Evolution of the structures and magnetic properties of the manganese dicarboxylates, Mn2(CO2(CH2)nCO2)(OH)2 and Mn4(CO2(CH2)nCO2)3(OH)2†
Paul J.
Saines
a,
Prashant
Jain
b and
Anthony K.
Cheetham
*a
aDepartment of Materials Science and Metallurgy, The University of Cambridge, Cambridge, CB2 3QZ, United Kingdom. E-mail: akc30@cam.ac.uk
bDepartment of Chemistry and Biochemistr, Florida State University, Tallahassee, Florida 32306, USA
First published on 15th July 2011
Abstract
Two new series of basic Mn dicarboxylate frameworks, Mn2(CO2(CH2)nCO2)(OH)2 (where n = 0, 2, 4 and 5) and Mn4(CO2(CH2)nCO2)3(OH)2 (where n = 3 and 5), have been synthesised and the evolution of their structures with changing length of the dicarboxylate ligand was examined. Compounds in the Mn2(CO2(CH2)nCO2)(OH)2 series contain 2 dimensionally inorganically connected layers, which are bridged in the third dimension by the dicarboxylate ligand. While the compounds in this series with n = 0 or 2 contain only octahedrally coordinated Mn, the frameworks with longer ligands, n = 4 or 5, have a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of octahedrally and trigonal bipyramidally coordinated cations. Structures in the Mn4(CO2(CH2)nCO2)3(OH)2 series contain inorganically connected double chains of MnOx polyhedra, which comprise an equal number of octahedra and trigonal bipyramids. Trigonal bipyramidal coordination environments are very rarely found in dicarboxylate frameworks and the roles of the longer dicarboxylate ligands and the d5 electronic configuration of Mn2+ in their formation are discussed. The magnetic properties of Mn2(CO2(CH2)2CO2)(OH)2 have also been examined. It undergoes two magnetic transitions. The higher temperature transition to a two dimensionally ordered antiferromagnetic phase occurs around 44 K, in low applied fields, and is followed by a transition to a three dimensionally ordered canted antiferromagnetic state near 36 K. The Néel temperature of this phase is unusually high for a transition metal dicarboxylate and the factors thought to support this are examined.
:1 ratio of octahedrally and trigonal bipyramidally coordinated cations. Structures in the Mn4(CO2(CH2)nCO2)3(OH)2 series contain inorganically connected double chains of MnOx polyhedra, which comprise an equal number of octahedra and trigonal bipyramids. Trigonal bipyramidal coordination environments are very rarely found in dicarboxylate frameworks and the roles of the longer dicarboxylate ligands and the d5 electronic configuration of Mn2+ in their formation are discussed. The magnetic properties of Mn2(CO2(CH2)2CO2)(OH)2 have also been examined. It undergoes two magnetic transitions. The higher temperature transition to a two dimensionally ordered antiferromagnetic phase occurs around 44 K, in low applied fields, and is followed by a transition to a three dimensionally ordered canted antiferromagnetic state near 36 K. The Néel temperature of this phase is unusually high for a transition metal dicarboxylate and the factors thought to support this are examined.
1 Introduction
Inorganic–organic frameworks have been the focus of extensive research due to the fascinating range of structures and wide variety of properties these versatile materials can exhibit.1,2 Recently more of this work has been focused on denser frameworks, with extensive inorganic (metal–oxygen–metal) connectivity, less bulky ligands and little or no porosity.1 This is primarily due to their ability to exhibit properties more commonly associated with metal oxides, such as co-operative magnetic behaviour, electronic conductivity and multiferrocity, but with the structure-directing properties of the ligand giving rise to unusual, often lower dimensional, behaviour.3 Amongst these denser frameworks, the structures of the first row transition metal linear dicarboxylates, where the organic ligand is of the type CO2(CH2)nCO2 (0 ≤ n ≤ 12), have been extensively studied.4–20 These materials often include hydroxy and aquo-ligands and adopt an intriguing variety of structures, ranging from simple 1D organically connected polymers to extensively 3D inorganically connected frameworks, depending on a wide range of factors such as reaction temperature, pH and composition, including cation and ligand present.4–20Despite this variation in structures, however, the vast majority of transition metal dicarboxylates contain principally octahedrally coordinated cations. Although some Zn2+ and Co2+ compounds have tetrahedral metal ions18–20 and several Jahn–Teller distorted Cu2+ frameworks have essentially square planar metal sites, with longer bonds to fifth and sixth ligands in a second coordination sphere,12,13,17 other geometries are exceptionally rare. The recent discovery of a fascinating new Mn succinate, Mn(CO2(CH2)2CO2), which has two alternating layers of edge-sharing chains and corner-sharing sheets of distorted octahedra, highlights the ability of high spin d5Mn2+ cations to adopt coordination environments that do not have regular octahedral symmetry.10 These layers order antiferromagnetically at 10 K and 6 K, respectively, with very different magnetic structures.9 The Néel temperature of this phase is comparable to the other known Mn transition metal dicarboxylates, none of which order magnetically above 13 K, emphasising the importance of tailoring these materials to achieve higher ordering temperatures.6,11,14,21
Given the intriguing structural and magnetic behaviour of Mn(CO2(CH2)2CO2) we were inspired to explore the structures adopted by two new families of Mn dicarboxylates, with a particular focus on the bonding environment of the Mn2+ cation and the physical properties this gives rise to. We describe the structures of six new compounds formed in solutions with higher pH than required to synthesise the Mn dicarboxylates known to date.6,10,11,14,22,23 These materials belong to one of two series, the Mn2(CO2(CH2)nCO2)(OH)2 (where n = 0, 2, 4 or 5) compounds and the Mn4(CO2(CH2)nCO2)3(OH)2 (where n = 3 or 5) frameworks.
2 Experimental
The novel phases discovered in this work were made during an extensive exploration of the phases formed, under hydrothermal conditions, by Mn(CO2(CH2)nCO2) dicarboxylates, where the ligand was 0 ≤ n ≤ 5 (oxalate, malonate, succinate, glutarate, adipate and pimelate). Conditions were varied around a standard mixture of MnCl2·4H2O, dicarboxylic acid, KOH and water in a 1:1.5:5:200 molar ratio, with a total of 9 mL of water used. These mixtures were heated for 3 days, at temperatures between 100 °C and 200 °C, in 23 mL teflon-lined Parr autoclaves, after which the products were recovered by filtration. The phases reported in this work were the only frameworks identified when the ratio of KOH to ligand was 5:1.5 or higher, although both frameworks 1, Mn2(CO2CO2)(OH)2, and 5, Mn4(CO2(CH2)3CO2)3(OH)2, were found to readily form at ratios as low as 3.75:1.5. The compounds generally formed over the whole temperature range explored, except phases 1 and 6, Mn4(CO2(CH2)5CO2)3(OH)2, which only formed in significant quantities in reactions carried out above 150 °C and 180 °C, respectively. All frameworks can be formed at the standard molar ratio apart from compound 6, which requires the ratio of MnCl2·4H2O used to be at least doubled for it to form in appreciable quantities. The precise conditions used to synthesise the single crystals used for structure determinations are described in Table S1 and details of the structural determinations, including crystallographic data in Table S2 and S3, are also presented in the ESI.†
X-ray powder diffraction patterns, collected on a Cu-Kα Bruker D8 Advance diffractometer, indicated that all samples made in this work contained small amounts of inorganic impurities. These were typically a mixture of divalent and trivalent MnOx(OH)y type salts or MnCO3 and are thought to form due to the high pH of the solution required to make these frameworks. Extensive attempts were made to eliminate the formation of these impurities by modifying the synthetic conditions, particularly lowering pH. This led to the formation of other, previously known phases, primarily the Mn2(CO2(CH2)nCO2)2(H2O), Mn(CO2(CH2)2CO2) or Mn(CO2CO2)(H2O)2 frameworks.10,11,22,23 Additionally, attempts were made to purify samples post-synthesis, either by washing with dilute acid or purification by flotation.24 These approaches, however, were also unsuccessful.
It was possible to obtain compound 2, Mn2(CO2(CH2)2CO2)(OH)2, as a pure phase by manual separation from the same sample used for structural determination. Microanalysis of this sample, obtained at the Department of Chemistry, University of Cambridge, indicated a composition of 18.38% C and 2.20% H (expected values of 18.48% C and 2.33% H) and its purity as a single phase was confirmed by powder X-ray diffraction (see Fig. S1 for Le Bail fitting done using the program Rietica25). Experimental details of infra-red spectroscopy, thermogravimetric analysis and magnetic susceptibility measurements are presented in the ESI.†
3 Results and discussion
3.1 Structures of the Mn2(CO2(CH2)nCO2)(OH)2 (n = 0, 2, 4 or 5) series
At first glance the four compounds in the series Mn2(CO2(CH2)nCO2)(OH)2 have very similar structures. They are all either P21/n or P21/a monoclinic, alternate settings of the same space group, and consist of sheets of MnOx polyhedra bridged by dicarboxylate ligands. In the notation of Cheetham et al.1 they are all I2O1, indicating they have inorganic bridging (Mn–O–Mn) in two dimensions but only organic bridging (Mn-ligand-Mn) in the third. The density of the crystal structure decreases as n increases due to the increased separation of the inorganically connected layers, caused by the longer ligand length. Examining the details of each structure in further depth allows significant, but often subtle, differences to be noted and the evolution of the structures in this series to be understood.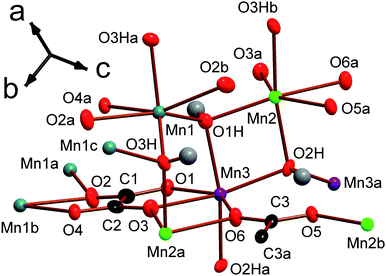 | ||
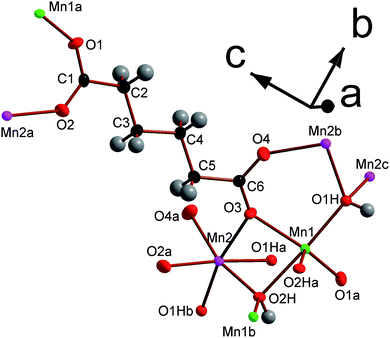
| Fig. 1 The asymmetric unit of framework 1 with 80% probability ellipsoids. The labels on the hydroxyl hydrogens are omitted for the sake of clarity; their numbering is the same as for their corresponding oxygen, which are marked O1H, O2H and O3H. Additional non-hydrogen atoms, included to illustrate the coordination sphere of all atoms in the structure, are shown and labelled alphabetically. In the online colour version the Mn1, Mn2 and Mn3 atoms are teal, light green and violet, respectively. The oxygen, carbon and hydrogen atoms are red, black and gray. | ||
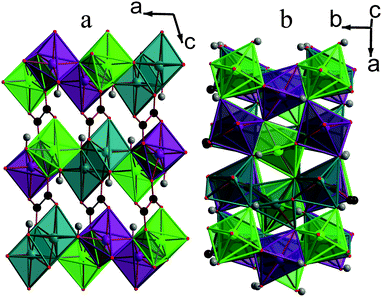 | ||
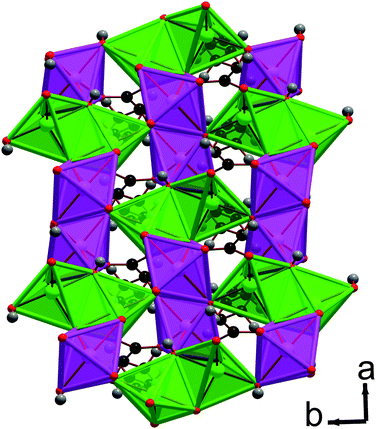
| Fig. 2 Structure of compound 1 displaying a) the ac plane and b) the ab plane. In the online colour version the MnO6 octahedra are teal, light green and violet for Mn1, Mn2 and Mn3, respectively, and all other colours are the same as for Fig. 1. | ||
Each Mn in compound 1 is coordinated to three carboxylate oxygen atoms, two of which are from either end of the same ligand, and three different hydroxide ligands, which are arranged in a meridional fashion. All hydroxide groups are coordinated to three different Mn atoms within the same layer. Two of the three distinct carboxylates are on the two ends of the same ligand while the third features on both ends of the other crystallographically distinct carboxylate. By analogy with the terminology used to describe coordination in phosphonate frameworks, the dicarboxylates in this phase are described as having (1212) connectivity.27,28 This indicates that the two carboxylate groups in the ligand have one oxygen atom that is bonded to one metal while the other oxygen atom is coordinated to two different Mn cations (see Fig. S2a†). The torsion angle between the planes of the two carboxylate groups in both crystallographically distinct ligands is close to zero.
Framework 1 is very similar to another phase, with the same empirical formula, discovered by Lethbridge et al.,22 which adopts the same space group and also consists of layers of MnO6 octahedra bridged by oxalate ligands. In this previously known structure, however, there is only one distinct Mn cation, one hydroxide and half a carboxylate group in the asymmetric unit. Additionally, octahedra in adjacent layers are not offset but rather lie directly below each other. Lethbridge et al.22 determined the structure of their phase at room temperature, while the structure of 1 was obtained at 120 K. Single crystal X-ray diffraction was therefore performed on crystals of 1 at 300 K, which confirmed that it retains the same crystal structure up to room temperature, signifying that it is a distinctly different framework.
It is clear that the phase discovered by Lethbridge et al.22 and 1 are polymorphs of the Mn2(CO2CO2)(OH)2 phase, which form at lower and higher synthetic temperatures, respectively. This is consistent with the lower synthesis temperature used in the previous work and the fact that 1 could only be formed at temperatures above 150 °C with reactions at lower temperatures appearing to favour the formation of the previously known phase. Since 1 is the denser phase, albeit only slightly, (cf. a crystallographic density at room temperature of 3.069 g cm−3 for framework 1 with 3.046 g cm−3 for the phase discovered by Lethbridge et al.22) it is tempting to consider it to be the more thermodynamically stable compound, consistent with previous studies of similar frameworks.16,29 This would imply that the phase discovered by Lethbridge et al.22 forms as a result of it being kinetically favoured at lower temperatures. The recent study of the relative stabilities of Li tartrate phases by Yeung et al.,30 however, indicates that the differences in energy between different polymorphic dicarboxylate phases is very small and difficult to establish precisely, even using high level computational methods. Therefore it is not possible to draw any clear conclusions regarding the relative stability of the two Mn oxalate phases examined in this study.
 | ||
| Fig. 3 The asymmetric unit of framework 2 with 80% probability ellipsoids. The labels on the hydrogen atoms are omitted for the sake of clarity. Additional non-hydrogen atoms, included to illustrate the coordination sphere of all atoms in the structure and a whole succinate ligand, are shown and labelled alphabetically. In the online version the Mn atoms are pink and all other colours are the same as for Fig. 1. | ||
 | ||
| Fig. 4 Structure of compound 2 displaying a) the bc plane and b) the ab plane. In the online version the MnO6 octahedra are pink and all other colours are the same as for Fig. 1. | ||
The Mn cations in framework 2 bond to three oxygen atoms from three different carboxylate groups, two of which are from the O1 positions and the third from the O2 site, and three hydroxide groups in a meridional arrangement. As in 1, all hydroxide groups bond with three Mn atoms while atoms O1 and O2, belonging to the one crystallographically distinct carboxylate group, bond to two and one Mn cation, respectively. This results in all carboxylate ligands co-ordinating to cations in a (1212) fashion and, similarly to compound 1, the torsion angle of the two carboxylate groups of the ligand is close to zero (see Fig. S2b†). Unlike 1, however, where the bond distance between the carboxylate oxygen atoms and the Mn to which they coordinate does not change greatly depending on whether the oxygen is one or two fold coordinated, in 2 the two Mn–O1 bonds are significantly longer than the Mn–O2 bond distances (see Table S4†).
:50 mixture of octahedral and trigonal bipyramidal coordination environments. The asymmetric units of both contain two Mn cations, two hydroxide groups and either an adipate or a pimelate ligand (see Fig. 5 and Fig. S3†). The layers of MnOx polyhedra consist of edge-sharing dimers of octahedra and trigonal bipyramids, which alternate within an edge-sharing polyhedral chain aligned along the a-axis (see Fig. 6). Additionally, one of the distinct hydroxide groups causes the trigonal bipyramids and octahedra that are next-nearest neighbours to share corners. The chains are linked by the other distinct hydroxide group, which bonds to one trigonally bipyramidally coordinated Mn in one chain and two octahedrally coordinated Mn in another, causing the trigonal bipyramids and octahedra in neighbouring chains to share corners. The inorganically connected sheets are bridged by either the adipate or pimelate ligand, in structures 3 and 4, respectively, with the larger chain lengths of these ligands increasing the interlayer spacings compared to compounds 1 and 2 to separations of approximately 10.0 Å and 10.9 Å, respectively. Similarly to 2, carboxylate groups on either end of the ligands bond to different layers. The one noticeable difference between frameworks 3 and 4 is that, while in 3 the carbon chains of the adipate ligands are essentially perpendicular to the inorganic sheets, in 4 the carbon backbone of the pimelate ligands are not (see Fig. 7). This is presumably a result of the difference in the number of carbons in the two ligands.
 | ||
| Fig. 5 The asymmetric unit of framework 3 with 80% probability ellipsoids. The labels on the hydrogen atoms are omitted for the sake of clarity. Additional non-hydrogen atoms, included to illustrate the coordination sphere of all atoms in the structure, are shown and labelled alphabetically. In the online version the five and six coordinate Mn are green and pink, respectively. All other colours are the same as for Fig. 1. | ||
 | ||
| Fig. 6 Structure of compound 3 displaying the ab plane. In the online version the MnO5 trigonal bipyramids are green, the MnO6 octahedra are pink and all other colours are the same as for Fig. 5. | ||
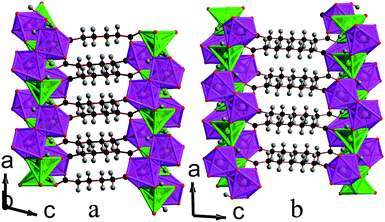 | ||
| Fig. 7 Structure of compounds a) 3 and b) 4 displaying the ac plane. The colours in the online version are the same as for Fig. 6. | ||
In compounds 3 and 4 the Mn1 cation is the five coordinated site and is bonded to three different hydroxide groups and two carboxylate oxygen atoms from different ligands, in a trans-arrangement. Mn2 is octahedrally coordinated to three different hydroxide groups, which are in a facial-arrangement and three carboxylate oxygen atoms from different ligands. Mn1 and Mn2 have bond valencies close to two confirming that all Mn in these structures are divalent (cf. values of 2.02 and 2.05 for Mn 1 and 2.05 and 2.04 for Mn 2 in frameworks 3 and 4, respectively).26 Three of the five Mn1–O bonds are less than 2.10 Å, significantly shorter than found for the octahedrally coordinated cations in this work, thereby compensating for their lower coordination number (see Table S4 for bond distances†). As in structures 1 and 2, both crystallographically distinct hydroxide groups bond to three different Mn cations. Unlike the oxalate and succinate structures, the dicarboxylate ligands in 3 and 4 exhibit (1112) connectivity with only one of the four distinct carboxylate oxygen atoms bonded to two Mn cations, while the other three coordinate to only one cation (see Fig. S2c†). Interestingly, O3, one of the two oxygen atoms from the carboxylate group where both oxygen atoms only bond to one Mn, has a bond length of 2.094(2) Å and 2.087(6) Å to Mn1 in 3 and 4, respectively. This is much shorter than is typical for the other bonds of this type encountered in this study and suggests it is shortened to compensate for the decrease in bonding between the Mn cations and the carboxylate group that occurs as a result of the lower coordination of Mn1. The torsion angles of the adipate and pimelate ligands in these structures are about 30°, much higher than in frameworks 1 and 2.
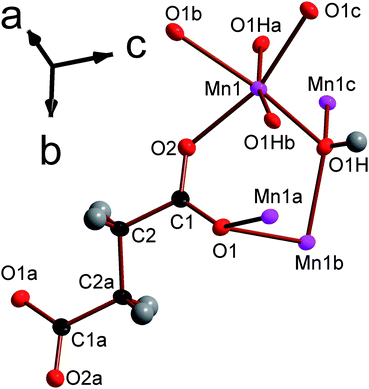
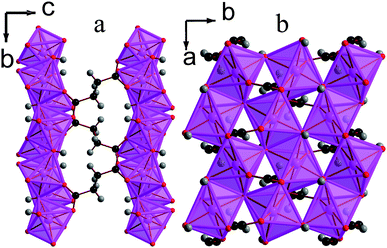
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) triclinic structure, double chains of MnOx polyhedra, which run parallel to the a-axis, are interconnected in the other two dimensions by the glutarate ligand, giving a structure with I1O2 connectivity (see Fig. 8). The asymmetric unit of 5 contains four Mn cations, three glutarate ligands and two hydroxide groups (see Fig. 9). Two of these Mn cations, Mn2 and Mn4, occupy severely distorted octahedral sites while the other two cations, Mn1 and Mn3, adopt trigonal bipyramidal sites. The polyhedra in the structure 5 are arranged such that the two distinct octahedra and trigonal bipyramids are in the different threads of the double chain and each thread has alternating octahedra and trigonal bipyramids (see Fig. 8a). Each polyhedron shares a corner with one of its neighbours in a chain and an edge with the other. Nearest neighbours in different threads of the double chain have different coordination numbers and are linked by a μ3-hydroxide, such that every polyhedron in a chain shares a corner with both an octahedron and a trigonal bipyramid in the neighbouring thread. Examining the inter-chain bonding it can be seen that each double chain is connected by glutarate ligands to four other double chains, with two connections for every four Mn cations to two of these chains and one to the other two (see Fig. 8b).
triclinic structure, double chains of MnOx polyhedra, which run parallel to the a-axis, are interconnected in the other two dimensions by the glutarate ligand, giving a structure with I1O2 connectivity (see Fig. 8). The asymmetric unit of 5 contains four Mn cations, three glutarate ligands and two hydroxide groups (see Fig. 9). Two of these Mn cations, Mn2 and Mn4, occupy severely distorted octahedral sites while the other two cations, Mn1 and Mn3, adopt trigonal bipyramidal sites. The polyhedra in the structure 5 are arranged such that the two distinct octahedra and trigonal bipyramids are in the different threads of the double chain and each thread has alternating octahedra and trigonal bipyramids (see Fig. 8a). Each polyhedron shares a corner with one of its neighbours in a chain and an edge with the other. Nearest neighbours in different threads of the double chain have different coordination numbers and are linked by a μ3-hydroxide, such that every polyhedron in a chain shares a corner with both an octahedron and a trigonal bipyramid in the neighbouring thread. Examining the inter-chain bonding it can be seen that each double chain is connected by glutarate ligands to four other double chains, with two connections for every four Mn cations to two of these chains and one to the other two (see Fig. 8b).
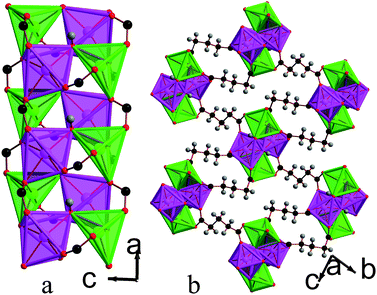 | ||
| Fig. 8 Structure of compound 5 in a) the ac plane and b) the bc plane. In the online version the colours are the same as Fig. 6. | ||
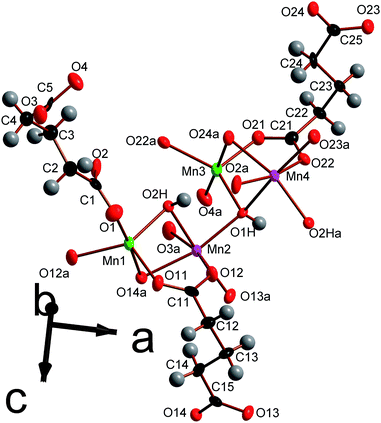 | ||
| Fig. 9 Depiction of the asymmetric unit of framework 5 with 80% probability ellipsoids. The labels on the hydrogen atoms are omitted for the sake of clarity. Additional non-hydrogen atoms, included to illustrate the coordination sphere of all cations in the structure, are shown and labelled alphabetically. In the online version the colours are the same as Fig. 6. | ||
In compound 5 every Mn is coordinated to four carboxylate oxygen atoms from four different ligands. The five and six fold coordinated Mn are bonded to one and two hydroxide anions, respectively, with the two hydroxide groups binding cisoid to each other in the later case (see Table S5 for bond distances†). The bond valencies of all four distinct Mn cations are very close to 2 (between 1.99 and 2.05), confirming that all the Mn in this structure is divalent.26 As was the case in compounds 3 and 4, the decreased number of ligands coordinated to the penta-coordinated Mn cations are compensated by these five bonds being significantly shorter than those of the octahedrally coordinated Mn. Similarly to the compounds in the Mn2(CO2(CH2)nCO2)(OH)2 series, all hydroxide ligands are coordinated to three distinct Mn. Of the distinct carboxylate ligands, two of the three exhibit (1212) connectivity (see Fig. S2†) while the third ligand bonds Mn in a (1111) fashion. These two types of carboxylate ligands both have torsion angles of approximately 80–90°, which is markedly higher than in any of the ligands in the Mn2(CO2(CH2)nCO2)(OH)2 series.
Apart from the substitution of the glutarate with the longer pimelate ligand, compound 6, Mn4(CO2(CH2)5CO2)3(OH)2, is almost isostructural with framework 5. The only significant difference is that the presence of the longer ligand leads to the inorganic chains being significantly further apart, resulting in a less dense crystal structure (see Fig. 10). It has an asymmetric unit that contains four Mn cations, two hydroxide anions and three pimelate ligands, and the cations have the same coordination environment as in framework 5 (see Fig. S4 and Table S5†). The bond valencies of the Mn cations are between 1.93 and 2.05, with Mn1 and Mn3 having significantly shorter bonds to make up for their lower coordination number.26 As with all other compounds examined in this work, the hydroxides are coordinated to three different Mn cations and the pimelate ligands have the same arrangement and number of bonds with the cations as in compound 5. The torsion angles of the pimelate ligands are all approximately 80–90°.
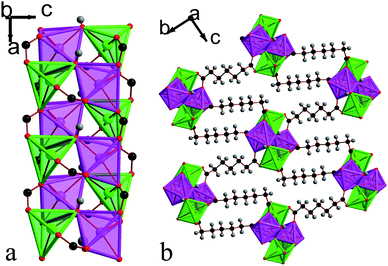 | ||
| Fig. 10 Structure of compound 6 in a) the ac plane and b) the bc plane. In the online version the colours are the same as Fig. 6. | ||
3.3 Structural evolution of the Mn2(CO2(CH2)nCO2)(OH)2 and Mn4(CO2(CH2)nCO2)3(OH)2 series
A comparison of the structures of the six compounds studied in this work allows the effect of increasing the length of the carbon chain of the ligand to be understood. Starting with the oxalate ligand, the shortest of the dicarboxylates with n = 0, it is clear that there are two frameworks which belong the Mn2(CO2(CH2)nCO2)(OH)2 series, the compound previously synthesised by Lethbridge et al.22 and compound 1. These are polymorphs that can be synthesised at low and higher temperatures, respectively. 1, the higher temperature polymorph, has a more complex asymmetric unit and distinctly different MnO6 polyhedra, which shift positions along the a-axis between alternate layers. Changing the ligand to the longer succinate chain (n = 2) in framework 2 appears to have three main effects. The first is that, as would be expected, the distance between the MnO6 layers increases significantly. This also changes the connectivity between the layers from featuring different oxygen atoms from the same carboxylate group bonding to two different layers, as was the case in both oxalate compounds, to the two carboxylate groups on opposite ends of the ligand bonding to two neighbouring layers. Finally lengthening the ligand appears to favour the formation of a structure in which the arrangement of the octahedra in the MnO6 layers is more akin to that of the low temperature polymorph of the oxalate composition. It is possible that a polymorph of 2 with a similar structure to 1 exists but, if so, it appears that lengthening the chain length of the ligand increases the temperature range over which the simpler structure is stable so such a high temperature polymorph is not observed in this study.Since there are significant, but subtle, differences between the structures of 1 and 2, attempts were made to make an analogous phase using the malonate (n = 1) ligand to increase the understanding of the evolution of structures in this series with changing ligand length. Unfortunately this was unsuccessful and, indeed, any attempts to make malonate frameworks under basic conditions, similar to those used to make the other phases discussed in this work, led to the formation of MnCO3; this is interpreted as an indication that the ligand has decomposed. Lengthening the carbon chain of the ligand to n = 3 seems to initially destabilise compositions of the type Mn2(CO2(CH2)nCO2)(OH)2, leading to the alternate Mn4(CO2(CH2)3CO2)3(OH)2 composition, framework 5, which will be discussed later in this section.
For the adipate and pimelate ligands (n = 4 and 5 respectively), however, the Mn2(CO2(CH2)nCO2)(OH)2 composition again becomes accessible, but in these cases only half the Mn positions are octahedrally coordinated while the remaining cations occupy trigonal bipyramidal sites. In contrast to the other changes in ligand length, the effect of changing between the adipate and pimelate ligands is very small. The arrangement of adjacent polyhedral layers in compounds 3 and 4 appears to be similar to that found in 2, although there are some subtle differences in the connectivity of the polyhedra. The type of coordination expressed by the ligands changes from (1212) in compounds 1 and 2 to (1112) in 3 and 4; this is thought to be intimately related to the presence of the trigonal bipyramids, since they require one less metal–ligand coordination bond. The torsion angles of the dicarboxylic acids are significantly higher in compounds 3 and 4, increasing to approximately 30° from close to 0° in 1 and 2. The longer linear dicarboxylates have previously been shown to be able to accommodate higher torsion angles in framework structures than their shorter analogues and this is also likely to play a significant role in the differences in the structures adopted by compounds in the Mn2(CO2(CH2)nCO2)(OH)2 series.28,31 Despite a number of differences in the four compounds discussed in this series, the MnOx polyhedra are either significantly distorted octahedra or have five-fold coordination. This suggests that, as was previously seen in the Mn(CO2(CH2)2CO2) compound synthesised by Saines et al.,10 the high spin d5 electronic configuration of Mn2+ may play an important role in allowing these unusual structures to form. Likewise, all hydroxide ligands in these compounds have μ3 coordination and it is likely this is important to stabilising structures with this composition.
The Mn4(CO2(CH2)3CO2)3(OH)2 composition adopted by the glutarate under basic conditions belongs to a second series of Mn dicarboxylate compounds that have cations in a trigonal bipyramidal coordination environment. It is not, however, generally favoured with any other ligand. It only forms for Mn pimelate (n = 5) under a very narrow range of conditions, usually in combination with either the Mn2(CO2(CH2)5CO2)2(H2O) or Mn2(CO2(CH2)5CO2)(OH)2 frameworks, and is not observed to form with the other dicarboxylates examined in this study. The presence of MnO5 trigonal bipyramids is also found to be associated with a decreased connectivity of the carboxylate ligands, as was the case for structures 3 and 4. Here, however, this occurs by a third of the ligands adopting (1111) connectivity while the other two-thirds maintain the (1212) connectivity found in the structures that only contain octahedrally coordinated Mn. The torsion angles of the carboxylates in compounds 5 and 6 are also significantly higher than those in other compounds examined in this work and the additional flexibility of these longer ligands is likely to be important in the formation of these frameworks.
The architecture of Mn4(CO2(CH2)nCO2)3(OH)2 is somewhat akin to that adopted by Fe4(CO2(CH2)3CO2)3(OH)2·H2O, which also has an I1O2 structure with double chains of FeOx polyhedra.32 An extensive search of the Cambridge Structural Database33 indicated that this is the only previously known transition metal dicarboxylate that features a transition metal in a trigonal bipyramidal site. The iron compound is somewhat different, however, in that it adopts P21/n symmetry and has a double chain with one strand containing only corner-sharing trigonal bipyramids and the other with only edge-sharing octahedra. The double chains, while having the same basic arrangement, are also spaced in such a way to create pores, which are occupied by water molecules. Examination of the crystal structures 5 and 6 using the SOLV routine in PLATON34 indicated that these compounds did not contain any accessible pore space. There is no evidence for a hydrated phase in the present study.
Fe4(CO2(CH2)3CO2)3(OH)2·H2O is interesting as it contains Fe2+, which indicates that the Fe is d6. This shows that, while the d5 electron configuration of Mn2+ may assist the stabilisation of a trigonal bipyramidal cation site in a transition metal dicarboxylate framework, it is not essential. There are other known transition metal dicarboxylates that contain penta coordinated cations. The majority of these are Jahn–Teller distorted Cu2+ cations in regular square planar arrangements with an additional, significantly longer bond to another ligand, often a water molecule, making a very distorted square pyramidal site.12,13,17 The only other example is Co6(C4H4O4)5(OH)2·H2O, which adopts a layered I2O1 structure with a quarter of its Co cations in square pyramidal sites and the remainder in octahedral positions.8
It is worth noting at this point that compound 6 is less dense than 4 and that, similarly, framework 5 is much less dense than might be expected by comparison with compounds 2 and 3. Since there is no significant pore volume in any of these compounds, the lower density can be attributed to the higher ratio of carboxylic acid ligands per cation in the structures of the Mn4(CO2(CH2)nCO2)3(OH)2 phases. The higher ratio of carboxylate ligands in the Mn4(CO2(CH2)nCO2)3(OH)2 compounds also leads to these phases having a larger number of oxygen atoms available to coordinate to the Mn cations (cf. 3.5 oxygen atoms per Mn in Mn4(CO2(CH2)nCO2)3(OH)2 with 3.0 oxygen atoms in the Mn2(CO2(CH2)nCO2)(OH)2 series). There is also a corresponding decrease in the number of μ3 co-ordinating hydroxide ligands per Mn cation in the Mn4(CO2(CH2)nCO2)3(OH)2 series. These two factors promote structures with oxygen atoms coordinated to a lower number of cations and lead to the Mn4(CO2(CH2)nCO2)3(OH)2 composition favouring architectures with lower inorganic connectivity, when compared to the Mn2(CO2(CH2)nCO2)(OH)2 compounds. The other three known Mn dicarboxylates whose composition can be expressed in the form Mn(CO2(CH2)nCO2)1−x(OH)2x, however, underline that the relationship between composition and structure is more complicated with other factors affecting the architectures adopted. Mn(CO2(CH2)2CO2), Mn5(CO2(CH2)2CO2)4(OH2)5 and Mn(CO2(CH2)3CO2) all exhibit significantly different structures yet, despite featuring a higher oxygen to metal ratio than the Mn4(CO2(CH2)nCO2)3(OH)2 series, they all have inorganic connectivity in more than one dimension.10,14,15 They appear to achieve this by featuring some combination of multiple layers with different dimensionalities of inorganic connectivity, having layers with 2D inorganically connected MnO6 rings and using more extensive corner-sharing connectivity, as opposed to edge-sharing, between MnO6 octahedra. All three of these factors enable structures with higher dimensional inorganic connectivity to form without increasing the coordination number of the average oxygen atom.
3.4 Thermal analysis
Thermogravimetric analysis of a pure sample of compound 2 reveals that it loses significant weight between 260 °C and 360 °C, indicating that it decomposes in this temperature range (see Fig. S5†). Subsequently there is a small weight gain between 420 °C and 580 °C which, by analogy with Mn(CO2(CH2)2CO2),10 and in light of the dark brown colour of the final residue, can be attributed to the oxidation of the initial decomposition product of Mn3O4 to Mn2O3. The weight of the sample stabilises above 580 °C at a net loss of 40.5% of initial weight, consistent with the complete decomposition of the ligand and formation of Mn2O3 (calculated 39.3%). The presence of small amounts of inorganic impurities present in the other samples synthesised in this study prevented a thorough analysis of their decomposition. All samples examined, however, have a significant weight loss, consistent with decomposition beginning between 230 °C and 260 °C. There is no significant difference in decomposition temperatures with different ligands or between the Mn2(CO2(CH2)nCO2)(OH)2 and Mn4(CO2(CH2)nCO2)3(OH)2 series.3.5 Magnetic properties of compound 2, Mn2(CO2(CH2)2CO2)(OH)2
Magnetic susceptibility measurements, carried out in an applied field of 100 Oe, suggest that compound 2 undergoes two magnetic transitions (see Fig. 11). As explained below, the first of these transitions, around 44 K, is attributed to two dimensional antiferromagnetic ordering within a layer. The second occurs at 36 K and is attributed to the onset of a three dimensional state with predominantly antiferromagnetic interactions. At higher temperatures compound 2 exhibits Curie–Weiss paramagnetism with fits to 1/χ yielding a Curie–Weiss theta of −112 K, and a magnetic moment, μeff, of 6.05 μB, close to the expected spin-only μeff for high spin Mn2+, 5.92 μB. The susceptibility increases significantly at 44 K, indicative of the first transition, while at 36 K the magnetic susceptibility increases sharply in the field cooled (FC) case and diverges greatly from the zero-field cooled (ZFC) measurement, suggesting that a transition to another magnetic phase occurs. Below this temperature the ZFC cooled measurement decreases to nearly zero while the FC curve rapidly increases, reaching a χ of 0.4 cm3 mol−1 at 1.8 K. This indicates a significant ferromagnetic component to its properties in the FC case. A plot of χmT versus temperature shows that the value of χmT increases sharply at both 44 K and 36 K, consistent with both magnetic phases having a ferromagnetic component, even in ZFC conditions (see Fig. S6†). The general trend of χmT with temperature is, however, to decrease from 3.3 cm3 mol−1 K at room temperature to nearly zero at 1.8 K, confirming that the dominant magnetic interactions in compound 2 are antiferromagnetic.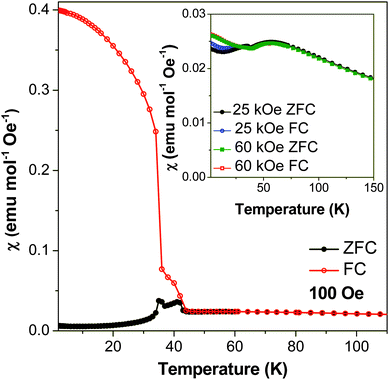 | ||
| Fig. 11 Plot of magnetic susceptibility of compound 2versus temperature at 100 Oe. Measurements in 25 kOe and 60 kOe are shown in the inset plot. | ||
The Curie–Weiss temperature is significantly greater than the temperature at which compound 2 orders magnetically, although not enough to indicate significant magnetic frustration. A plot of C/(χ|Θ|) − 1 as a function of T/|Θ| (where C = the Curie constant, χ = the magnetic susceptibility, and Θ is the Curie–Weiss theta) shows positive deviation from ideal Curie–Weiss behaviour near to the Curie–Weiss temperature, confirming that compound 2 has significant antiferromagnetic interactions well above the temperature at which magnetic ordering occurs (see Fig. 12).35 The magnetic interactions within a layer are likely to be much stronger in compound 2 than those between the layers due to the shorter intra-layer distances between Mn cations (cf. a Mn–O–Mn superexchange pathway of 4.3 Å within a layer to a Mn–ligand–Mn distance of 11.5 Å between layers). Therefore the antiferromagnetic interactions above the ordering temperature are most likely short range interactions within the layers.
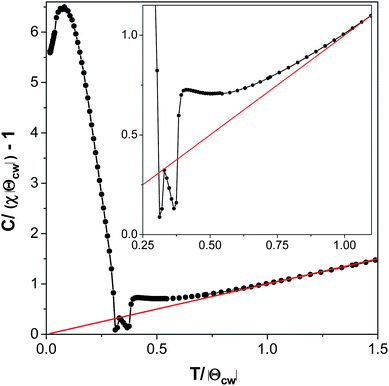 | ||
| Fig. 12 Plot of C/(χ|Θ|) − 1 versus T/|Θ| of compound 2 measured in a 100 Oe field in the zero-field case. The inset highlights the behaviour near the Néel temperature. | ||
The transition around 44 K appears to be somewhat broader than the transition near 36 K. While AC measurements do not indicate any frequency dependence of the ordering temperature of either transition, the intensity of the feature at 36 K has significant frequency dependence in both χ′ and χ′′, while that at 44 K does not (see Fig. S7†). This is consistent with the higher and lower temperature transitions being related to two dimensional and three dimensional ordering, respectively. The magnetic behaviour of compound 2 is consistent with the low temperature phase being a spin-canted antiferromagnet, which requires an applied magnetic field to induce the ferromagnetic component of their spins to align in the same direction throughout the material. This is most likely a result of weak magnetic coupling between layers.
Examination of an isothermal magnetisation loop collected at 1.8 K indicates clear, but very weak, magnetic hysteresis with very little remnant magnetisation (see Fig. 13a). This indicates that the antiferromagnetic canting angle must be very small, as would be expected for a magnetically isotropic cation. The magnetisation does not saturate, even under an applied field of 70 kOe. The derivative of the magnetisation loop shows two clear discontinuities at 10 kOe and 50 kOe with increasing magnetic field, suggesting the presence of field-induced magnetic transitions (see Fig. 13b). Magnetic susceptibility measurements carried out at 25 kOe and 60 kOe indicate that the compound retains behaviour similar to a spin canted antiferromagnet, although the FC and ZFC curves no longer diverge as significantly (see Fig. 11). The two dimensional ordering appears to occur at a slightly higher temperature in higher fields, around 55 K, while the temperature at which three dimensional ordering occurs remains the same.
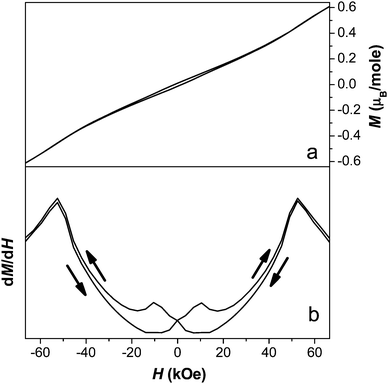 | ||
| Fig. 13 Plot of a) the isothermal magnetisation and b) the derivative of magnetisation measured at 1.8 K for compound 2. | ||
That compound 2 appears to exhibit low dimensional magnetic ordering at higher temperature before complete three dimensional ordering occurs around 36 K is consistent with the structure of this compound and studies of several other layered systems, particularly the transition metal phosphonates which have been studied extensively.36Neutron diffraction, particularly in combination with calorimetry measurements, would be a powerful technique for determining the precise nature of the magnetic phases with changing temperature and applied magnetic field. This is highlighted by several recent papers examining the magnetic structures of inorganic–organic hybrid frameworks.9,37 Unfortunately it was not possible to obtain either large single crystals or a large amount of a pure polycrystalline sample, which would be required for such a study. Similarly the morphology of the crystals of the other samples examined in this study prevents crystal picking from being used to obtain single phase samples suitable for studying their magnetic properties. This precludes a comparison of the magnetic properties of the different types of structures obtained in this work.
The temperature at which compound 2 orders magnetically is much higher than was found for the two previously known Mn succinates. One of these, Mn5(CO2(CH2)2CO2)4(OH)2, is highly magnetically frustrated and only exhibits weak antiferromagnetic interactions, while Mn(CO2(CH2)2CO2) is a purely compensated antiferromagnet and orders at around 10 K.9,10,14 Similarly to compound 2, Mn(CO2(CH2)2CO2) is known to undergo a number of magnetic phase transitions with both temperature and applied field; however, the transitions present in framework 2 have a more significant affect on the macroscopic magnetic properties of this compound than is the case in Mn(CO2(CH2)2CO2), where the changes are more subtle, and Mn(CO2(CH2)2CO2) does not show any obvious indications of lower dimensional magnetic order despite also featuring a layered structure. Compound 2 also orders at significantly higher temperatures than the other known Mn transition metal dicarboxylates, including the tartrates, which all order below 13 K.6,11,14,21,38 It is also higher than most other transition metal (CO2(CH2)nCO2) dicarboxylates, with n > 0.7,13,14,19,39
There are, however several Co, Ni and Cu frameworks that are reported to have comparable magnetic ordering temperatures.5,12,20,40 These include Ni7(CO2(CH2)CO2)4(OH)6(H2O)3·7H2O,5 which reportedly orders ferrimagnetically near 20 K, and Ni5(CO2(CH2)4CO2)2(OH)6, Ni5(CO2(CH2)6CO2)2(OH)6 and Co5(CO2(CH2)6CO2)2(OH)6,40 which are antiferromagnets below 27 K and 19 K and ferrimagnets below 16 K, respectively. Ni7(CO2(CH2)CO2)4(OH)6(H2O)3·7H2O has I1O1 connectivity and its layers contain chains with edge- and face-sharing NiO6 octahedra, while the related Ni5(CO2(CH2)4CO2)2(OH)6, Ni5(CO2(CH2)6CO2)2(OH)6 and Co5(CO2(CH2)6CO2)2(OH)6 phases have I2O1 connectively with layers of edge-sharing MO6. This suggests that the high ordering temperature of 2 may be related to the presence of the edge-sharing chains and hydroxide ligands, since it has these features in common with these relatively high-ordering compounds. Although the difference in cations in these compounds undoubtedly plays a major role in their magnetic properties, it is likely the Néel temperature of compound 2, 36 K, is further enhanced by the shorter connectivity between its layers. This reduces the difference in the strength of its intra- and inter-layer interactions, by strengthening the latter, and leads to it having a higher ordering temperature than the other four compounds despite its Curie–Weiss temperature being lower than the Ni containing compounds (cf. a value of Θ/TN of 3.1 for compound 2 with 5.1, 8.4 and 13.1 from previous reports of Ni7(CO2(CH2)CO2)4(OH)6(H2O)3·7H2O, Ni5(CO2(CH2)4CO2)2(OH)6 and Ni5(CO2(CH2)6CO2)2(OH)6).5,40
4 Conclusions
This study has explored the evolution of the structures of the transition metal linear dicarboxylates of the type Mn2(CO2(CH2)nCO2)(OH)2 and Mn4(CO2(CH2)nCO2)3(OH)2 (where 0 ≤ n ≤5) with changing ligand length. It was found that the Mn2(CO2(CH2)nCO2)(OH)2 series is stable for n = 0, 2, 4 and 5 and that all four compounds can be described as two dimensionally inorganically connected MnOx polyhedral layers bridged by dicarboxylate ligands in the third dimension. The structures adopted by this composition change significantly, but often subtly, with increasing n. While the oxalate and succinate members contain only octahedrally coordinated Mn, the adipate and pimelate compounds are the first examples of Mn linear dicarboxylates that contain trigonal bipyramidally coordinated Mn. As such this series represents a novel case where changing the ligand length can alter the coordination environment of the metal cations in a framework structure. It is thought that both the different connectivity and ability of the longer carboxylate ligands to accommodate higher torsion angles play significant roles in the formation of the different structures. In all cases the inorganic layers can be described as consisting of edge-sharing chains of MnOx polyhedra, which are interconnected in a corner-sharing fashion by μ3-coordinated hydroxide ligands. The Mn4(CO2(CH2)nCO2)3(OH)2 composition is only stable for n = 3 and 5 and consists of one dimensional inorganically connected double chains of MnOx polyhedra, which are bridged in the other two dimensions by the dicarboxylate ligands. Each thread of the chain contains alternating octahedrally and trigonal bipyramidally coordinated Mn and, similarly to the Mn2(CO2(CH2)nCO2)(OH)2 series, the two strands of a chain are interconnected viahydroxide ligands. It is thought that the ability of the longer-carboxylate ligands to accommodate higher torsion angles and their expression of (1111) ligand connectivity is also important to the formation of the Mn4(CO2(CH2)nCO2)3(OH)2 structure.Examination of the magnetic properties of Mn2(CO2(CH2)2CO2)(OH)2 indicates that this compound undergoes two dimensional antiferromagnetic ordering at 44 K before adopting a three dimensionally ordered spin canted antiferromagnetic state at 36 K. Compound 2 also undergoes two magnetic transitions with increased applied magnetic field at 25 kOe and 60 kOe, although the low temperature phase retains canted antiferromagnetic order at all fields examined. The magnetic ordering in this compound occurs at a significantly higher temperature than in most other known transition metal dicarboxylates.
Acknowledgements
The authors acknowledge the European Research Council for financial support. The authors thank the EPSRC National Crystallographic Service for obtaining the single crystal data from compounds 2–5 used in this study. We would also like to thank Dr Eun Sang Choi of the National High Magnetic Field Laboratory, Tallahassee, for some help with DC magnetic measurements and Sir Harold W. Kroto for access to these facilities.References
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 2006, 4780–4795 Search PubMed.
- (a) A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491–2510 CrossRef CAS; (b) G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC; (c) J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- (a) A. K. Cheetham and C. N. R. Rao, Science, 2007, 318, 58–59 CrossRef CAS; (b) C. N. R. Rao, A. K. Cheetham and A. Thirumurugan, J. Phys.: Condens. Matter, 2008, 20, 083202 CrossRef; (c) P. Jain, V. Ramachandran, R. J. Clark, H. D. Zhou, B. H. Toby, N. S. Dalal, H. W. Kroto and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 13625–13627 CrossRef CAS; (d) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353–1379 RSC.
- (a) Y. Kim and D.-Y. Jung, Bull. Korean Chem. Soc., 1999, 20, 827–830 CAS; (b) C. Livage, C. Egger and G. Férey, Chem. Mater., 2001, 13, 410–414 CrossRef CAS; (c) P. M. Forster and A. K. Cheetham, Angew. Chem., Int. Ed., 2002, 41, 457–459 CrossRef CAS; (d) N. Guillou, C. Livage and G. Férey, Eur. J. Inorg. Chem., 2006, 2006, 4963–4978 Search PubMed; (e) D. Ghoshal, A. K. Ghosh, G. Mostafa, J. Ribas and N. R. Chaudhuri, Inorg. Chim. Acta, 2007, 360, 1771–1775 CrossRef CAS.
- N. Guillou, C. Livage, W. van Beek, M. Noguès and G. Férey, Angew. Chem., Int. Ed., 2003, 42, 643–647 CrossRef.
- Y. Kim, Y. Park and D.-Y. Jung, Inorg. Chem. Commun., 2004, 7, 347–349 CrossRef CAS.
- C. Livage, C. Egger and G. Férey, Chem. Mater., 1999, 11, 1546–1550 CrossRef CAS.
- L.-S. Long, X.-M. Chen, M.-L. Tong, Z.-G. Sun, Y.-P. Ren, R.-B. Huang and L.-S. Zheng, J. Chem. Soc., Dalton Trans., 2001, 2888–2890 RSC.
- P. J. Saines, J. R. Hester and A. K. Cheetham, Phys. Rev. B: Condens. Matter Phys., 2010, 82, 144435 Search PubMed.
- P. J. Saines, B. C. Melot, R. Seshadri and A. K. Cheetham, Chem.–Eur. J., 2010, 16, 7579–7585 CrossRef CAS.
- Y. Kim, E. Lee and D.-Y. Jung, Chem. Mater., 2001, 13, 2684–2690 CrossRef CAS.
- E. G. Bakalbassis, M. Korabik, A. Michailides, J. Mrozinski, C. Raptopoulou, S. Skoulika, A. Terzis and D. Tsaousis, J. Chem. Soc., Dalton Trans., 2001, 850–857 RSC.
- (a) C. Ruiz-Perez, J. Sanchiz, M. H. Molina, F. Lloret and M. Julve, Inorg. Chem., 2000, 39, 1363–1370 CrossRef CAS; (b) F. S. Delgado, J. Sanchiz, C. Ruiz-Perez, F. Lloret and M. Julve, CrystEngComm, 2004, 6, 443–450 Search PubMed.
- Y. Kim, D.-Y. Jung, K.-P. Hong and G. Demazeau, Solid State Sci., 2001, 3, 837–846 CrossRef CAS.
- R. Vaidhyanathan, S. Natarajan and C. N. R. Rao, Dalton Trans., 2003, 1459–1464 RSC.
- (a) P. M. Forster, A. R. Burbank, C. Livage, G. Férey and A. K. Cheetham, Chem. Commun., 2004, 368–369 RSC; (b) P. M. Forster, N. Stock and A. K. Cheetham, Angew. Chem., Int. Ed., 2005, 44, 7608–7611 CrossRef CAS.
- (a) B. H. O'Connor and E. N. Maslen, Acta Crystallogr., 1966, 20, 824–835 CrossRef CAS; (b) Y.-Q. Zheng and J.-L. Lin, Journal of Coordination Chemistry, 2008, 61, 3420–3437 Search PubMed.
- (a) Y.-Q. Zheng, J.-L. Lin and H.-L. Zhang, Z. Kristallogr. - New Cryst. Struct., 2000, 215, 535 Search PubMed; (b) Y.-Q. Zheng, A.-Y. Pan and J.-L. Lin, Z. Kristallogr. - New Cryst. Struct., 2001, 216, 267 Search PubMed; (c) T. A. Bowden, H. L. Milton, A. M. Z. Slawin and P. Lightfoot, Dalton Trans., 2003, 936–939 RSC.
- E. Lee, Y. Kim and D.-Y. Jung, Inorg. Chem., 2002, 41, 501–506 CrossRef CAS.
- C. Livage, C. Egger, M. Nogues and G. Férey, C.R. Acad. Sci. IIC - Chem., 2001, 4, 221–226 Search PubMed.
- (a) A. Murasik and P. Fischer, J. Phys. C: Solid State Phys., 1987, 20, 2247–2259 CrossRef CAS; (b) I. Sledzinska, S. Mitsuda, S. Simizu, J. Lukin, S. A. Friedberg, B. X. Yang and G. Shirane, Phys. Rev. B, 1988, 38, 9035–9039 Search PubMed; (c) Y.-G. Wei, S.-W. Zhang, M.-C. Shao, Q. Liu and Y.-Q. Tang, Polyhedron, 1996, 15, 4303–4305 CrossRef CAS; (d) T. K. Maji, S. Sain, G. Mostafa, T.-H. Lu, J. Ribas, M. Monfort and N. R. Chaudhuri, Inorg. Chem., 2003, 42, 709–716 CrossRef CAS.
- Z. A. D. Lethbridge, A. F. Congreve, E. Esslemont, A. M. Z. Slawin and P. Lightfoot, J. Solid State Chem., 2003, 172, 212–218 CrossRef CAS.
- J. Soleimannejad, H. Aghabozorg, S. Hooshmand, M. Ghadermazi and J. Attar Gharamaleki, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, E63, m2389–m2390 Search PubMed.
- O. K. Farha, K. L. Mulfort, A. M. Thorsness and J. T. Hupp, J. Am. Chem. Soc., 2008, 130, 8598–8599 CrossRef CAS.
- B. A. Hunter and C. J. Howard, A computer program for Rietveld analysis of X-ray and neutron powder diffraction patterns, Lucas Heights Laboratories, 1998 Search PubMed.
- (a) I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Sci., 1985, B41, 244–247 CrossRef CAS; (b) N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, B47, 192–197 CrossRef CAS.
- D. Massiot, S. Drumel, P. Janvier, M. Bujoli-Doeuff and B. Bujoli, Chem. Mater., 1997, 9, 6–7 CrossRef CAS.
- A. Thirumurugan and C. N. R. Rao, J. Solid State Chem., 2008, 181, 1184–1194 CrossRef CAS.
- (a) C. Lee, C. Mellot-Draznieks, B. Slater, G. Wu, W. T. A. Harrison, C. N. R. Rao and A. K. Cheetham, Chem. Commun., 2006, 2687–2689 RSC; (b) L. N. Appelhans, M. Kosa, A. V. Radha, P. Simoncic, A. Navrotsky, M. Parrinello and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 15375–15386 CrossRef CAS.
- H. H. M. Yeung, M. Kosa, M. Parrinello, P. M. Forster and A. K. Cheetham, Cryst. Growth Des., 2011, 11, 221–230 Search PubMed.
- (a) Y.-Y. Lin, Y.-B. Zhang, J.-P. Zhang and X.-M. Chen, Cryst. Growth Des., 2008, 8, 3673–3679 CrossRef CAS; (b) X.-J. Zhang, Y.-H. Xing, J. Han, X.-Q. Zeng, M.-F. Ge and S.-Y. Niu, Cryst. Growth Des., 2008, 8, 3680–3688 CrossRef CAS.
- Y. Kim, H. Jin Nam and D.-Y. Jung, Chem. Lett., 2009, 38, 72–74 Search PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, B58, 380–388 CrossRef.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS; (b) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, D65, 148–155 CrossRef CAS.
- B. C. Melot, J. E. Drewes, R. Seshadri, E. M. Stoudenmire and A. P. Ramirez, J. Phys.: Condens. Matter, 2009, 21, 216007 CrossRef.
- (a) D. Visser, S. G. Carling, P. Day and J. Deportes, J. Appl. Phys., 1991, 69, 6016–6018 CrossRef CAS; (b) S. G. Carling, P. Day, D. Visser and R. K. Kremer, J. Solid State Chem., 1993, 106, 111–119 CrossRef CAS; (c) S. G. Carling, P. Day and D. Vissen, Inorg. Chem., 1995, 34, 3917–3927 CrossRef CAS; (d) E. Fanucci, J. Krzystek, M. W. Meisel, L.-C. Brunel and D. R. Talham, J. Am. Chem. Soc., 1998, 120, 5469–5479 CrossRef CAS; (e) J. T. Culp, G. E. Fanucci, B. C. Watson, A. Nicole Morgan, R. Backov, H. Ohnuki, M. W. Meisel and D. R. Talham, J. Solid State Chem., 2001, 159, 362–370 CrossRef CAS.
- (a) R. A. Mole, J. A. Stride, A. S. Wills and P. T. Wood, Physica B, 2006, 385–386, 435–437 CrossRef CAS; (b) R. A. Mole, J. A. Stride, P. F. Henry, M. Hoelzel, A. Senyshyn, A. Alberola, C. J. Gómez-García, P. R. Raithby and P. T. Wood, Inorg. Chem., 2011, 50, 2246–2251 Search PubMed; (c) P. J. Saines, H. H. M. Yeung, J. R. Hester, A. R. Lennie and A. K. Cheetham, Dalton Trans., 2011, 40, 6401–6410 RSC.
- (a) A. Beghidja, P. Rabu, G. Rogez and R. Welter, Chem.–Eur. J., 2006, 12, 7627–7638 CrossRef CAS; (b) E. Coronado, J. R. Galàn-Mascarós, C. J. Gómez-García and A. Murcia-Martínez, Chem.–Eur. J., 2006, 12, 3484–3492 CrossRef CAS.
- (a) N. Guillou, C. Livage, M. Drillon and G. Férey, Angew. Chem., Int. Ed., 2003, 42, 5314–5317 CrossRef; (b) F. S. Delgado, M. Hernandez-Molina, J. Sanchiz, C. Ruiz-Perez, Y. Rodriguez-Martin, T. Lopez, F. Lloret and M. Julve, CrystEngComm, 2004, 6, 106–111 RSC.
- A. Mesbah, A. Carton, L. Aranda, T. Mazet, F. Porcher and M. François, J. Solid State Chem., 2008, 181, 3229–3235 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic conditions for crystal preparation and details of structural determination including CIFs; plots of powder diffraction, thermogravimetric and magnetic data, and pictures of the carboxylate ligand coordination and the asymmetric unit of compounds 4 and 6 are available as supplementary information. CCDC reference numbers 823424–823429. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00253h |
| This journal is © The Royal Society of Chemistry 2011 |