ion is expected to introduce a chiral center in the formed complex, analogous to the well-known chiral tris(ethylenediamine)-Co complexes. The aforementioned structural characteristics of the ligand molecules upon coordination to Co ions permit utilization of NMR spectroscopy to probe and assess ligand binding to Co ions. The 1H NMR spectrum obtained upon dissolving crystals of 1 in D2O at room temperature (referred to as 1S), Fig. 4, shows a distinct spin–spin splitting pattern for the proton signals of thp rings, characteristic of diasteriotopic protons. Observation of diasteriotopic methylene protons in coordinated L1 molecules can be ascribed to the presence of chiral centers (CoIII–N6 tris-chelates)13 in the solvated species, and thus suggests the presence of solvated MOCs in the aqueous solution. The 1H NMR spectrum of 1S exhibits seven distinct chemical shifts attributed to the six diasteriotopic methylene protons, 1H NMR (D2O, 399.78 MHz): δ = 6.09 (s, 1H), 3.45–3.56 (m, 2 H), 3.36–3.44 (m, 2H), 3.20–3.31 (m, 2H), 2.80–2.84 (m, 2H), 2.05–2.18 (m, 2H), 1.61–1.75 ppm (m, 2H), Table 1. The proposed assignment for the proton peaks in the 1H NMR spectrum of 1S, indicating bis-bidentate coordination of L1 to cobalt ion centres, is further supported by the 13C NMR spectrum (D2O, 100.53MHz): δ = 155.7, 143.6, 135.9, 43.7, 39.2, 21.4 ppm. The 13C spectrum reflects chemical shift equivalence between imidazolate C4 and C5 atoms (143.6 ppm) and chemically non-equivalent C4′ (43.7 ppm) and C6′(39.2 ppm) of the thp rings. The majority of 1H and 13C NMR signals of 1S are shifted toward higher chemical shift values relative to those of H-L1, indicating metal ion chelation by the ligand molecules. However, the 13C NMR signal of the imidazolate C2 atom appears at higher shielding in 1S compared to H-L1 (Δδ = −0.987 ppm), most probably due to an increase in electron density caused by deprotonation of acidic imidazole proton prior to second Co coordination by the imidazolate ring. Distortionless enhancement through polarization transfer (DEPT), 1H-{1H} correlation (gCOSY) and 1H-{13C} heteronuclear single quantum correlation (gHSQC) NMR spectra for 1S further support the chemical shifts and spin systems assignments, Fig. S3–S5.† In addition, the relative integrals of 1H signals strongly indicate maintained integrity of the MOC and absence of uncoordinated ligands in solution. The absence of paramagnetic proton signals in 1H NMR spectrum of 1S confirms that the aqueous solution contains the diamagnetic, low-spin CoIII complex(es), in agreement with the crystallographically determined structure of CoIII-MOCs in 1.
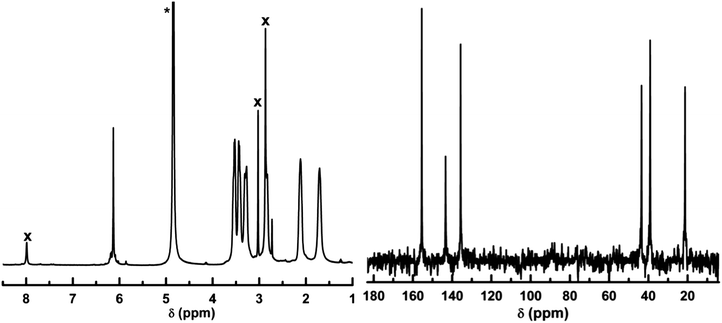 | ||
| Fig. 4 1H NMR spectrum (left) and 13C NMR spectrum (right) of 1S in D2O, spectra acquired at 298 K, proton spectrum referenced to the DSS singlet at 0 ppm as internal standard. * = Solvent peak, x = DMF peaks. | ||
| Nucleus | H-L1 | 1S | ||
|---|---|---|---|---|
| 1H | 13C | 1H | 13C | |
| H(2)Im/C2 Im | 7.15 (s, 1 H) | 144.41 | 6.09 (s, 1H) | 143.6 |
| H6′(a*) thp/C6′ thp | 3.35 (t, J = 5.65 Hz, 2H) | 39.67 | 3.36–3.44 (m, 2H) | 39.2 |
| H6′(e*) thp/C6′ thp | 3.35 (t, J = 5.65 Hz, 2H) | 39.67 | 3.45–3.56 (m, 2 H) | 39.2 |
| H5′(a) thp/C5′ thp | 1.75 (quin, J = 5.70 Hz, 2H) | 20.16 | 1.61–1.75 (m, 2H) | 21.4 |
| H5′(e) thp/C5′ thp | 1.75 (quin, J = 5.70 Hz,2H) | 20.16 | 2.05–2.18 (m, 2H) | 21.4 |
| H4′(a) thp/C4′ thp | 3.35 (t, J = 5.65 Hz, 2H) | 39.67 | 2.80–2.84 (m, 2H) | 43.7 |
| H4′(e) thp/C4′ thp | 3.35 (t, J = 5.65 Hz, 2H) | 39.67 | 3.20–3.31 (m, 2H) | 43.7 |
Furthermore, addition of CoII ions to 1S did not result in observation of paramagnetic 1H signals, expected upon coordination of L1 to paramagnetic CoII ions, demonstrating that 1S is inert towards metal ion-exchange in aqueous solution. To further assess the structural features of solvated species, the hydrodynamic radius of the molecular entity was calculated from measurement of the diffusion coefficient obtained through 1H 2D-DOSY NMR experiment, Fig. S6.† The NMR diffusion measurements were performed using a 1.2 mM D2O solution of 1 on a Varian Inova-500MHz spectrometer equipped with 5 mm Performa II pulsed-field gradient probe where the sample was held at 298 ± 0.1 K using a variable temperature control unit. For the determination of self-diffusion coefficients, the calibrated 1H 90° pulse of 8 μs at a power level setting of 59dB was employed and the sample temperature was equilibrated at 298 ± 0.1 K (calibrated with neat methanol sample) for at least 10 min prior to data acquisition. The number of scans was fixed at 256, preceded by 4 dummy scans in all measurements. The 1H 2D-DOSY spectra were recorded using the convection-corrected bipolar pulse pairs stimulated echo pulse sequence (Dbppste_cc) implemented in the Varian Vnmrj2.2D software package. The natural logarithm of the ratio of signal integrals, A and A0, in the presence and absence of the pulsed-field gradient, respectively, is proportional to the square, G2, of the gradient pulse magnitude according to eqn (1)
| ln(A/A0) = −Dγ2(Δ − δ/3) δ2G2 | (1) |
Formation of the MOC presumably proceeds through coordination of CoII ion by the pyridine-type nitrogen of imidazole, facilitating deprotonation of the pyrrole-type nitrogen16 and thus permitting subsequent coordination to a second CoII ion. Chelation of the two cobalt ions occurs through coordination to the pyridine-type nitrogen atoms (N3′) of the thp rings in mono-deprotonated L1. Subsequent redox process involving molecular oxygen, as the oxidant, results in the formation of the CoIII-MOCs. The chemically-balanced equation for the overall reaction is described in eqn (2).
| 8 CoCl2 + 12 H-L1 + 2 O2 → [Co8L112]Cl12 + 4 HCl + 4 H2O | (2) |
The observation of CoII/III transformation, under the aerobic synthesis conditions of 1, is presumably facilitated through deprotonation of H-L1 and thus indicates a proton-coupled electron transfer reaction. In related CoII-imidazole complexes, where no deprotonation of the imidazole ring occurred, aerobic oxidation of CoII ions was not detected.17 Several examples of aerobic CoII/III transformations have been reported for octahedral Co–N6 complexes containing the imidazolate ligands.17a,18
To gain insight into the nature of the species present in the initial reaction mixture, shortly after mixing, 1H NMR spectrum of a freshly prepared solution of CoCl2·6H2O (0.0237 g, 0.1 mmol) and H-L1 (0.0348 g, 0.15 mmol) in 1 mL of D2O under anaerobic conditions was acquired. The spectrum obtained exhibits a well-resolved pattern of isotropically-shifted proton signals, due to coordination of ligand molecules to paramagnetic CoII ions, Fig. 5. An exponential function with a line broadening of 10 Hz was used to process the spectrum, 1H NMR (D2O, 399.78 MHz): δ = 51.3 (br. s., 1H), 23.546 (br. s., 1H), 21.969 (br. s., 1H), 17.105 (br. s., 1H), 13.452 (br. s., 1H), 12.763 (br. s., 1H), 10.608 (br. s., 1H), 10.168 (br. s., 1H), 9.319 (br. s., 1H), 8.68 (br. s., 1H), 7.652 (br. s., 1H), −14.4 (br. s., 1H), −20.7 ppm (br. s., 1H). The 1H NMR spectrum acquired for the reaction mixture revealed 13 different chemical shifts characteristic isotropic shifts to the 1H signals of CoII-coordinated ligand molecules. As the ligand molecule possess two chelation sites, coordination to one site was considered as a possible explanation for the observed pattern in the 1H NMR spectrum of the anaerobic reaction mixture. This was expected, especially in aqueous solution, due to competitive protonation of the basic amidine functionality. However, no changes were observed in the spectrum upon addition of NaOCH3 (0.3 mmol) as a base, suggesting that the observed spectrum corresponds to bis-bidentate, mono-deprotonated L1 molecules bridging two CoII ions. Interestingly, the number of observed proton signals matches the total number of non solvent-exchangeable proton nuclei present in the ligand molecule. This finding confirms that each proton is chemically distinguishable in the obtained CoII complex(es). One plausible explanation of this finding is a non-equivalency of chemical environments in the two thp rings, due to nonplanar conformation of the two rings with respect to the central imidazole ring. This molecular conformation results in different ring proximity to the coordinated CoII ions. Indeed, the geometrically-optimized model of the simple fragment [Co2(L1)(H2O)8]3+, Fig. 6, indicates presence of an intra-molecular hydrogen bond interaction between N3′ atoms of thp rings.
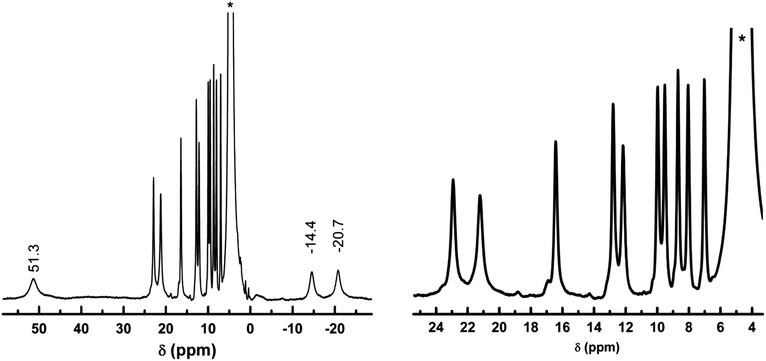 | ||
| Fig. 5 (Left) 1H NMR spectrum of the reaction mixture of CoCl2·6(H2O) (0.1 mmol) and H-L1 (0.15 mmol) in 1 mL D2O after mixing at 298 K, (right) the 4–24 ppm region of the spectrum magnified. * = HDO solvent peak used as internal reference at 4.76 ppm. | ||
![The B3LYP/LANL2DZ geometrically-optimized model for the [Co2(L1)(H2O)8]3+ fragment showing the different thp rings proximity to coordinated CoII ions.](/image/article/2011/SC/c1sc00269d/c1sc00269d-f6.gif) | ||
| Fig. 6 The B3LYP/LANL2DZ geometrically-optimized model for the [Co2(L1)(H2O)8]3+ fragment showing the different thp rings proximity to coordinated CoII ions. | ||
This interaction, along with unfavourable steric hindrance for coplanar thp rings conformation, appears to induce out-of-plane displacement of the two thp rings and promotes detectable differences in proximity of the rings' proton nuclei to the coordinated CoII ions. Such an effect can cause noticeable differences in the spin–lattice relaxation times of thp rings' protons.
Relative metal–nucleus distance, rM–H, can be easily obtained with respect to a reference nucleus employing the relation rM–H = [(T1/T1(ref))1/6 × rM–H(ref)], where T1 and T1(ref) are the experimental spin–lattice relaxation times for the nucleus of interest and a reference nucleus, respectively.19 Proton spin–lattice relaxation times for the majority of isotropically shifted proton signals were determined using the inversion recovery technique (D1-180°-τ-90°-FID) with 15 different τ values (Table S2, supporting information†). The signal at 8.69 ppm (T1 = 0.071 s) was assigned to the equatorial proton on C6′ atom in thp ring furthest from CoII, and employed as the reference nucleus. The rCo–H distances obtained from experimental T1 measurements correlate well with those from the model, Fig. 7, further supporting the argument for formation of bis-bidentate, mono-deprotonated L1 molecules, bridging two CoII ions. Over a period of 12 h under aerobic conditions and at r.t., a gradual decrease in paramagnetic proton integrals with concomitant appearance of several proton signals in the diamagnetic region of the spectrum indicates a CoII/III transformation due to aerobic oxidation of coordinated CoII ions. During the aerobic oxidation process, no changes in the number nor chemical shifts of the isotropically shifted proton signals were observed, suggesting conservation of the initial complex geometry while undergoing transformation into diamagnetic products. Under anaerobic conditions, the characteristic spectrum of paramagnetic CoII-complex(es) is maintained with no detectable changes, confirming the identity of the oxidant as atmospheric molecular oxygen.
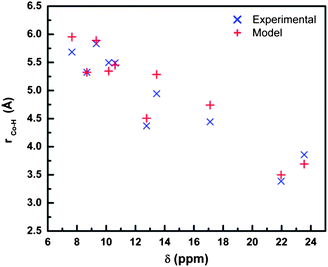 | ||
| Fig. 7 Cobalt-to-proton, rCo–H, distances obtained through T1 measurements, referenced to the signal at 8.68 ppm (T1 = 0.071 s, rCo–H = 5.324 Å as extracted from the geometrically-optimized model). | ||
The observed diamagnetic 1H NMR spectrum obtained upon aerobic oxidation of the reaction mixture, in absence of the base, confirms the formation of the MOCs and presence of uncoordinated protonated ligands[H2-L1]+. The acidic reaction medium (pH = 5.76) allows equilibrium between Co-coordinated and protonated [H2-L1]+ In fact, the spectrum obtained upon addition of NaOCH3 base under aerobic conditions matches 1S spectrum with no observed signal of free ligand, indicating quantitative formation of the MOCs in basic solution upon reactants mixing at room temperature in a correct stoichiometry, Fig. S7–S12.†
UV-vis solution studies
The freshly prepared aqueous solution of CoCl2·6H2O (4.14 mM) and H-L1 (6.25 mM) exhibits a characteristic absorption band in the visible 400∼600 nm range (ε481 = 175 M−1 cm−1). Under aerobic conditions, a gradual red-shift with an increase in molar extinction coefficient (ε493 = 205 M−1 cm−1) is observed, Fig. 8. This observation is ascribed to a CoII/III aerobic oxidation process, as confirmed by the 1H NMR study, vide supra. The presence of isosbestic points at 475 and 575 nm in the absorption spectra indicate an absence of intermediates, confirming direct transformation from paramagnetic to diamagnetic complexes. Spectrophotometric titration of CoCl2 (2.5 mM) with H-L1 (0 to 6 mM in 0.5 mM increments), was conducted and followed through measurements of A493, Fig. 9a. Incubation of the reaction mixture after each addition of H-L1 at ambient temperature for 24 h was necessary to ensure complete oxidation of coordinated CoII ions. Simultaneously, measurements of the aforementioned solutions pH indicate suppressed protonation of added ligand molecules due to preferential coordination to Co ions. Furthermore, the noticeable pH drop, dependent on time and concentration of added ligand, strongly suggests deprotonation of ligand molecules (generating imidazolate anions) due to coordination to Co ions. The A493 increases linearly with increasing amounts of H-L1 and reaches a pseudo plateau at 2 equiv, accompanied by an abrupt increase in solution pH.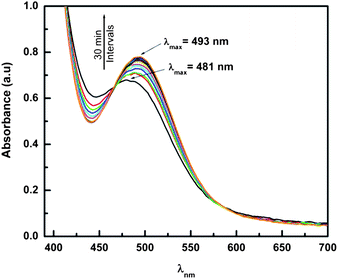 | ||
| Fig. 8 UV-vis absorption spectra for the reaction mixture of H-L1 (6.25 mM) and CoCl2 (4.15 mM) in aqueous solution, 296 K under aerobic conditions. Spectra accumulated at 30 min intervals. | ||
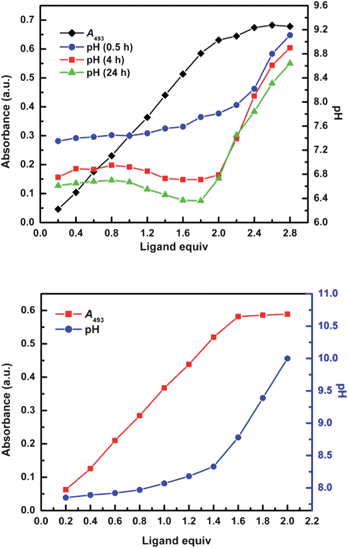 | ||
| Fig. 9 (a) Changes in A493 and solution pH for the spectrophotometric titration of CoCl2 (2.5 mM) with increasing concentration of H-L1 (0.5∼6.0 mM) in 20 m L aqueous solution. (b) Changes in A493 and pH for the titration mixture after addition of 2 equiv (in terms of H-L1 added) NaOCH3. Absorption measurements acquired after standing at room temperature for 24 h; measurements conducted at 296 K. | ||
This can be interpreted by formation of ligand-to-cobalt complexes in the presence of non-coordinating ligand, predominantly in conjugate acid forms after 2 equiv. However, the formation of MOCs, 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of ligand-to-cobalt, implies that a fraction of H-L1 molecules introduced into solution are not available for metal chelation due to competitive protonation, [H2-L1]+ and/or [H3-L1]2+. In order to eliminate the formation of protonated species, we carried similar measurements in the presence of NaOCH3, introduced as proton scavenger to control the pH, Fig. 9b. Indeed, the onset of the A493 plateau is shifted to the expected H-L1 mole equivalents, ∼1.5:1 (ligand-to-cobalt), supporting the quantitative formation of MOCs in solution upon reacting ligand to metal in the correct stoichiometry. In support of the spectrophotometric titration findings, a Job's plot constructed for the two binding species exhibits a maximum absorbance at 0.4 mole fraction of Co ions, corresponding to the expected 1.5:1 (ligand-to-cobalt) stoichiometry, Fig. S7.† These findings are further confirmed by solution NMR spectroscopy conducted on a mixture of H-L1 (0.15 mmol), CoCl2·6H2O (0.1 mmol), and NaOCH3 (0.3 mmol) in D2O, 1 mL, under aerobic conditions. The observed 1H NMR spectrum corresponds to the formation of solvated MOCs (1S) with no detectable signals characteristic of free or partially-coordinated ligand molecules, Fig. S8–S13.†
:1 stoichiometry of ligand-to-cobalt, implies that a fraction of H-L1 molecules introduced into solution are not available for metal chelation due to competitive protonation, [H2-L1]+ and/or [H3-L1]2+. In order to eliminate the formation of protonated species, we carried similar measurements in the presence of NaOCH3, introduced as proton scavenger to control the pH, Fig. 9b. Indeed, the onset of the A493 plateau is shifted to the expected H-L1 mole equivalents, ∼1.5:1 (ligand-to-cobalt), supporting the quantitative formation of MOCs in solution upon reacting ligand to metal in the correct stoichiometry. In support of the spectrophotometric titration findings, a Job's plot constructed for the two binding species exhibits a maximum absorbance at 0.4 mole fraction of Co ions, corresponding to the expected 1.5:1 (ligand-to-cobalt) stoichiometry, Fig. S7.† These findings are further confirmed by solution NMR spectroscopy conducted on a mixture of H-L1 (0.15 mmol), CoCl2·6H2O (0.1 mmol), and NaOCH3 (0.3 mmol) in D2O, 1 mL, under aerobic conditions. The observed 1H NMR spectrum corresponds to the formation of solvated MOCs (1S) with no detectable signals characteristic of free or partially-coordinated ligand molecules, Fig. S8–S13.†
The rate of development of the 483 nm band, characteristic of the metal–ligand complex formation, is monitored by mixing a solution containing L1 (5 mM) and NaOCH3 (10 mM), in a 1:1 mixture of DMF and H2O, with CoCl2 (3.3 mM in water) in a stopped-flow mixer. The data collected at four different sampling periods (10 μs, 100 μs, 500 μs, and 5 ms) are fitted by exponential decay functions with lifetimes of τ1 ≤ 2 ms, τ2 = 48.16 ± 0.09 ms, τ3 = 101.86 ± 0.68 ms, τ4 = 292.43 ± 1.67 ms, τ5 = 1.29 ± 0.01 s, τ6 = 17.75 ± 0.02 s, Fig. S14, S15.† The smallest lifetime τ1 ≤ 2 ms (within mixing time) indicates a diffusion controlled process, potentially the formation of the simplest building unit (A), containing a metal ion chelated by a neutral ligand molecule. A part of the population of fragment (A) can subsequently undergo further chelation by ligand molecule due to the presence of ligand molecules at 50% excess in the initial mixture, forming (B). Due to the concentration dependence of all processes with lifetimes larger than τ1, a fragment condensation mechanism is proposed. The MOC formation, according to the proposed mechanism, can proceed through sequential or simultaneous fragment condensation processes. In a sequential fragment condensation mechanism, deprotonation of one of the two coordinated ligand molecules in (B) is expected to precede its association with (A) to form fragment (C). Deprotonation of coordinated ligands in (C) followed by bimolecular condensation generates fragment (D). Condensation of deprotonated (C) with (D) generates larger fragment (E), leading eventually to formation of the MOC (F), Fig. 10. The proposed plausible fragments are in agreement with those observed in the MALDI-TOF/MS spectra of 1.
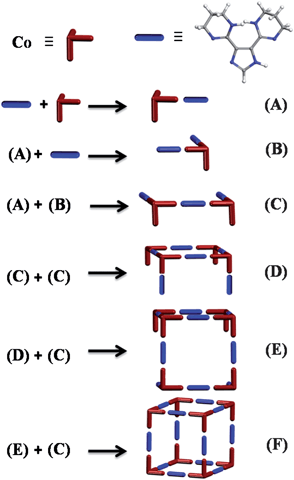
Counterions effects
The ability to construct cationic MOCs starting from different CoII salts offers the opportunity to investigate effect(s) of counterions on the assembly process. Reactions of H-L1 and CoBr2·6H2O, Co(SO4)·6H2O, Co(NO3)2·6H2O, or Co(BF4)2·6H2O, under similar reaction conditions generating 1, resulted in cationic MOCs crystallizing in presence of corresponding counterions, 1a, 1b, 1d, and 1e, respectively. The MOCs in presence of bromide, sulfate, nitrate, and tetraflouroborate adopt different packing patterns as compared to 1. Interestingly, reactions of H-L1 in the presence of NH4Cl, as a source of chloride ions, and either Co(SO4)·6H2O or Co(NO3)2·6H2O resulted in a crystalline material characterized through single crystal X-ray diffraction to be identical to 1, i.e. cationic MOCs with Cl− as counterions. Evidently, all explored counterions have permitted the construction of the targeted MOC, and thus precluding any specific counterion impact on the formation of the MOC. Nonetheless, the nature of counterion contributes substantially to the crystallization process; specifically controlling the packing pattern of MOCs in the resulting crystalline solids via intermolecular H–bond interactions.Solvent effect(s)
The successful construction of predesigned MOMs is closely dependent on the proper choice of a solvent system, permitting generation in situ of pre-programmed MBBs and their assembly into a targeted structure. In the solvothermal synthesis of 1, a water/DMF (1:1) solvent system facilitated isolation of crystalline material. In fact, solvothermal reactions of CoCl2·6H2O and H-L1 in pure DMF consistently resulted in amorphous precipitate while hydrothermal reactions of CoCl2·6H2O and H-L1 resulted in clear red solutions. The important role of DMF (a weak base, aprotic, polar solvent with relatively high boiling point, ca. 153 °C) in the crystallization of 1 is thus validated.
In an attempt to assess the relative significance of its several characteristics as a co-solvent, DMF was substituted with dimethylsulfoxide (DMSO) and hexamethylphosphoramide (HMPA) in two separate reactions. The alternate solvents were selected for their potential to provide solvent systems with different range of boiling points, basicity, hydrophobicity, and ability to act as hydrogen bond acceptors. In fact, the solvothermal reaction in water/DMSO (polar, aprotic solvent with high boiling point, ca. 189 °C) solvent mixture resulted in brown rectangular crystals of 1c, only after considerable loss of water solvent molecules through evaporation. Similarly, compound 1f was isolated upon substitution of DMF by HMPA (aprotic, polar solvent with relatively high boiling point ∼232 C) as a co-solvent, and only after significant water evaporation. The decrease of water content in the reaction mixture promotes the crystallization of cationic MOCs in the presence of various counterions, due to the anticipated decreased MOCs solubility in aprotic solvents. The importance of water evaporation in the crystallization process was further confirmed for reactions at room temperature where extended evaporation time is needed. Reactions at room temperature of H-L1 and Co(NO3)2·6H2O in H2O/DEF (1 mL each) mixture, in an open 25 mL scintillation vial, have sat for one week prior to appearance of any crystalline materials, i.e. formation of the MOCs (1d) following the reaction mixture volume decrease to roughly 1 mL through water evaporation.
In order to decipher the role of water in the assembly of MOCs, we followed the CoII complexation with H-L1 ligands in DMF as the sole solvent. The UV-vis absorption spectrum of CoCl2·6H2O dissolved in DMF indicates presence of the tetrahedral Co(DMF)2Cl2 species, characterized by an absorption band in the 560∼720 nm range assigned to the 4T1(P) ← 4A2 transition. Addition of H-L1 promotes ligand exchange and the formation of a plausible metal ion complex Co(H-L1)Cl2, as evidenced by observed changes in the absorption spectrum, broadening of the 560∼720 nm band and appearance of a second absorption band at 493 nm, Fig. 11. In contrast, dissolution of CoCl2·6H2O in H2O, a solvent with higher dielectric constant, permits the initial formation of the octahedral cobalt complex [Co(H2O)6]Cl2 that presumably undergoes facile exchange of coordinated water molecules upon addition of H-L1, facilitating the construction of MOCs. It is appropriate to conclude, in the case of cobalt chloride salt, that the higher nucleophilicity of Cl− ions in DMF, relative to water, precludes reaction progress towards formation of MOCs. It is to mention that the interdependence between counterion nucleophilicity and solvent nature on the rate of ligand exchange dynamics has been addressed in the case of coordination supermolecular polygons.20
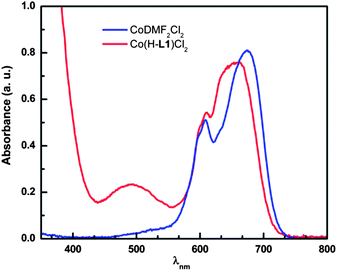 | ||
| Fig. 11 UV-vis absorption spectra for the CoCl2 (2.5 mM) solution in DMF (blue) and the reaction mixture of H-L1 (3.75 mM) and CoCl2 (2.5 mM) in DMF solution (red), directly after mixing at 296 K. | ||
In conclusion, water is indeed needed to initiate the formation of MOCs and addition of aprotic co-solvent such as DMF is necessary to reduce the solubility, in this case of the cationic MOC, and promote crystallization.
Metal ions effect(s)
Attempts to crystallize isostructural MOCs based on various divalent metal ions were unsuccessful for chloride based salts. Reactions of H-L1 with NiCl2 and CdCl2, under similar reaction conditions employed to form 1, did not result in any crystalline material. Furthermore, solvothermal reactions of Ni(NO3)2·4H2O and equimolar amounts of H-L1 in DMF resulted in molecular squares, 2. Each of the four NiII ions is octahedrally-coordinated to two ligand molecules in bis-bidentate fashion and one nitrate ion acting as a capping bidentate agent, Fig. 12. Attempts to construct MOCs, in various solvent mixtures, by reacting Ni(NO3)2 with 1.5 equiv of H-L1 were unsuccessful, resulting in amorphous precipitates. It is possible that complete coordination of the Ni(II) ions by three ligands, necessary for the formation of NiII-based MOCs, is precluded by the capping nitrate ion as observed in 2. Similarly, solvothermal reactions of equimolar amounts of Cd(NO3)2·4H2O and H-L1 in DMF resulted in 1D coordination polymer (3), Fig. 13. Similar to the case of NiII molecular squares, the coordination of nitrate ions to CdII appears to limit the coordination of CdII, precluding isolation of crystalline material containing the targeted CdII–N6 MOCs. Analysis of the CSD database revealed that only ∼4.8% and ∼5.7% of octahedral NiII–N6 and CdII–N6 based complexes, respectively, were isolated in presence of (NO3)− ions. In addition to the apparent preferred hetero-coordination spheres for octahedral NiII and CdII complexes in presence of nitrate ions, it appears that the stable +2 oxidation state of Cd and Ni might also hinder the possible construction and/or crystallization of the respective MOCs. Further investigations are underway to assert the presence of MII–MOCs in solution as well as their possible crystallization in the presence of various counterions. It is apparent that the oxidation state of the metal ion and its preferred coordination sphere are directly correlated to the successful design and formation of the respective MOC. Indeed, solvothermal reactions of In(NO3)3·5H2O and H-L1 resulted in the formation of the targeted cationic InIII-based MOCs, 4. The similarity in oxidation state between InIII and CoIII appears to facilitate construction of the isostructural cationic MOCs based on H-L1.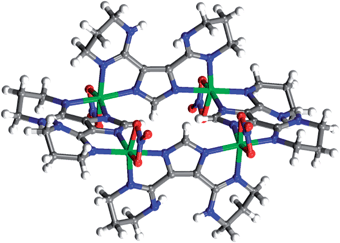 | ||
| Fig. 12 Crystal structure of 2, NiII molecular square. Carbon (gray), nickel (green), nitrogen (blue), oxygen (red), hydrogen (white). DMF solvent molecules omitted for clarity. | ||
![Crystal structure of 3, molecular chains of [Cd(L1)(NO3)]n, Carbon (gray), cadmium (buff), nitrogen (blue), hydrogen (white), oxygen (red). DMF solvent molecules omitted for clarity.](/image/article/2011/SC/c1sc00269d/c1sc00269d-f13.gif) | ||
| Fig. 13 Crystal structure of 3, molecular chains of [Cd(L1)(NO3)]n, Carbon (gray), cadmium (buff), nitrogen (blue), hydrogen (white), oxygen (red). DMF solvent molecules omitted for clarity. | ||
Structural stability of MOCs in aqueous solution
The MOCs maintain their structural integrity in aqueous solution under a wide range of solution pH and temperatures. The crystallized MOCs, 1, was dissolved in D2O and studied by solution 1H NMR spectroscopy at different temperature and pH.The maintained structural integrity of solvated MOCs in a wide solution pH range (2.03–8.07) and various temperatures (298–333 K) was demonstrated by 1H NMR studies. The characteristic pattern for the solvated MOCs is maintained, under the above mentioned solution conditions, indicating preserved structural integrity, Fig. S16.† In addition, no detectable disintegration of the MOCs was noticed over extended period of time for a solution of 1 in D2O, as confirmed by both 1H and 2D-DOSY NMR spectroscopy, Fig. S6.†
Conclusion
The assembled MOCs in solution from pre-programmed MBBs were characterized using NMR spectroscopy, UV-vis absorption spectroscopy, solution pH potentiometry, and MALDI-TOF mass spectrometry. Aqueous dissolution of the isolated crystalline material results in formation of solvated MOCs with demonstrated structural stability under a wide range of solution temperature and pH.In this study, a systematic investigation is conducted to assess the roles and effects associated with a wide range of reaction variables (nature of metal ions, counterions, solvent systems, solution pH, and temperature) on the nature of isolated products. It is demonstrated that the assembly of MOCs from various CoII salts proceeds readily in aqueous solution, simply upon reactants mixing. The reaction is driven to completion by inhibition of competitive protonation of the ligand molecules through careful adjustment of solution pH. Crystallization of MOCs was observed only in the presence of polar aprotic co-solvent with elevated boiling point (DMF, DEF, DMSO, and HMPA) after considerable loss of water molecules through evaporation. The role associated with the nature of counterions in inducing crystallization of cationic MOCs, through intermolecular hydrogen bond interactions, was evaluated.
The present study clearly indicates that formation of a given targeted MBB requires careful choice of solvent mixture, metal ion, counterion, and pH. The ligand–solvent–counterion coordination to the metal cation is governed by HSAB concept, and should be primarily taken into consideration for selecting the metal salt, polytopic ligand and reaction solvent for the construction of a given targeted MOM. A proper chosen solvent system should promote desired ligand-to-metal coordination as well as facilitating nucleation and crystallization. Counterions with limited interference to ligand coordination are vital for the metal–ligand directed assembly of the desired MBB necessary for the formation of a given MOM.
The present systematic study, methodology, adopted herein offer potential to gain better understanding of MOMs (i.e. MOPs and MOFs) assembly, leading to the developments of new pathways for the design and construction of made-to-order MOMs with desired physical and chemical properties.
Acknowledgements
We gratefully acknowledge the financial support of the NSF Career Award, DMR 0548117 (M. E.) and King Abdullah University of Science and Technology (KAUST).Notes and references
- (a) J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef CAS; (b) M. Dinca, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419–1422 CrossRef CAS; (c) K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604–9605 CrossRef CAS; (d) Y. Liu, J. F. Eubank, A. J. Cairns, J. Eckert, V. Ch. Kravtsov, R. Luebke and M. Eddaoudi, Angew. Chem., Int. Ed., 2007, 46, 3278–3283 CrossRef CAS; (e) H. Hayashi, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501–506 CrossRef CAS; (f) P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. DeWeireld, J. Chang, D. Hong, Y. K. Hwang, S. H. Jhung and G. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef; (g) R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS; (h) So-Hye Cho, T. Gadzikwa, M. Afshari, SonBinh T. Nguyen and T. J. Hupp, Eur. J. Inorg. Chem., 2007, 4863–4867 CrossRef CAS; (i) P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS; (j) R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS.
- (a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef; (b) G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef CAS; (c) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS; (d) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS.
- (a) B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962–5964 CrossRef CAS; (b) B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS; (c) B. F. Abrahams, B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727–729 CrossRef CAS.
- (a) M. O'Keeffe, M. Eddaoudi, H. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3–20 CrossRef CAS; (b) N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS; (c) L. R. MacGillivray and J. L. Atwood, Angew. Chem., Int. Ed., 1999, 38, 1018–1033 CrossRef CAS.
- (a) S. Li, H. Yan, L. Wan, H. Yang, B. H. Northrop and P. J. Stang, J. Am. Chem. Soc., 2007, 129, 9268–9269 CrossRef CAS; (b) L. Zhao, B. H. Northrop and P. J. Stang, J. Am. Chem. Soc., 2008, 130, 11886–11888 CrossRef CAS; (c) Y. Zheng and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 3487–3489 CrossRef CAS; (d) T. Yamamoto, A. M. Arif and P. J. Stang, J. Am. Chem. Soc., 2003, 125, 12309–12317 CrossRef CAS; (e) R. W. Larsen, G. J. McManus, J. J. Perry, E. Rivera-Otero and M. J. Zaworotko, Inorg. Chem., 2007, 46, 5904–5910 CrossRef CAS; (f) R. W. Larsen, J. Am. Chem. Soc., 2008, 130, 11246–11247 CrossRef CAS; (g) M. Tonigold and D. Volkmer, Inorg. Chim. Acta, 2010, 363, 4220–4229 CrossRef CAS; (h) C. Sgarlata, J. Mugridge, M. Pluth, B. Tiedemann, V. Zito, G. Arena and K. Raymond, J. Am. Chem. Soc., 2010, 132, 1005–1009 CrossRef CAS.
- (a) M. Eddaoudi, J. Kim, J. B. Wachter, H. K. Chae, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 4368–4369 CrossRef CAS; (b) H. Abourahma, A. W. Coleman, B. Moulton, B. Rather, P. Shahgaldian and M. J. Zaworotko, Chem. Commun., 2001, 2380–2381 RSC; (c) D. J. Tranchemontagne, Z. Ni, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2008, 47, 5136–5147 CrossRef CAS; (d) S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS; (e) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369–378 CrossRef CAS; (f) D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- (a) M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC; (b) K. Nakabayashi, Y. Ozaki, M. Kawano and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 2046–2048 CrossRef CAS; (c) K. Umemoto, K. Yamaguchi and M. Fujita, J. Am. Chem. Soc., 2000, 122, 7150–7151 CrossRef CAS; (d) M. Fujita, M. Aoyagi, F. Ibukuro, K. Ogura and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 611–612 CrossRef CAS; (e) K. Umemoto, H. Tsukui, T. Kusukawa, K. Biradha and M. Fujita, Angew. Chem., Int. Ed., 2001, 40, 2620–2622 CrossRef CAS; (f) M. Dinca, T. David Harris, A. T. Iavarone and J. R. Long, J. Mol. Struct., 2008, 890, 139–143 CrossRef CAS.
- (a) F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS; (b) A. J. Cairns, J. A. Perman, L. Wojtas, V. C. Kravtsov, M. H. Alkordi, M. Eddaoudi and M. J. Zaworotko, J. Am. Chem. Soc., 2008, 130, 1560–1561 CrossRef CAS.
- (a) Y. Liu, V. Kravtsov, R. D. Walsh, P. Poddar, H. Srikanth and M. Eddaoudi, Chem. Commun., 2004, 2806–2807 RSC; (b) M. H. Alkordi, J. A. Brant, L. Woitas, V. Kravtsov, A. J. Cairns and M. Eddaoudi, J. Am. Chem. Soc., 2009, 131, 17753–17755 CrossRef CAS.
- A. de la Hoz, Á Díaz-Ortiz, M. del Carmen Mateo, M. Moral, A. Moreno, J. Elguero, C. Foces-Foces, M. L. Rodríguez and A. Sánchez-Migallón, Tetrahedron, 2006, 62, 5868–5874 Search PubMed.
- (a) M. R. Grimmett in Advances in Imidazole Chemistry; A. R. Katritzky and A. J. Boulton, Ed.; Advances in Heterocyclic Chemistry; Academic Press: 1981; Vol. 27, pp 241–326 Search PubMed; (b) D. Nolting, N. Ottosson, M. Faubel, I. V. Hertel and B. Winter, J. Am. Chem. Soc., 2008, 130, 8150–8151 CrossRef CAS; (c) H. Green and A. R. Day, J. Am. Chem. Soc., 1942, 64, 1167–1173 Search PubMed; (d) J. D. Halliday, E. A. Symons and P. D. Binder, Can. J. Chem., 1978, 56, 1470–1476 CAS; (e) H. Komber, H. Limbach, F. Bohme and C. Kunert, J. Am. Chem. Soc., 2002, 124, 11955–11963 CrossRef CAS; (f) R. J. Sundberg and R. B. Martin, Chem. Rev., 1974, 74, 471–517 CrossRef CAS.
- A. Werner, Ber. Dtsch. Chem. Ges., 1912, 45, 121–130 CrossRef CAS.
- J. E. Tanner and E. O. Stejskal, J. Chem. Phys., 1968, 49, 1768–1777 CrossRef.
- (a) M. Holz, S. R. Heil and A. Sacco, Phys. Chem. Chem. Phys., 2000, 2, 4740–4742 RSC; (b) E. H. Hardy, A. Zygar, M. D. Zeidler, M. Holz and F. D. Sacher, J. Chem. Phys., 2001, 114, 3174–3181 CrossRef CAS; (c) H. Weingartner and M. Holz, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2002, 98, 121–156 RSC.
- N. R. Brodsky, N. M. Nguyen, N. S. Rowan, C. B. Storm, R. J. Butcher and E. Sinn, Inorg. Chem., 1984, 23, 891–897 Search PubMed.
- (a) D. M. Boghaei, E. Askarizadeh and A. Bezaatpour, Spectrochim. Acta, Part A, 2008, 69, 624–628 Search PubMed; (b) G. Stupka, L. Gremaud and A. Williams, Helv. Chim. Acta, 2005, 88, 487–495 CrossRef CAS; (c) K. Kurdziel and T. Glowiak, Polyhedron, 2000, 19, 2183–2188 Search PubMed; (d) Z. Wang, F. Jian, Y. Zhang, F. Li, H. Fun and K. Chinnakali, J. Chem. Crystallogr., 1999, 29, 885–890 Search PubMed; (e) Q. Zhao, H. Li, X. Wang and Z. Chen, Chem. Lett., 2002, 31, 988–989 Search PubMed.
- (a) M. Tadokoro, H. Kanno, T. Kitajima, H. Shimada-Umemoto, N. Nakanishi, K. Isobe and K. Nakasuji, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4950–4955 CrossRef CAS; (b) F. Tuna, M. R. Lees, G. J. Clarkson and M. J. Hannon, Chem.–Eur. J., 2004, 10, 5737–5750 CrossRef CAS; (c) V. Sánchez, A. Storr and R. C. Thompson, Can. J. Chem., 2002, 80, 133–140 CrossRef CAS; (d) Y. Tian, C. Cai, Y. Ji, X. You, S. Peng and G. Lee, Angew. Chem., Int. Ed., 2002, 41, 1384–1386 CrossRef CAS; (e) K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS.
- (a) J. D. Epperson and L. Ming, Biochemistry, 2000, 39, 4037–4045 Search PubMed; (b) S. H. Koenig, J. Mag. Res., 1978, 31, 1–10 Search PubMed.
- Y.-R. Zheng and P. J. Stang, J. Am. Chem. Soc., 2009, 131, 3487–3489 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRPD, NMR spectra, and X-ray crystallographic data. CCDC reference numbers 824164–824173. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00269d |
| This journal is © The Royal Society of Chemistry 2011 |