Influence of structure on exchange strength and relaxation barrier in a series of FeIIReIV(CN)2 single-chain magnets†
Xiaowen
Feng
,
T.
David Harris
and
Jeffrey R.
Long
*
Department of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: jrlong@berkeley.edu
First published on 23rd June 2011
Abstract
Cyano-bridged single-chain magnets of the type L4FeReCl4(CN)2, where L = diethylformamide (DEF) (1), dibutylformamide (DBF) (2), dimethylformamide (DMF) (3), dimethylbutyramide (DMB) (4), dimethylpropionamide (DMP) (5), and diethylacetamide (DEA) (6), have been synthesized to enable a systematic study of the influence of structural perturbations on magnetic exchange and relaxation barrier. Across the series, varying the amide ligand leads to Fe–N–C bond angles ranging from 154.703(7)° in 1 to 180° in 6. Variable-temperature dc magnetic susceptibility data indicate ferromagnetic exchange coupling in all compounds, with the strength of exchange increasing linearly, from J = +4.2(2) cm−1 to +7.2(3) cm−1, with increasing Fe–N–C bond angle. Ac magnetic susceptibility data collected as a function of frequency reveal that the relaxation barrier of the chain compounds rises steeply with increasing exchange strength, from 45 cm−1 to 93 cm−1. This examination demonstrates that subtle tuning of orbital overlap, and thus exchange strength, can engender dramatic changes in the relaxation barrier. Indeed, the perfectly linear Fe–N–C bond angle in 6 leads to one of the highest barriers and coercive fields yet observed for a single-chain magnet.
Introduction
Over the past decade, a number of magnetic chain compounds have been shown to retain their magnetization upon removal of an applied field,1 a phenomenon predicted by Glauber nearly half a century ago for chains of ferromagnetically coupled anisotropic spins.2 This dynamic behavior is analogous to that observed in single-molecule magnets,3 and, consequently, chain compounds that exhibit slow magnetic relaxation have come to be known as single-chain magnets.1b While the relaxation dynamics of single-molecule and single-chain magnets share several key characteristics, one pronounced difference is that single-chain magnets tend to display much higher relaxation barriers than do their molecular counterparts. As a result, these chain compounds have received considerable recent attention, owing to their potential utility in applications such as spin-based high-density information storage.4The propensity for single-chain magnets to exhibit higher relaxation barriers than single-molecule magnets can be attributed to short range magnetic correlation along individual chains. In a single-molecule magnet, the magnitude of the relaxation barrier, ΔA, is governed by the equation ΔA = S2|D|, where S is the spin ground state and D is the axial zero-field splitting parameter. In a single-chain magnet, the barrier similarly depends on S and D of the repeating spin unit, but additionally this value scales with the strength of magnetic exchange, J, between spin units. Indeed, the overall relaxation has been theoretically predicted1g,2,5 and experimentally demonstrated1f,5b,6 to follow the expression Δτ = (8J + D)S2 for systems falling within the Ising limit.7 Thus, increasing the strength of exchange along a chain represents an important route toward achieving high magnetic relaxation barriers in single-chain magnets.
An ideal system for installing strong magnetic exchange along a chain would feature a synthetically adjustable component to enable facile tuning of J. Along these lines, cyano-bridged chain compounds offer themselves as attractive candidates, owing to the predictability of both structure and magnetic exchange afforded by the M–CN–M′ linkage.8 In particular, numerous investigations have shown that the strength of magnetic exchange between two metal centers through cyanide stems directly from the M′–N–C angle and can often be explained through simple molecular orbital considerations.9 As such, controlling the M′–N–C angle should provide a convenient handle through which to adjust and maximize the magnitude of the relaxation barrier.
Recently, we reported the synthesis of a series of cyano-bridged single-chain magnets of the type (DMF)4MReCl4(CN)2 (M = Mn, Fe, Co, Ni).10 The structures of these compounds feature considerably bent M–N–C angles (155.8(1)°–159.4(1)°), resulting from solid-state packing effects. We were intrigued by the possibility of constructing a series of related chain compounds, where only the identity of the amide ligand is varied across the series. The different steric and electronic properties of the amides, along with the associated alterations to solid-state packing, should provide a means through which to adjust the M–N–C angle and thereby enhance significantly the intrachain exchange strength and relaxation barrier. Herein, we demonstrate that this is indeed feasible through the synthesis and characterization of a series of L4FeReCl4(CN)2 chain compounds, where L = diethylformamide (DEF) (1), dibutylformamide (DBF) (2), dimethylformamide (DMF) (3), dimethylbutyramide (DMB) (4), dimethylpropionamide (DMP) (5), and diethylacetamide (DEA) (6).
Results and discussion
Syntheses and structures
In general, the chain compounds were prepared by combining [ReCl4(CN)2]2− and [Fe(amide)6]2+ in neat amide (see Scheme 1). In the case of 1, the reaction solution produced single crystals upon standing. In contrast, for 2 and 4–6, direct combination of solutions containing the precursor complexes resulted in the immediate precipitation of product mixtures. In each of these cases, a layering technique was employed to obtain single crystals. Here, a solution containing [ReCl4(CN)2]2− in the appropriate amide was carefully added to the top of a solution containing [Fe(amide)6]2+ ions in the amide. Over the course of days, each layering produced single crystals of the intended chain compounds.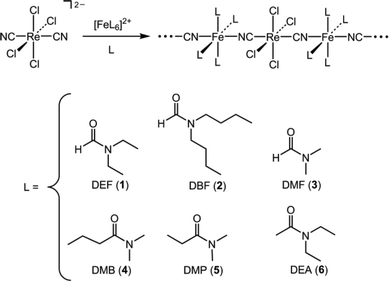 | ||
| Scheme 1 Syntheses of Compounds 1–6 | ||
Single-crystal X-ray analyses of compounds 1–6 revealed the structures to consist of parallel one-dimensional chains, where each chain features alternating [ReCl4(CN)2]2− and [Fe(amide)4]2+ units connected through Re–CN–Fe linkages (see Fig. 1 and S1–S4 in the Supplementary Information†). The local coordination environment of the ReIV center is preserved across the series of compounds and does not significantly deviate from that observed in the structure of (Bu4N)2[ReCl4(CN)2]·2DMA.10 Similarly, each FeII center resides in an approximate octahedral coordination environment, with slight variations in the Fe–N and Fe–O distances (see Table 1). Specifically, the Fe–N distances range from 2.0753(3) to 2.1736(4) Å, consistent with a high-spin (S = 2) electron configuration for FeII.11 In addition, the Fe–O distances range from 2.1095(4) to 2.1339(1), consistent with other high-spin FeII complexes featuring amide ligands.12 Within each structure, individual chains are well separated, with the shortest interchain metal–metal distances ranging from 8.3027(6) Å in 3 to 9.976(6) Å in 5. Additionally, no significant hydrogen bonding contacts between chains are evident in any of the structures.
| Compound | ∠Fe–N–C | ∠Re–C–N | Shortest interchain M–M | Re–C | Fe–N | Fe–O |
|---|---|---|---|---|---|---|
| 1 | 154.703(7) | 173.265(7) | 8.6226(8) | 2.1124(2) | 2.1529(2) | 2.1253(2) |
| 2 | 157.562(6) | 173.233(5) | 8.9348(5) | 2.1182(5) | 2.1443(5) | 2.1138(6) |
| 3 10 | 157.968(5) | 175.259(6) | 8.3027(6) | 2.1177(1) | 2.1546(1) | 2.1339(1) |
| 4 | 164.374(4) | 179.685(4) | 9.477(6) | 2.1217(4) | 2.1736(4) | 2.1095(4) |
| 5 | 170.579(3) | 176.564(3) | 9.976(6) | 2.1132(3) | 2.1293(3) | 2.1227(3) |
| 6 | 180 | 180 | 9.902(4) | 2.1084(3) | 2.0753(3) | 2.1170(2) |
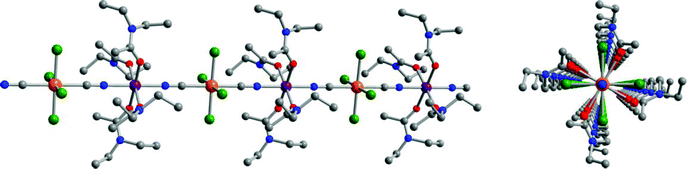 | ||
| Fig. 1 Left: Crystal structure of (DEA)4FeReCl4(CN)2 (6). Orange, purple, green, red, blue, and gray spheres represent rhenium, iron, chlorine, oxygen, nitrogen, and carbon atoms, respectively; hydrogen atoms are omitted for clarity. Right: View down the four-fold crystallographic axis of the chain, demonstrating linear Re–C–N and Fe–N–C angles. | ||
Across the series of compounds, only a subtle variation is observed in the mean Re–C–N angle, which changes from 173.233(5)° in 2 to 180° in 6. Note that the space groupP4/n adopted by 6 positions the Re–CN–Fe linkages along a crystallographic four-fold axis, thereby imposing perfectly linear Re–C–N and Fe–N–C angles. In contrast, the Fe–N–C angle varies much more drastically across the series, from 154.703(7)° in 1 to 180° in 6. Such deviation from linearity is common for Fe–N–C angles in metal–cyanide compounds and is thought to stem primarily from crystal packing and steric conflicts imposed by ligands.13 However, perfectly linear angles are rare in Fe–N–C linkages and have only been observed in several compounds featuring AuI-CN-FeII linkages.14 Here, no clear correlation is observed between Fe–N–C angle and extent of ligand steric bulk or electronic character across the series, indicating that slight differences in crystal packing likely give rise to the bent angles.
Magnetic exchange interactions
With crystallographic data for the FeRe chain compounds in hand, we set out to examine the influence of structure on exchange coupling between the FeII and ReIV centers through the cyanide bridge. Toward this end, variable-temperature dc magnetic susceptibility data were collected for compounds 1–6 under an applied field of 1000 Oe. The corresponding plots of χMT vs. T, as exemplified in the data for 6 shown in Fig. 2, exhibit a common general profile (see also Figs. S5–S8)†. At 300 K, χMT = 5.09, 4.33, 5.46,10 5.09, 5.04, 4.56 cm3 K mol−1 for 1–6, respectively, slightly higher than the value of χMT = 4.28 cm3 K mol−1 that would be expected for magnetically isolated ReIV (S = 3/2, g = 1.66) and FeII (S = 2, g = 2.00) ions. Across the series, χMT rises with decreasing temperature, gradually at first, then more abruptly below 50 K. This temperature dependence indicates the presence of intrachain ferromagnetic coupling between neighboring ReIV and FeII centers. Finally, below 20 K, χMT for each compound undergoes a precipitous downturn, likely due to a combination of magnetic anisotropy of the ReIV and FeII centers and weak interchain antiferromagnetic interactions.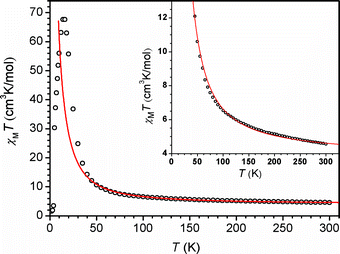 | ||
| Fig. 2 Variable-temperature dc magnetic susceptibility data for 6, collected in an applied field of 1000 Oe. The solid red line corresponds to a fit to the data, as described in the text. Inset: Expanded view of the data and fit, highlighting the presence of intrachain ferromagnetic coupling. | ||
In order to quantify the exchange between neighboring ReIV and FeII centers in each chain, the χMT vs. T data were modeled according to the following spin Hamiltonian for an alternating classical-spin Heisenberg chain:
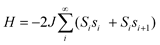
| ∠Fe–N–C | g | J (cm−1) | Δ τ (cm−1) | τ 0 (s) | |
|---|---|---|---|---|---|
| 1 | 154.703(7) | 2.21 | 4.2(2) | 45 | 4.2 × 10−10 |
| 2 | 157.562(6) | 1.84 | 4.5(2) | 55 | 9.0 × 10−11 |
| 3 | 157.968(5) | 1.96 | 4.8(4) | 56 | 1.0 × 10−10 |
| 4 | 164.374(4) | 2.14 | 5.6(3) | 49 | 8.6 × 10−10 |
| 5 | 170.579(3) | 1.84 | 6.3(2) | 53 | 3.9 × 10−10 |
| 6 | 180 | 1.78 | 7.2(3) | 93 | 8.7 × 10−11 |
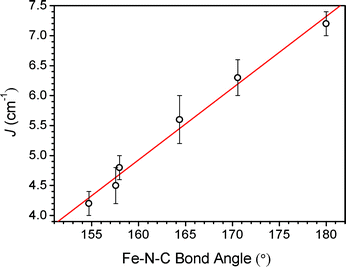 | ||
| Fig. 3 Dependence of exchange strength (J) on Fe–N–C angle in 1–6 (left to right data points, respectively). The solid red line corresponds to a line of best fit through the data. | ||
Based on simple molecular orbital considerations, magnetic exchange through cyanide between octahedral metal centers with t2g4eg2 and t2g3 electronic configurations is expected to be governed by multiple exchange pathways.17,18 First, two π–π interactions occur in symmetry compatible ReIVt2g orbitals through cyanide π* orbitals with the FeII orbitals of t2g symmetry, resulting in antiferromagnetic coupling. In opposition to this, two σ–π interactions between electrons in orthogonal eg and t2g orbitals, respectively, should give rise to ferromagnetic exchange. Such competitive interactions often lead to net ferromagnetic coupling, as σ-type interactions are invariably stronger than π-type interactions. Indeed, ferromagnetic exchange has been observed in several compounds containing CrIII-CN-FeII19 and ReIV-CN-FeII10,20 linkages. In the case of the CrIII-CN-FeII linkage, this behavior has been attributed to a kinetic exchange mechanism, where partial electron transfer from an FeIIt2 orbital into a CrIIIt2 orbital enforces a parallel alignment of spins.21 Given the isoelectronic nature of CrIII and ReIV ions, such a mechanism may also dominate in the FeRe chain compounds. If a t2g → t2g electron transfer is responsible for the ferromagnetic interaction, then the magnitude of the exchange should be maximized when overlap between the cyanide π* and FeIIt2g orbitals is maximized. Indeed, this is exactly the case in 1–6, where the compound exhibiting a linear Re–CN–Fe fragment (6) demonstrates the strongest ferromagnetic interaction. Accordingly, the ferromagnetic interaction decreases in magnitude from 7.2(3) cm−1 to 4.2(2) cm−1 as the Fe–N–C angle undergoes subsequent bending, as overlap between the π-type orbitals is progressively reduced. These values of J are similar to those previously reported for molecular clusters featuring CrIII-CN-FeII19c and ReIV-CN-FeII.20 Additionally, while no J value has been extracted, the Prussian blue analogue Fe3[Cr(CN)6]2·15H2O undergoes ferromagnetic ordering below TC = 21 K.19a We note that this is the first example of a magnetostructural correlation observed through cyanide between metal centers with t2g4eg2 and t2g3 electronic configurations. Similar dependence of J on M–N–C angle has been reported in compounds featuring FeIII-CN-MnIII, MIII-CN-CuII (M = Cr, Mn and Fe) and CrIII-CN-NiII linkages.9a,d,g For instance, in the series of FeIII-CN-MnIII compounds, J was shown to scale linearly with Mn–N–C angle, with a crossover from positive to negative J occurring at a critical angle.9g This phenomenon was attributed to increasing overlap between magnetic FeIII and MnIII dπ orbitals.
Magnetization dynamics
To probe slow magnetic relaxation in the chain compounds, variable-frequency ac susceptibility data were collected for 1–6 at multiple temperatures under zero applied dc field. Data collected for all compounds show frequency dependence in both the in-phase (χM′) and out-of-phase (χM′′) components of the susceptibility. Moreover, plots of χM′ vs. ν and χM′′ vs. ν show a feature that shifts position as the temperature is varied (see Fig. 4 and S9–S13†). From these data, Cole-Cole plots of χM′′ vs. χM′ were constructed and fit to a generalized Debye model to obtain α values and relaxation times (τ) at each temperature (see Figs. S14–S18†).22 Here, α provides a quantitative measure of how closely the Cole-Cole plot resembles a semicircle, and this value provides some insight into the distribution of relaxation times. Across the series, α ranges from 0.16 to 0.20, indicating a relatively narrow distribution of relaxation times. In the cases of 1–3, 5, and 6, Arrhenius plots of ln τ vs. 1/T each show a linear arrangement of data, as expected for a single-chain magnet (see Fig. 5, S19, S20, and S23†). Accordingly, considering the expression τ = τ0exp(Δτ/kBT), fits to the data provide relaxation barriers of Δτ = 45, 55, 56, 53, and 93 cm−1 for compounds 1–3, 5, and 6, respectively (see Table 2). In addition, values of attempt time vary from τ0 = 8.7 × 10−11 s for 6 to 4.2 × 10−10 s for 1, within the range typically observed for single-chain magnets.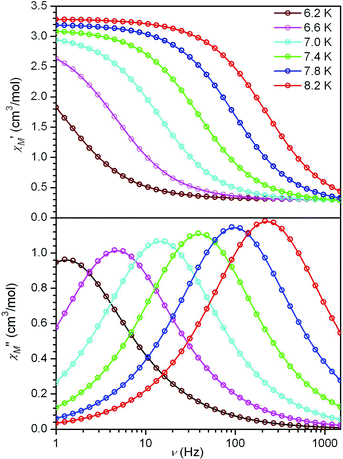 | ||
| Fig. 4 Variable-frequency in-phase (upper) and out-of-phase (lower) components of the ac magnetic susceptibility data for 6, collected in a 4 Oe ac field at selected temperatures. Solid lines correspond to fits to the data, as described in the text. | ||
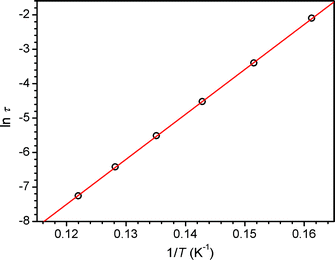 | ||
| Fig. 5 Arrhenius plot of relaxation time for 6. The solid red line corresponds to a linear fit to the data, giving Δτ = 93 cm−1. | ||
Notably, the relaxation barrier observed for 6 is among the highest yet observed for single-chain magnets.1a,c In comparison, the radical bridged chain compound Co(hfac)2(p-butoxyphenyl-NN) was shown to exhibit a relaxation barrier of Δτ = 243 cm−1, but with a much smaller attempt time (τ0 = 6.8 × 10−13 s).23 The authors note that the small τ0 may be diagnostic of spin glass behavior caused by interchain interactions. Indeed, this hypothesis is supported by a lack of frequency dependence in the ac susceptibility upon application of a 500 Oe dc field. Another compound, (bpy)2(H2O)CoIIFeIII2(CN)8, was reported to exhibit a barrier of Δτ = 106 cm−1.24 However, this large barrier is misleading, as it is associated with an extremely small attempt time (τ0 = 1.5 × 10−17 s). As the authors note, this value is much smaller than is to be expected, and this compound deserves further study. Considering chain compounds that show attempt times within the range typically observed for superparamagnets and do not exhibit anomalous relaxation behavior, the highest barrier yet reported belongs to the radical-bridged chain compound Co(hfac)2(NITPhOMe).1a This compound, which was also the first reported example of a single-chain magnet, displays a relaxation barrier of Δτ = 107 cm−1.1a,25 In addition, the double-zigzag chain compound [FeIII(bpy)(CN)4]2CoII(H2O)2, the first example of a cyano-bridged single-chain magnet, exhibits ferromagnetic coupling between FeIII and CoII centers and an overall relaxation barrier of Δτ = 99 cm−1.1c As such, compound 6 exhibits, to our knowledge, the second highest barrier for a cyano-bridged single-chain magnet and the third highest barrier among all single-chain magnets.
Finally, note that plots of χM′ vs. T for the compounds exhibit maxima at ca. 10 K, likely stemming from weak interchain interactions and/or a magnetic phase transition (see Figs. S24, S25, S27, and S28†). Nevertheless, the above analysis of the ac susceptibility clearly shows the presence of single-chain magnet behavior, which may occur within either an ordered or a simply paramagnetic phase.26
In the case of compound 4, the relaxation time does not follow Arrhenius behavior under zero applied dc field (see Fig. S21†). Moreover, the plot of χM′ vs. T exhibits a sharp maximum at ca. 10 K, which may be the mark of a magnetic phase transition as described above (see Fig. S26†). As such, the ac susceptibility data were recollected under an applied dc field of 1000 Oe in order to suppress the phase transition. Indeed, relaxation times acquired from these data clearly exhibit Arrhenius behavior, and a linear fit to the data provides values of Δτ = 49 cm−1 and τ0 = 8.6 × 10−10 s (see Fig. S22†).
Table 2 summarizes the correlation between Fe–N–C angle, exchange strength, and relaxation barrier. Remarkably, the barrier is more than doubled across the series, from Δτ = 45 cm−1 in 1 to 93 cm−1 in 6. This dramatic increase in barrier is directly linked to the enhancement in magnetic correlation afforded by increasing exchange strength. Overall, the 71% increase in J leads to a 107% increase in Δτ. Inspection of Fig. 6 reveals that for compounds 1–3 and 6, the relaxation barrier increases with increasing J in a moderately linear progression. In contrast, the barriers observed for compounds 4 and 5 do not fall along this line. The anomalous behavior in 4 may arise due to the application of a dc field in the data collection to suppress interchain effects. Along those lines, interchain interactions in 5 may also affect the overall barrier. It is not immediately clear why such similar interchain interactions would affect the chain compounds disproportionately. Note, however, that while interchain exchange may affect the relaxation barrier, J values were obtained from data fit well above the temperatures at which maxima in χ′ are observed. As such, these interactions should not bear a significant effect on determinations of exchange strength.
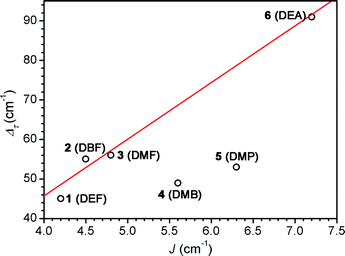 | ||
| Fig. 6 Dependence of relaxation barrier (Δτ) on exchange strength (J) for 1–6. The solid red line corresponds to a line of best fit through data for compounds 1–3 and 6. | ||
Considering the high relaxation barrier observed for 6 in ac susceptibility experiments, variable-field magnetization data were collected for the compound to probe for classical magnet-like behavior. The resulting plot of M vs. H, constructed from data that were collected at a sweep-rate of 100 Oe min−1 between −7 and 7 T, shows substantial magnetic hysteresis at 1.8 K (see Fig. 7). Indeed, under these experimental conditions, the hysteresis loop features a remnant magnetization of MR = 3.34 μB and a coercive field of HC = 2.8 T. We note that this coercive field is the largest yet reported for a cyano-bridged single-chain magnet and among the largest for any single-chain magnet.1a
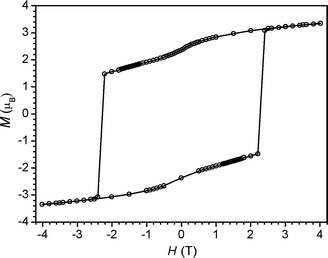 | ||
| Fig. 7 Variable-field magnetization data for 6, collected at 1.8 K under a sweep-rate of 100 Oe min−1. The solid line is a guide to the eye. | ||
Conclusion and outlook
The foregoing results demonstrate that subtle structural perturbations can give rise to dramatic increases in exchange strength and relaxation barrier in single-chain magnets. Specifically, a series of cyano-bridged FeRe chain compounds has been synthesized in which the Fe–N–C angle varies from 154.703(7)° to 180°. Fits to dc susceptibility data across the series reveal that the strength of ferromagnetic exchange between ReIV and FeII ions increases linearly with increasing Fe–N–C angle, from J = +4.2(2) to +7.2(3) cm−1. Moreover, ac susceptibility measurements show a pronounced effect of exchange strength on relaxation barrier, with Arrhenius fits of relaxation time providing values ranging from Δτ = 45 to 93 cm−1. Notably, the high relaxation barrier, along with the significant magnetic hysteresis at low temperature, establishes compound 6 as one of the strongest low-dimensional magnets yet observed.Efforts are underway to pursue other routes toward magnetic chain compounds with strong intrachain exchange. Indeed, we recently reported a related CuRe chain compound that exhibits the strongest ferromagnetic coupling (29 cm−1) yet observed through cyanide.27 Unfortunately, the zig-zag arrangement of the chain acts to minimize the overall anisotropy. As an extension of that discovery, future work will seek to construct linear chain compounds, such as the ones presented above, that feature Re–CN–Cu linkages. In addition to the Re–CN–Cu system, we will also target chain compounds where [ReCl4(CN)2]2− units are linked to second- and third-row transition metals, in an effort to impart stronger single-ion anisotropy and exchange coupling.
Acknowledgements
This research was supported by NSF Grant No. CHE-0617063. We thank Tyco Electronics for providing T.D.H. with a graduate fellowship, Dr. R. Clérac for helpful discussions, and Mr. J. M. Zadrozny for experimental assistance.Notes and references
- (a) A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760 CrossRef CAS; (b) R. Clérac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837 CrossRef CAS; (c) R. Lescouëzec, J. Vaissermann, C. Ruiz-Pérez, F. Lloret, R. Carrasco, M. Julve, M. Verdaguer, Y. Dromzée, D. Gatteschi and W. Wernsdorfer, Angew. Chem., Int. Ed., 2003, 42, 1483 CrossRef CAS; (d) T. F. Liu, D. Fu, S. Gao, Y. Z. Zhang, H. L. Sun, G. Su and Y. J. Liu, J. Am. Chem. Soc., 2003, 125, 13976 CrossRef CAS; (e) S. Wang, J.-L. Zuo, S. Gao, Y. Song, H.-C. Zhou, Y.-Z. Zhang and X.-Z. You, J. Am. Chem. Soc., 2004, 126, 8900 CrossRef CAS; (f) M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K. Sugiura, M. Yamashita, C. Coulon and R. Clérac, J. Am. Chem. Soc., 2005, 127, 3090 CrossRef CAS; (g) C. Coulon, H. Miyasaka and R. Clérac, Struct. Bonding, 2006, 122, 163 CAS; (h) K. Bernot, L. Bogani, A. Caneschi, D. Gatteschi and R. Sessoli, J. Am. Chem. Soc., 2006, 128, 7947 CrossRef CAS; (i) H. Miyasaka, T. Madanbashi, K. Sugimoto, Y. Nakazawa, W. Wernsdorfer, K. Sugiura, M. Yamashita, C. Coulon and R. Clérac, Chem.–Eur. J., 2006, 12, 7028 CrossRef CAS; (j) H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420 CrossRef CAS and references therein; (k) T. S. Venkatakrishnan, S. Sahoo, B. Brefuel, C. Duhayon, C. Paulsen, A. Barra, S. Ramasesha and J. Sutter, J. Am. Chem. Soc., 2010, 132, 6047 CrossRef CAS.
- R. J. Glauber, J. Math. Phys., 1963, 4, 294 CrossRef.
- (a) R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 804, 115 Search PubMed; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS; (c) D. Gatteschi, R. Sessoli, J. Villain, Molecular Nanomagnets, Oxford University Press: New York, 2006 and references therein Search PubMed; (d) E. J. Schelter, A. V. Prosvirin and K. R. Dunbar, J. Am. Chem. Soc., 2004, 126, 15004 CrossRef CAS; (e) C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 2754 CrossRef CAS; (f) D. E. Freedman, D. M. Jenkins, A. T. Iavarone and J. R. Long, J. Am. Chem. Soc., 2008, 130, 2884 CrossRef CAS; (g) D. Yoshihara, S. Karasawa and N. Koga, J. Am. Chem. Soc., 2008, 130, 10460 CrossRef CAS; (h) P.-H. Lin, T. J. Burchell, L. Ungur, L. Chibotaru, W. Wernsdorfer and M. Murugesu, Angew. Chem., Int. Ed., 2009, 48, 9489 CrossRef CAS.
- (a) D. A. Garanin and E. M. Chudnovsky, Phys. Rev. B: Condens. Matter, 1997, 56, 11102 CrossRef CAS; (b) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS; (c) M.-H. Jo, J. E. Grose, W. Liang, K. Baheti, M. M. Deshmukh, J. J. Sokol, E. M. Rumberger, D. N. Hendrickson, J. R. Long, H. Park and D. C. Ralph, Nano Lett., 2006, 6, 2014 CrossRef CAS; (d) A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2007, 98, 57201 Search PubMed; (e) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS; (f) P. C. E. Stamp and A. Gaita-Arino, J. Mater. Chem., 2009, 19, 1718 RSC; (g) S. Loth, K. von Bergmann, M. Ternes, A. F. Otte, C. P. Lutz and A. J. Heinrich, Nat. Phys., 2010, 6, 340 CrossRef CAS.
- (a) M. Suzuki and R. Kubo, J. Phys. Soc. Jpn., 1968, 24, 51; (b) C. Coulon, R. Clérac, L. Lecren, W. Wernsdorfer and H. Miyasaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 132408 CrossRef.
- A. Saitoh, H. Miyasaka, M. Yamashita and R. Clérac, J. Mater. Chem., 2007, 17, 2002 RSC.
- This equation applies in the infinite-size regime for an Ising-type single-chain magnet considering the following Hamiltonian:
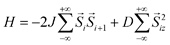 . See Ref. 5b.
. See Ref. 5b. - (a) K. R. Dunbar and R. A. Heintz, Prog. Inorg. Chem., 1997, 45, 283 CAS , and references therein; (b) L. M. C. Beltran and J. R. Long, Acc. Chem. Res., 2005, 38, 325 CrossRef CAS; (c) M. Shatruk, C. Avendano, K. Dunbar, In Progress in Inorganic Chemistry; K. D. Karlin, John Wiley & Sons: Amsterdam, 2009; 56, pp 155, and references therein Search PubMed.
- (a) A. Rodriguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Inorg. Chem., 2001, 40, 5868 CrossRef CAS; (b) A. D. Bond, S. Derossi, C. J. Harding, E. J. L. McInnes, V. McKee, C. J. McKenzie, J. Nelson and J. Wolowska, Dalton Trans., 2005, 2403 RSC; (c) H. Miyasaka, H. Takahashi, T. Madanbashi, K. Suguira, R. Clérac and H. Nojiri, Inorg. Chem., 2005, 44, 5969 CrossRef CAS; (d) M. Atanasov, P. Comba and C. A. Daul, J. Phys. Chem. A, 2006, 110, 13332 CrossRef CAS; (e) J. Gu, L. Jiang, M. Tan and T. Lu, J. Mol. Struct., 2008, 890, 24 Search PubMed; (f) D. Visinescu, M. Toma, J. Cano, O. Fabelo, C. Ruiz-Perez, A. Labrador, F. Lloret and M. Julve, Dalton Trans., 2010, 39, 5028 RSC; (g) H. Y. Kwak, D. W. Ryu, J. W. Lee, J. H. Yoon, H. C. Kim, E. W. Koh, J. Krinsky and C. S. Hong, Inorg. Chem., 2010, 49, 4632 CrossRef CAS.
- T. D. Harris, M. V. Bennett, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2010, 132, 3980 CrossRef CAS.
- (a) B. Soula, A. M. Galibert, B. Donnadieu and P. L. Fabre, Dalton Trans., 2003, 2449 RSC; (b) P. Guionneau, M. Marchivie, G. Bravic, J. F. Letard and D. Chasseau, Top. Cuur. Chem., 2004, 234, 97 Search PubMed; (c) E. Lefebvre, F. Conan, N. Cosquer, J. Kerbaol, M. Marchivie, J. Sala-Pala, M. M. Kubicki, E. Vigier and C. J. G. Garcia, New J. Chem., 2006, 30, 1197 RSC.
- O. Baumgatner, Z. Kristallogr., 1986, 174, 253 CAS; F. Marchetti, F. Marchetti, B. Melai, G. Pampaloni and S. Zacchini, Inorg. Chem., 2007, 46, 3378 CrossRef CAS; A. Albinati, F. Calderazzo, F. Marchetti, S. A. Mason, B. Melai, G. Pampaloni and S. Rizzato, Inorg. Chem. Commun., 2007, 10, 902 Search PubMed.
- (a) M. Shatruk, A. Draguloscu-Andrasi, K. E. Chambers, S. A. Stoian, E. L. Bominaar, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2007, 129, 6104 CrossRef CAS; (b) V. Niel, M. C. Munoz, A. B. Gaspar, A. Galet, G. Levchenko and J. A. Real, Chem.–Eur. J., 2002, 8, 2446 CrossRef CAS; (c) Y. Zhang, T. Liu, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2009, 131, 7942 CrossRef CAS; (d) V. Jacob, G. Huttner, E. Kaifer, P. Kircher and P. Rutsch, Eur. J. Inorg. Chem., 2001, 2783 Search PubMed; (e) M. G. Hilfiger, M. Chen, T. V. Brinzari, M. Shatruk, D. T. Petasis, J. L. Musfeldt, C. Achim and K. R. Dunbar, Angew. Chem. Int. Ed., 2010, 49, 1410 CAS.
- (a) M. Seredyuk, H. Haukka, I. O. Fritsky, H. Kozlowski, R. Kramer, V. A. Pavlenko and P. Gutlich, Dalton Trans., 2007, 3183 RSC; (b) A. Galet, M. C. Manoz, V. Martinez and J. A. Real, Chem. Commun., 2004, 2268 RSC.
-
(a) M. Drillon, E. Coronado, D. Beltran and R. Georges, Chem. Phys., 1983, 79, 449 CrossRef CAS;
(b)
R. Georges, J. J. Borras-Almenar, E. Coronado, J. Curely, M. Drillon, Magnetism: Molecules to Materials I: Models and Experiments, ed. J. S. Miller, M. Drillon, Wiley-VCH: Verlag, 2002 Search PubMed. The expression used to describe the magnetic susceptibility follows:
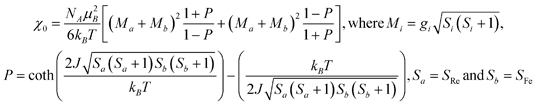 .
. - The χMT vs. T data were fit applying the constraint gRe = gFe, as the fitting procedure was unable to independently and accurately determine the two g parameters independently.
- Here, Oh symmetry is considered to approximate the local coordination environments of the ReIV and FeII ions.
- W. R. Entley, C. R. Trentway and G. S. Girolami, Mol. Cryst. Liq. Cryst., 1995, 273, 153 CrossRef; H. Weihe and H. U. Gudel, Comments Inorg. Chem., 2000, 22, 75 CrossRef CAS.
- (a) S. Ohkoshi, S. Yorozu, O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Appl. Phys. Lett., 1997, 70, 1040 CrossRef CAS; (b) B. Gao, J. Yao and D. J. Xue, J. Magn. Magn. Mater., 2010, 322, 2505 Search PubMed; (c) Y.-Z. Zhang, B.-W. Wang, O. Sato and S. Gao, Chem. Commun., 2010, 46, 6959 RSC.
- T. D. Harris, H. Soo, C. J. Chang and J. R. Long, Inorg. Chim. Acta., 2011, 369, 91 Search PubMed.
- A. V. Palii, B. S. Tsukerblat and M. Verdaguer, J. Chem. Phys., 2002, 117, 7896 CrossRef CAS.
- (a) K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef CAS; (b) C. J. F. Boettcher, Theory of Electric Polarisation; Elsevier: Amsterdam, 1952 Search PubMed; (c) S. M. Aubin, Z. Sun, L. Pardi, J. Krzystek, K. Folting, L.-J. Brunel, A. L. Rheingold, G. Christou and D. N. Hendrickson, Inorg. Chem., 1999, 38, 5329 CrossRef CAS.
- N. Ishii, Y. Okamura, S. Chiba, T. Nogami and T. Ishida, J. Am. Chem. Soc., 2008, 130, 24 CrossRef CAS.
- L. M. Toma, R. Lescouëzec, F. Lloret, M. Julve, J. Vaissermann and M. Verdaguer, Chem. Commun., 2003, 1850 RSC.
- Note that the term “single-chain magnet” was not initially used to describe this compound. Rather, the term first appeared in Ref. 1b.
- (a) C. Coulon, R. Clérac, W. Wernsdorfer, T. Colin and H. Miyasaka, Phys. Rev. Lett., 2009, 102, 167204-1–4 Search PubMed; (b) H. Miyasaka, K. Takayama, A. Saitoh, S. Furukawa, M. Yamashita and R. Clérac, Chem.–Eur. J., 2010, 3656 Search PubMed.
- T. D. Harris, C. Coulon, R. Clérac and J. R. Long, J. Am. Chem. Soc., 2011, 133, 123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 820653–820657. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00220a |
| This journal is © The Royal Society of Chemistry 2011 |